 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anders Brahme | -- | 5205 | 2023-09-01 04:12:48 |
Video Upload Options
The new biological interaction cross-section-based repairable–homologically repairable (RHR) damage formulation for radiation-induced cellular inactivation, repair, misrepair, and apoptosis was applied to optimize radiation therapy. This new formulation accurately describing the cell survival and apoptosis and implies renewed thinking about biologically optimized radiation therapy, suggesting that most TP53 intact normal tissues are low-dose hypersensitive (LDHS) and low-dose apoptotic (LDA). This generates a fractionation window in LDHS normal tissues, indicating that the maximum dose to organs at risk should be ≤2.3 Gy/Fr, preferably of low LET. This calls for biologically optimized intensity modulated treatments using a few high tumor dose photon or light ion beams, preferably lithium to boron ion thereby avoiding secondary cancer risks and generating true tumor cure without risk for caspase-3-induced accelerated tumor cell repopulation.
1. TP53 Damage Response
The new increased flexibility of describing the shape of the cell survival curve has resulted in a significantly improved description of low- and high-dose- apoptosis and LET radiation induced cell survival in mutant, wild-type, and repair-gene knockout cells. It is well known that the nonhomologous end-joining (NHEJ, [1][2]) pathway is the dominating DNA repair process of low LET, and it is very fast. The Ku70, Ku80, and DNApkcs hetero dimer bind together the broken DNA ends in a few seconds and simultaneously recruit p53 [3], such that high-dose and LET local damage can also be correctly repaired. This process is essential also for high LET ions where the MRN dimer complex often replaces the Ku-DNApk hetero dimer especially if the cell is in the late S or G2 phases of the cell cycle, and it may need the homology-searching mechanism of homologous recombination (HR) for high-fidelity repair [4][5]. This makes HR more important than NHEJ at very high LETs, partly also as less low-LET-type damage is induced ([2] with three ions (B, C, and N) and three cell lines, [6]: C, [7]: C). Most interestingly, the new repair formulation allows for the approximate quantification of concurrent independent NHEJ and HR repair, as well as the HR repair of NHEJ misrepair, alone or concurrent with other ongoing HR repair processes. These newly established DNA repair terms make it possible to describe cellular repair far beyond the conventional linear quadratic model (LQ). The new DNA repair based formulation thus works almost as a kind of Kepler’s low for cell survival, allowing to estimate the interaction of NHEJ and HR based on observed cell survival (similar to how continuously new outer planets were discovered based on deviations in the orbits of the inner ones). Interestingly, the new DNA-repair-based formulation inherently describes LDHS and LDA as they are linked to the DNA repair system of most, if not all, normal tissues, as described in more detail in Figure 1. This figure also illustrates how the TP53 gene works as a complex cellular controller by determining how the DNA damage structures should best be repaired, and whether senescence and apoptosis are needed, as shown in Figure 1 and Figure 2 [1][3][8][9][10][11]. After DNA damage by radiation, it works principally through the augmented phosphorylation of three serine 15, 20, 46 sites [10] following the increase in the extent of damage through some of its key upstream proteins such as ATM, CHK2, and p38K as well as 53BP1 or BRCA1,2 to signal whether the currently most optimal repair process is NHEJ or HR. Most tumor cell lines suffer from a TP53 mutant gene, making the early/low dose (0.25–1 Gy) radiation response more gradual without LDHS and low-dose LDA, as shown in the cell survival insert in Figure 1. As TP53 intact cells are irradiated after about ¼ Gy, ATM is auto-phosphorylated and in turn phosphorylates the serine 15 site on p53 to initiate NHEJ repair. After a total of ½ Gy or about 18 DSBs, CHK2 is also phosphorylated and phosphorylates the serine 20 site on p53 to achieve fully efficient DNA repair with both HR and NHEJ [1][2][12] from 0.5-2 Gy. As seen from the lower left insert in Figure 1, this last step results in a switch in normal tissue sensitivity from an initial LDHS and LDA stage, before the full serine 15 and 20 phosphorylation of p53 generates a more radiation-tolerant state. After that, the cellular repair system is fully activated and functional, with reduced cell loss and almost a survival plateau around 1-2 Gy [1][2][8][13][14][15]. It is clear from Figure 1 and Figure 2 and the Graphical Abstract that tumor doses well above 2 GyE are needed for cure, and the normal tissue dose should be just above 2 Gy or below to ensure optimal radiation recovery, as clearly shown in Figure 2 by the pale dotted tangent line to the survival curve through the origin. The LDA and LDHS of normal tissues cause about 5–15% acute low-dose apoptosis (Figure 1 and Figure 2 and [8][16]), but interestingly, most likely, due to the compensating measure of caspase-3-induced cellular repopulation [17], late effects are few.
