Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Juan Pablo Muñoz | -- | 3313 | 2023-08-27 10:58:07 | | | |
| 2 | Jessie Wu | -5 word(s) | 3308 | 2023-08-28 04:57:04 | | | | |
| 3 | Jessie Wu | Meta information modification | 3308 | 2023-08-28 05:00:22 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Muñoz, J.P.; Basei, F.L.; Rojas, M.L.; Galvis, D.