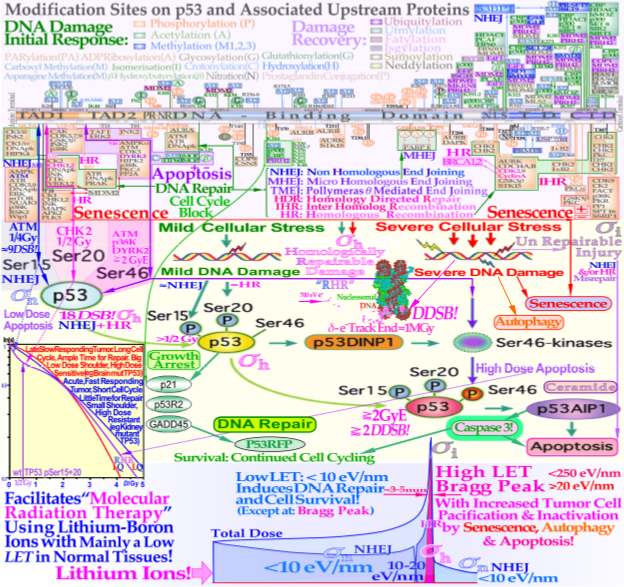
Figure 1. The complex response of the TP53 gene (upper third) to mild and severe genetic stress largely determines the cellular response to radiation [1][2][3][8][9][10][12][16]. Mild stress phosphorylates the serine 15 and 20 sites on p53 via ATM and CHK2, resulting in cell cycle block and DNA repair. This results in LDHS in normal tissues, but generally not in tumors, often with a mutant TP53 gene (as seen in the cell-survival insert (simplified middle third)). Local high doses or high ionization densities resulting in DDSBs (dual double-strand breaks [1][4][18]) increase the severity of the damage, also phosphorylating the serine 46 site, e.g., via p38K or ATM, and a high-dose apoptotic (HDA) response may be triggered. Lithium ions allow for unique therapeutic use by inducing a massive apoptotic–senescent tumor cell response, mainly within the Bragg peak (σh homologically repairable damage and σi direct inactivation cross-sections [1][2]). However, in front of and beyond the Bragg peak, the LET is low, and rapidly and easily repairable nonhomological damage is mainly induced (lower third, σn cross-section [1][2][19][20]). The upper third shows the inner workings of p53 in its complex downstream pathways (cf. [21](Figure 1)).
This will re-establish homeostasis in normal tissues and thus minimize late normal tissue damage, but it may sometimes also repopulate malignant tumor clonogens if they are not entirely eradicated by the treatment [17]. This means that LDA really protects normal tissues from potential low-dose mutations before NHEJ and HR are fully functional and can address the damage. Furthermore, in terms of radiation therapy, it means that the fully functional DNA repair system should be optimally utilized until the more severe high-dose apoptosis by serine 46 phosphorylation of p53, sets in after about 2–2.5 GyE, as seen in Figure 1.
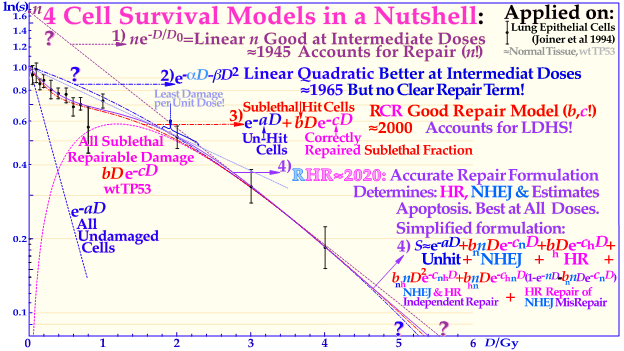
Figure 2. The development of models defining the shape of the cell survival curve during the last ≈ hundred years, from the linear exponential model with back extrapolated effective initial cell number (n, Ln) to the currently dominating linear quadratic formula (LQ), which does not even accurately account for cell repair as Ln does. The more recent repairable–conditionally repairable model handles the cellular repair considerably better and separates it from un-hit survival, whereas the most recent repairable homologically–repairable (RHR) formulation further accounts separately for nonhomologous and homologous recombination repair, as shown in the lower right corner, and can estimate the apoptotic fraction and individual repair processes. The least damage per unit dose is obtained between 1.8 and 2.3 Gy/Fr, as indicated by the fine dotted blue line with the shallowest slope possible through the unit survival point. For further details see [4].
2. Cell Survival
Interestingly, a recent publication on DNA repair [2] demonstrated that this early low-dose cell loss before establishing full repair efficiency is mainly due to p53 LDA induction, in general agreement with [8][16] and the present study’s Figure 1 for a TP53 intact tumors. This direct LDA and LDHS is most likely a general property of intact normal tissues and the cell’s natural way to protect itself from low-dose, potentially cancerous, mutations before full repair efficiency is established [2][8][15]. Standard therapeutic 2 Gy fractions generate 75 DSBs and possibly one or two dual DSBs (DDSBs) that may be inducing high-dose apoptosis (HDA) via serine 46. As seen from the insert in Figure 1, many tumor cell lines, and more than 50% of all tumors generally, have a mutant TP53 gene that commonly eliminates most of LDHS and early LDA to obtain a radiation-resistant LQ-like shoulder. As shown for mouse embryo fibroblasts with key repair genes knocked out, both CHK2-/- and particularly ATM-/- cells lose all the LDHS of wild-type (wt) cells, highlighting the importance of the low-dose phosphorylation steps [2] and [22]. Obviously, there are also some wt TP53 and wt ATM tumor cell lines that may show the LDHS property. The most recent RHR paper also investigated LDHS and LDA for light ions, demonstrating that it generally peaks at low-LET ions (≈20 eV/nm, see Figure 3), where, for reasons related to ion interaction physics, the highest low-energy δ-electron production per unit dose occurs, as the LET is low but sufficient to induce apoptosis, and the ion fluence per unit dose is highest. Thus, apoptosis peaked here (31% measured at 3 Gy of 40 eV/nm B5+ [2]), and a clear but small LDHS was observed [23] mainly due to the early dual NHEJ only and HR misrepair (5% at 0.5 Gy, calculated [2]. The 31% value was not just LDA but mainly serine-46-induced HDA. So, even if there is a weak low-LET ion LDHS and LDA, it is most likely not sufficient to establish a real light ion fractionation window with carbon ions but surely with the entrance plateau and fragmentation tail of lower-LET lithium ions (in Figure 1 [2][19], in Figure 2 [2][13] (with 60Co) and Figure 4 (with 60Co and mut p53-reactivated)), as well as for lowest-LET boron ions and 60Co. The Bragg peak should always be reserved for target tissues. Thus, HDA is therapeutically very valuable [24], and even more so, the often associated senescence [1][2][25][26][27], also described in Figure 1, since it does not cause later problems with caspase-3 [17]. To further elucidate the development of the shapes of the cell survival curve, some key steps are summarized in Figure 2, considering the normal lung epithelial cell data [1][2][13]. During the 1940s to 1960s, the logarithmic linear model (Ln) was established, with a back extrapolated (slope D0) initial cell number (n) larger than 1 to account for cellular division and repair during irradiation. Alternatively, the so-called quasi-threshold dose (Dq) was also used to indicate that the linear extrapolation seemed to start from a dose that was wasted due to tumor cell repopulation and repair. Clearly, this was a rather crude way to describe the early cell survival that often was of low clinical importance at the time but not really today [4]. From the late 1960s, the still currently dominating linear quadratic model could better describe the slight curvature of the quasi-linear high-dose cell survival by its α and β factors of most tumors but did not account so well for the low- and high-dose survival, at least for most normal tissues, as seen in Figure 2. It also misses a true repair term, as a high repair requires a high β, but that means less survival since the term is negative, so α needs to be reduced to better describe the survival, which may only work in a small-dose region and α loses its original meaning. This illogical effect, and the fact that it gives a poor description of LDHS normal tissue survival, has misled two generations of radiation biologists to trust it rather uncritically. The beauty of the third repairable–conditionally repairable (RCR) model is that it is very simple and describes what occurs at large to the cells using Poisson statistics with a simple exponential term for missed cells and a linear exponential term for the correctly repaired fraction of sublethally hit cells. Thus, it is a logical continuation of the Ln expression, as seen in Figure 2 and [28]. It therefore can describe the LDHS quite well and solves the repair and overkill problem at high doses seen with the LQ model. The repairable–homologically repairable formulation (RHR) goes a few steps further by separately accounting for the two major DNA repair pathways, as mentioned above, and their associated misrepair processes, as seen in the lower right corner of Figure 2. This model can therefore handle, e.g., cell lines with mutant and/or knocked-out repair genes, high and low LETs, and apoptosis induction [1][2]. It is thus extremely important to consider the significant differences between the cell survival of most tumors and generally LDHS normal tissues when designing optimal radiation therapy protocols. It is unfortunate that the bulk of established tumor cell lines mostly suffer from TP53 and associated mutations, so they can easily grow in the lab and lead to the assumption that all cell lines have LQ-like shoulders, almost making the LQ model a dogmatic model of true cell survival. In fact, it is most likely that all intact normal tissues have wild-type TP53 and ATM genes, and thus are linked to LDHS and LDA, as recently indicated [1][2][8], and it is probably an inherited growth advantage to avoid cancerous transformations after low-level genetic damage that may not be correctly repaired. The intriguing reason why it went undetected for such a long time is that too few studies were conducted on live normal tissues at low doses and with sufficient accuracy (until Joiner [29]), but also because the associated accelerated repopulation via caspase-3 tries to compensate for LDA and LDHS at the end of irradiation to re-establish tissue homeostasis [17]. The relative apoptotic effectiveness (RAE) value of about 3.4 has been noted for low-LET boron ions around 40 eV/nm, whereas the peak relative biological effectiveness (RBE) was found to be about 3.5 but closer to an LET of about 160 eV/nm, as seen in Figure 3.
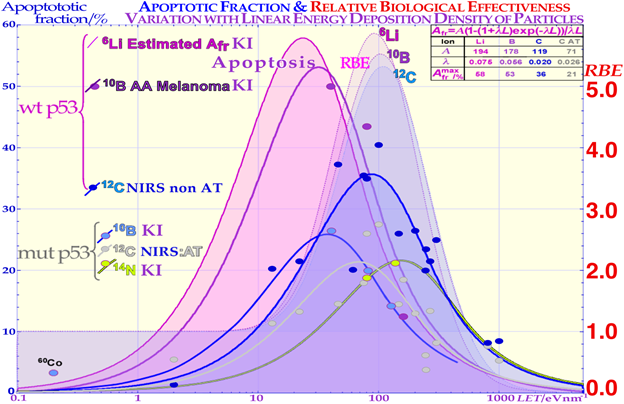
Figure 3. Comparison of the induced apoptosis (programmed cell death shown with solid lines and experimental dots) and the clonogenic survival measured by the RBE (pale-shaded dashed areas) as a function of increasing LET values. The induction of apoptotic cell kill depending on the status of the p53 pathway of the cells is also shown. When some part of the p53 pathway is mutant, AFr is reduced to about half its value for normal wt p53 and non-AT cell lines that are intact on the ATM gene upstream of p53. Interestingly, the AFr peaks occur at lower LET values than the RBE peaks due to the higher flux density of ions and apoptotic events per unit-absorbed dose. KI is Karolinska Institutet Stockholm Sweden, and NRIS is National Institute of Radiological Sciences, Chiba, Japan, and the table is used for extrapolation to lithium ions and discussed in the text.
3. Apoptosis Induction
The new increased flexibility to describe the shape of the cell survival curve and the major forms of interaction between HR and NHEJ, such as homologous repair of nonhomologous misrepair, are accounted for, and so is the probability of inducing apoptosis as a result of nine different types of misrepair processes [2]. The increase in apoptosis with doses at varying LETs provides a very good fit to the experimental data [2], mean error <≈0.5%). Interestingly, dual concurrent HR and NHEJ misrepair mechanisms in the same cell nuclei dominate at medium LETs, whereas concurrent and only HR misrepair mechanisms fully dominate at severely high LETs, as practically all types of such damage require HR processing [2][4][19]. In low-LET 60Co, almost all misrepair processes contribute via NHEJ that now dominate cell repair (see [2] for more details). It is indeed seen that low-LET ions produce the highest LDA and HDA per unit dose since the fluence and the number of ions are reduced by half each time the LET is doubled, and so is the apoptosis, as described in more detail in [2]. The upper right corner of Figure 3 includes the theoretical expression for apoptosis induction showing the increase by lowering the LET by its last 1/LET term that breaks down when the LET is too low, so very litle apoptosis can be induced. From Figure 3, it is seen that lower LETs (≈20 eV/nm) are preferable to optimize tumor apoptosis and senescence simultaneously, as they are also lowered in normal tissues, and this may generally be better than optimizing the total relative biological effectiveness (RBE) and LET in the tumor and consequently maximizing the hypoxic cell kill. Unfortunately, this is directly associated with adverse effects on normal tissue damage in front of and behind the tumor [1][2][9][30], which posed clinical challenges when using neutron and Neon ions [31] but also to some extent Carbon ions. The optimal therapeutic choice may also depend on the use of adjuvant therapies, e.g., to enhance antitumor immune reactions or ROS effects [2] as discussed next.
4. Reactivation of Mutant TP53
The cell survival effect of the mutant p53-reactivating compound PRIMA-1 on the TP53 mutant SCLC cell line U1690 after exposure to 60Co γ-rays is shown in detail in Figure 4. The curves with PRIMA-1 are here shown as solid lines, and the plain SCLC cell line U1690 without PRIMA-1 are shown as dashed lines, with arrows indicating the change in survival by the presence of PRIMA-1 during irradiation. At higher doses, the relative survival loss is quite large (almost 27% at 4 Gy, see Figure 4). Therefore, there would be a clinical advantage of using high doses per fraction with PRIMA-1. There is an increased HR repair alone (fh by almost 50%) but also to fix NHEJ misrepair (l is more than doubled), indicating that the HR pathway is largely improved through p53 reactivation, partly also increasing the very low-dose apoptosis [2] which is an advantage as it mainly increase tumor cell kill. The recovery of the tumor apoptosis with
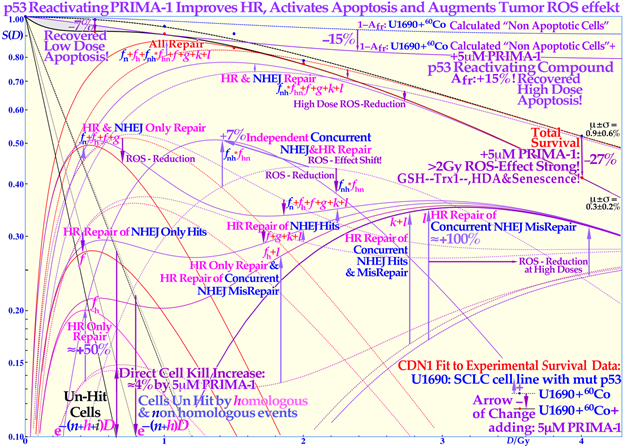
Figure 4. Increased LDA and HDA via mutant TP53 reactivation. The change in fractional cell survival, S(D), and major DNA repair processes with PRIMA-1 (solid lines) and without (dotted lines) for U1690 SCLC cells with mutant TP53 irradiated using 60Co γ-rays are shown. Arrows indicate the change after adding 5 μM PRIMA-1 for 14 h (10 h before to 4 h post-irradiation).
Even if the apoptotic survival change is small, about 7% at low doses (LDA) and up to 15% at high doses (HDA), there are large changes in the reparability of radiation damage with PRIMA-1 added, as seen in the HR-only repair (fh ≈ +50%) and the HR repair of NHEJ misrepair (l ≈ +100%) and sum of all repair terms that even compensate somewhat for the increased apoptosis via the reactivated mutant p53. The shaded low-dose area between the calculated apoptotic cell survival and measured clonogenic survival is due to apoptotic loss before the full activation of p53 at serine 15 and 20 via the checkpoint kinases ATM and Chk2 (cf. Figure 1 and [1][2][12]). The high-dose loss in cell survival is most likely due to PRIMA-1-induced augmented toxicity through the increasing associated ROS production (PRIMA-1 inhibits the enzyme thioredoxin reductase 1 and thioredoxin and decreases cellular glutathione levels), and increasing HDA and senescence [2][14][15][22]. The mean error μ and standard deviation σ are also shown all below ≈1% but obviously much higher in some of the individual components (≈10%). The volume of data that can be estimated using the new, more flexible, and thus probably more accurate, cell survival and DNA repair formulation is striking. Updated from [2] with LDA and HDA and the effects of ROS. CDN1: one-dimensional closest distance norm (not least square, see [1]). The original U1690 SCLC cell survival data were provided by Margareta Edgren at KI 2003, and the new RHR formulation significantly helped the interpretation of the wide range of effects of PRIMA-1 (f, g, k, l are repair fractions in which HR fixes various NHEJ misrepair and damage, as indirectly explained in this Figure, fh plain HR, and fn plain NHEJ; see [2][4] for further details).
PRIMA-1 is more than tenfold increased below 0.15 Gy, six fold at about ½ Gy (LDA), fourfold at 1 Gy, and more than doubled at high doses (HDA) with PRIMA-1. Due to the wide therapeutic effect spectrum of the active component MQ of APR 246 and PRIMA-1 [2], both on reactive oxygen species (ROS), inhibiting the enzyme thioredoxin reductase 1 and thioredoxin and decreasing cellular glutathione levels and increasing apoptosis, it is difficult to say exactly what caused the increase in HDA apoptosis by ≈15% in the current study. Even if PRIMA-1 increased the HR only, and its repair of NHEJ misrepair, this probably occurred due to improved HR initiation via TP53, there are also indications that apoptosis could be caused by direct mitochondrial effects or via caspase-3, and the cell cycle block via p21 is not generally restored either [32][33]. This latter fact may even be an advantage for cancer treatment as tumor cells will continue cycling and incorporate damaged DNA in their genomes without repair, and they may finally end up in a mitotic catastrophe situation like in classical radiation therapy. The first very interesting cell culture study of using APR 246 on colorectal cancer combined with radiation was recently published, and mut TP53 showed almost wt TP53 response with 5 μM APR 246, whereas TP53 Null cells were only half as responsive [34] generally consistent with Figure 4. At 7.5 μM, the mut cell line was even more responsive than the wt, which should be a valuable treatment property. In a tumor growth assay, 20 μM APR 246 and 6 Gy both halved the mut xenograft size, whereas the combination brought it down to ≈1/5, but wt showed only a 30% APR 246 reduction, and null and mut cells were similar [35]. Interesting Venn diagrams of significantly enriched pathways and genes for combined and radiation-alone treatments were also included for wt, mut, and TP53 null cells.
5. The Fractionation Window
According to Figure 1, the fractionation window is to a significant part due to the LDA of most normal tissues, and it is essential to maximize the use of it in cancer treatment to maximize the normal tissue tolerance. Normally, the 2 Gy dose is prescribed to the tumor, and more specifically the internal target volume [36], as the fractionation widow was established in the era of parallel-opposed beams. However, the recognition that most normal tissues are low-dose hypersensitive, and most tumors are not, generates a fractionation window between about 1.8 and 2.3 Gy, shown in Figure 1, Figure 2 and Figure 5 (for details, see [1][2][37][38]), which necessitates the reconsideration of the 2 Gy/Fr prescription, to ensure that normal tissues surrounding a tumor should get less than 2,3 Gy for minimal high dose damage (HDA). Furthermore, it is important to point out that the existence of a ≈ 2 Gy/Fr established optimal treatment regiment, which was identified some 80 years ago and may be expressed differently as the existence of a “fractionation window” where radiation therapy works well. This is in fact the most significant clinical proof for the general existence of low-dose hypersensitivity in most normal tissues not least because it was established in the era of parallel-opposed beams, with almost equal doses to the tumor and organs at risk. In addition, the molecular mechanisms seen in Figure 1 explain how it works in finer molecular details, and the new radiation biology in Figure 2 shows how the ≈2 Gy/Fr tangent to the survival curve from the origin (pale dotted line in Figure 2) reflects the shallowest and least damaging irradiation effect on normal tissues when having to deliver high doses to deep-seated tumors. In Figure 5, the effect of varying the dose per fraction on normal tissue damage is illustrated based on the experimental survival data in Figure 2 and more clearly indicates the function of the clinical fractionation window in reducing normal tissue injury. Both lower (yellow) and higher (violet) doses per fraction cause a higher probability of damage, so it is preferable to stay within 1.8–2.3 Gy/Fr, at least when passing through the lungs. Unfortunately, the conventional LQ model is blind to and does not describe this low-dose-per-fraction damage in normal tissues (except for Joiner’s nice but complex modification, handled better using the much simpler RCR formula, Figure 2) and of course also with the more complex and exact RHR formulation [1][2], which explains what really occurs and also eliminates other LQ shortcomings.
The fractionation window is important to consider in radiation therapy optimization in order to eradicate a tumor with minimal normal tissue damage: a too-low dose/Fr (<1.5 Gy) causes more accumulated LDHS and LDA in normal tissues, and a significantly higher dose than ≈ 2.5 Gy may result in more “double trouble” with both a high dose and high dose/Fr, considering the fact that the same curative dose need to be imparted to the gross tumor in both cases.
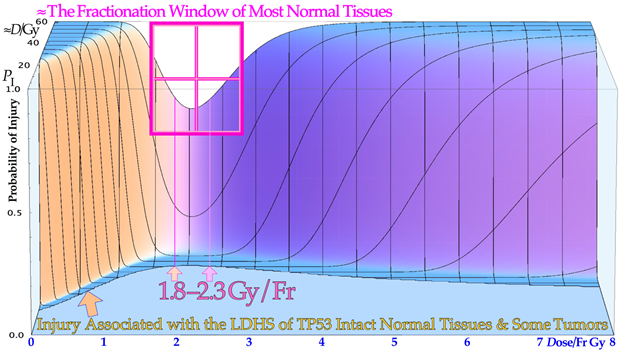
Figure 5. The effect of varying the doses per fraction on normal tissue damage is illustrated based on the experimental survival data in Figure 2 for lung epithelial cells. The low-dose hypersensitivity of most normal tissues establishes a therapeutic fractionation window of opportunity to cure cancer with minimal normal tissue damage. The new cell survival models discussed here can fine-tune the dose range to 1.8–2.3 Gy, as shown here for the lungs, a common organ at risk in the thorax region.
6. Optimal Weekly Time Dose Fractionation
To further minimize normal tissue damage as far as possible, it is desirable to introduce an optimal weekly dose fractionation schedule where the DNA repair of normal tissues is really taken into account to minimize their injury and optimally recover from weekly radiation damage. Up to about 50% higher tumor doses should preferably be delivered Monday morning, Wednesday midday, Friday evening, and the last evening of treatment. This will allow optimal use the weekend and end of therapy for maximal normal tissue recovery (see the dashed line in the Graphical Abstract, [1][39] and preferably still staying below the 2.3 Gy/Fr to organs at risk. This will especially optimize the weekly HR recovery towards ≈72+ hrs since NHEJ achieves its repair quite well in the 24+ h from day to day, as shown in the lower right part of the Graphical Abstract. This fractionation window advantage works well for low-LET radiations but also for the lightest ions with mainly a low LET in normal tissues. A gain in complication free cure of up to about 12 % should be possible by this approach as shown on the right hand scale! Interestingly, a somewhat similar thinking was recently presented by Yarnold and coworkers [40]. There are therefore definitely many reasons to re-evaluate the time–dose fractionation along the current ideas and those presented in a previous study on DNA repair [1] to really introduce a major paradigm shift in curative radiation therapy thinking
7. Secondary Cancer Induction
To further illustrate the power of quantifying apoptosis, in Figure 6 the probability of inducing a secondary cancer is illustrated based on experimental cell survival and apoptosis data [2]. It is unlikely that the apoptotic fraction will contribute to secondary cancer induction (except possibly in TP53 mutant cell lines), so it is useful that this fraction can be estimated using the new RHR formula and be removed from other forms of misrepair to more accurately describe the cells that are potentially capable of generating secondary cancer. This cell fraction, as seen in Figure 6, has its peak in the 1–4 Gy/Fr range, so in radiation therapy optimization, it is desirable to minimize this volume as much as possible in normal tissues. Figure 6 also shows that the maximal risk is the smallest for low-LET ions (blue-shaded) largely due to their high apoptotic induction. The real secondary cancer risk may be in the order of 5% of the maximal values in Figure 6 or less. Obviously, these experimental data are not really relevant for the surrounding normal tissues that may receive a fair amount of dose and are at risk for secondary cancer. However, the present tumor cell line is at least wt TP53 so probably not the most extremely mutated one and may, in first approximation, be assumed to represent both normal and tumor tissue risks. Furthermore, the dose axis is clearly the dose per fraction, so it means that the total dose is increased by the number of fractions. Interestingly, the curves can also be used to optimize the dose per fraction, and then a higher dose per fraction of higher LETs (e.g., 2 Gy at 160 eV/nm corresponding to ≈6 GyE) may even be better than the lowest peak value at 40 eV/nm. In fact, if the plot is instead drawn as a function of dose equivalent (dose x RBE; the 50% survival RBEs are given in the figure), all the maximal doses align very well with their present maximum values unchanged at about 3 GyE, showing that the ion with the lowest LET will always minimize the secondary cancer risk for a given delivered dose equivalent. Notably, this secondary cancer risk is a contraindication for large, low-dose volumes with many beam portals in intensity-modulated radiation therapy using methods such as “rapidarc”, “volumetricarc”, and “tomotherapy” on non-seniors, which may have time to develop a secondary cancer after some 20 years [41][42].
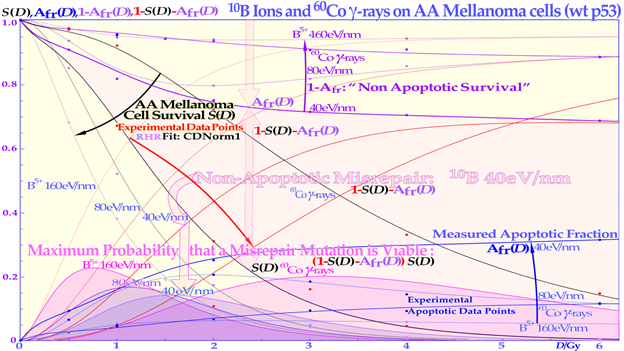
Figure 6. The secondary cancer induction probability as a function of the dose delivered to normal tissues. At low doses, the risk of inducing a mutation is small, whereas at high doses, the probability of generating a mutation is higher, but so is the probability of eliminating it via treatment. The risk is highest in normal tissues between 1 and 4 Gy, so this volume in patients should really be minimal. The LDA and LDHS of this TP53 intact cell line are clear from the curve shape for the two lowest LET beams. Interestingly, the risk is the smallest for the lowest-LET boron ions due to their high LDA and HDA. The upper shaded area is due to nonapoptotic misrepair for 40 eV/nm 10B ions. CDN1: the one-dimensional closest distance norm (not least square; for details, see [1]).
8. Conclusions on Optimal tumor Cure
Based on the function of the TP53 gene, present research introduces a new way to consider the classical 2 Gy/Fr mean tumor dose range for low-LET radiations mainly based on the recent observation that most normal tissues are low-dose hypersensitive. To minimize normal tissue damage, a dose of around 2 Gy/Fr implies optimal tolerance in normal tissues, as seen in Figure 2. This is where the least damage is generated in normal tissues, with about 1.5 Gy delivered with full NHEJ and HR repair activity, making NHEJ normal tissue recovery close to optimal, in time for the next day’s treatment fraction. Furthermore, it means avoidance of the more severe high-dose apoptosis that sets in after about 2–2.5 GyE, as seen in Figure 1. To really introduce a major paradigm shift in curative radiation therapy thinking, as described above, a dose far below or around 2 GyE/Fr should be delivered to most organs at risk to reach >≈3 Gy/Fr to the internal target volume [36]. To further increase the biologically effective tumor dose delivery, a few light ion beam portals should be used preferably in the range from helium to boron ions only with their Bragg peaks located in the gross tumor volume, to keep the LET low (<10 eV/nm) and the dose within 1.8–2.3 Gy/fraction in organs at risk [1]. Organs at risk have to be passed through with beams to reach the target volume, and with the lightest ions (He-B), this can be carried out using a fairly low LET (<10 eV/nm). To maximize the complication-free cure, it is best to switch to electrons or photons in the last 10–15 GyE, and for bulky tumors, possibly a light ion concomitant gross tumor boost should be used in the last 5 GyE before the final plain 10 GyE low-LET round-up [43][44][45]).The resultant increase in complication-free cure is likely to achieve improvements by as much as 10–25% and more for many tumor sites, e.g., using PRIMA-1 and APR-246 [35] for the problematic TP53-mutant tumors. About half this improvement alone was estimated to result from the improved fractionation schedule mentioned above, and described in more detail in a recent study on DNA repair (see the Graphical Abstract and [1]).
References
- Brahme, A. A DNA Repair-based model of cell survival with important clinical consequences. Radiat. Res. 2020, 194, 202–235.
- Brahme, A. Quantifying Cellular Repair, Misrepair and Apoptosis Induced by Boron Ions, Gamma Rays and PRIMA-1 Using the RHR Formulation. Radiat. Res. 2022, 198, 271–296.
- Wang, Y.H.; Ho, T.L.F.; Hariharan, A.; Goh, H.C.; Wong, Y.L.; Verkaik, N.S.; Lee, M.Y.; Tam, W.L.; van Gent, D.C.; Venkitaraman, A.R.; et al. Rapid recruitment of p53 to DNA damage sites directs DNA repair choice and integrity. Proc. Natl. Acad. Sci. USA 2022, 119, e2113233119.
- Brahme, A.; Lorat, Y. Dual Nucleosomal Double Strand Breaks are the Key Effectors of Curative Radiation Therapy. Int. J. Mol. Sci. 2023, In press.
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898.
- Gerelchuluun, A.; Manabe, E.; Ishikawa, T.; Sun, L.; Itoh, K.; Sakae, T.; Suzuki, K.; Hirayama, R.; Asaithamby, A.; Chen, D.J.; et al. The major DNA repair pathway after both proton and carbon ion radiation is NHEJ, but the HR pathway is more relevant in carbon ions. Radiat. Res. 2015, 183, 345–356.
- Takahashi, A.; Kubo, M.; Ma, H.; Nakagawa, A.; Yoshida, Y.; Isono, M.; Kanai, T.; Ohno, T.; Furusawa, Y.; Funayama, T. et al. Nonhomologous End-Joining Repair. Plays a More Important Role than Homologous Recombination Repair. in Defining Radiosensitivity after Exposure to High-LET Radiation. Radiat. Res. 2014, 182, 338–344.
- Enns, L.; Bogen, K.T.; Wizniak, J.; Murtha, A.D.; Weinfeld, M. Low-dose radiation hypersensitivity is associated with p53-dependent apoptosis. Mol. Cancer Res. 2004, 2, 557–566.
- Nakamura, Y. Isolation of p53-target genes and their functional analysis. Cancer Sci. 2004, 95, 7–11.
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070.
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714.
- Buscemi, G.; Perego, P.; Carenini, N.; Nakanishi, M.; Chessa, L.; Chen, J.; Khanna, K.; Delia, D. Activation of ATM and Chk2 kinases in relation to the amount of DNA strand breaks. Oncogene 2004, 23, 7691–7700.
- Singh, B.; Arrand, J.E.; Joiner, M.C. Hypersensitive response of normal human lung epithelial cells at low radiation doses. Int. J. Radiat. Biol. 1994, 65, 457–464.
- Krueger, S.A.; Collis, S.J.; Joiner, M.C.; Wilson, G.D.; Marples, B. Transition in Survival from low-dose hyper-radiosensitivity to increased radioresistance is independent of activation of ATM Ser1981 activity. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 1262–1271.
- Turesson, I.; Nyman, J.; Qvarnström, F.; Simonsson, M.; Book, M.; Hermansson, I.; Sigurdardottir, S.; Johansson, K.-A. A low-dose hypersensitive keratinocyte loss in response to fractionated radiotherapy is associated with growth arrest and apoptosis. Radiot. Oncol. 2010, 94, 90–101.
- Turesson, I.; Nyman, J.; Qvarnström, F.; Simonsson, M.; Book, M.; Hermansson, I.; Sigurdardottir, S.; Johansson, K.-A. A low-dose hypersensitive keratinocyte loss in response to fractionated radiotherapy is associated with growth arrest and apoptosis. Radiot. Oncol. 2010, 94, 90–101.
- Zhao M, Wang Y, Zhao Y, He S, Zhao R, Song Y; Cheng, J.; Gong, Y.; Xie, J.; Wang, Y.; et al. Caspase-3 knockout attenuates radiation-induced tumor repopulation via impairing the ATM/p53/Cox-2/PGE2 pathway in non-small cell lung cancer. Aging 2020, 12, 21758–21776.
- Brahme, A.; Rydberg, B.; Blomqvist, P. Dual spatially correlated nucleosomal double strand breaks in cell inactivation. In Microdosimetry: An Interdisciplinary Approach; Goodhead, D.T., O’Neill, P., Menzel, H.G., Eds.; The Royal Society of Chemistry: Cambridge, UK, 1997; pp. 125–128.
- Brahme, A. High resolution molecular radiation therapy and tumor imaging for the 21st century. J. Nucl. Med. Radiat. Ther. 2016; 7, 1-11. https://doi.org/10.4172/2155-9619.1000311+.
- Brahme, A. Development of Highly Specific Molecular Cancer Therapy with the Lightest Ions. In Proceedings of the 5th Takahashi Memorial International Symposium, Japan, 2007; Volume 57; Book of Abstract Japan.
- Kastenhuber, E.R. Lowe SW. Putting p53 in Context. Cell 2017, 170, 1062–1078.
- Riballo, E.; Kuhne, M.; Rief, N.; Doherty, A.; Smith, G.C.M.; Recio, M.J.; Reis, C.; Dahm, K.; Fricke, A.; Krempler, A.; et al. A pathway of double-strand break rejoining dependent upon ATM, artemis, and proteins locating to γ-H2AX foci. Mol. Cell 2004, 16, 715–724.
- Xue, L.; Yu, D.; Furusawa, Y.; Cao, J.; Okayasu, R.; Fan, S. ATM-dependent hyper-radiosensitivity in mammalian cells irradiated by heavy ions. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 235–243.
- Vreede, P.; Brahme, A. Development of biologically optimized radiation therapy: Maximizing the apoptotic cell kill. Radiol. Sci. 2009, 52, 31–52. Available online: http://www.nirs.go.jp/info/report/rs-sci/pdf/200907.pdf (accessed on 08092014).
- Schmitt, C.A. Senescence, apoptosis and therapy—Cutting the lifelines of cancer. Nat. Rev. Cancer 2003, 3, 283–295.
- d’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522.
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511.
- Lind, B.K.; Persson, L.M.; Edgren, M.R.; Hedlöf, I.; Brahme, A. Repairable-conditionally repairable damage model based on dual Poisson processes. Radiat. Res. 2003, 160, 366–375.
- Joiner, M.C.; Johns, H. Renal damage in the mouse: The response to very small doses per fraction. Radiat. Res. 1988, 114, 385–398.
- Brahme, A. Optimal use of light ions for radiation therapy. Radiol. Sci. 2010, 53, 35–61. Available online: http://www.nirs.go.jp/publication/rs-sci/pdf/201008.pdf (accessed on 08092014).
- Brahme, A. Physical, Biological and Clinical Merits of High Energy Boron Ions for Radiation Therapy. In Boron, Boron Compounds and Boron-Based Materials and Structures; Aydin, M., Ed.; IntechOpen: London, UK, 2023. https://doi.org/10.5772/intechopen.111485.
- Perdrix, A.; Najem, A.; Saussez, S.; Awada AJourne, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1MET (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers 2017, 9, 172.
- Furukawa H, Makino T, Yamasaki M; Tanaka, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Nakajima, K.; Takiguchi, S.; Mori, M.; et al. PRIMA-1 induces p53-mediated apoptosis by upregulating Noxa in esophageal squamous cell carcinoma with TP53 missense mutation. Cancer Sci. 2018, 109, 412–421. https://doi.org/10.1111/cas.13454.
- Xie, X.; Fan, C.; Luo, B.; Zhang, J.; Jensen, L.D.; Burman, J.; Jönsson, C.; Ljusberg, A.; Larsson, P.; Zhao, Z.; et al. APR-246 Enhances Colorectal Cancer Sensitivity to Radiotherapy. Available online: http://aacrjournals.org/mct/article-pdf/doi/10.1158/1535-7163.MCT-22-0275/3333376/mct-22-0275.pdf (accessed on 27072023).
- Xie, X.; Fan, C.; Luo, B.; Zhang, J.; Jensen, L.D.; Burman, J.; Jönsson, C.; Ljusberg, A.; Larsson, P.; Zhao, Z.; et al. APR-246 Enhances Colorectal Cancer Sensitivity to Radiotherapy. Available online: http://aacrjournals.org/mct/article-pdf/doi/10.1158/1535-7163.MCT-22-0275/3333376/mct-22-0275.pdf (accessed on 27072023).
- Aaltonen, P.; Brahme, A.; Lax, I.; Levernes, S.; Näslund, I.; Reitan, J.V.; Turesson, I. Specification of dose delivery in radiation therapy. Recommendations by the NACP. Acta Oncol. 1997, 36 (Suppl. S10),1–32.
- Brahme, A. Biologically based treatment planning. Acta Oncol. 1999, 38, 61–68.
- Brahme, A. Individualizing cancer treatment: Biological optimization models in treatment planning and delivery. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 327–337.
- Siddiqi, M.; Lind, B.K.; Brahme, A. Optimal dose fractionation of lung cancer using biologically optimized IMRT. 525 Poster. Radiother. Oncol. 2004, 73, S235. Available online: https://bit.ly/2LCN1m7 (accessed on 04052006).
- Brand, D.H.; Kirby, A.M.; Yarnold, J.R.; Somaiah, N. How Low. Can. You Go? The Radiobiology of Hypofractionation. Clin. Oncol. 2022, 34, 280–287.
- Brahme, A. Optimization of radiation therapy and the development of multileaf collimation. Editorial. Int. J. Rad. Oncol. Biol. Phys. 1993, 25, 373–375.
- Hall, E.J.; Wuu, C.S. Radiation-Induced Second Cancers: The Impact of 3D-CRT and IMRT. Int. J. Radiat. Oncol. Biol. Phys. 2003, 56, 83–88.
- Brahme, A. TP53 and the Ultimate Biological Optimization Steps of Curative Radiation Oncology. Cancers 2023,15,4286. https:// doi.org/10.3390/cancers15174286.
- Brahme, A. Biologically Optimized Lightion Therapy. In: Comprehensive BioMedical Physics; Major Reference Work; Brahme A. Ed. In Chief; Elsevier: Oxford, UK, 2014; Volume 9, pp. 529–554.
- Brahme, A.; Svensson, H. Physical, biological and clinical background for the development of biologically optimized light ion therapy. In Biologically Optimized Radiation Therapy; Brahme A., Ed.; World Scientific Publishing: Singapore, 2014; pp. 499–648.

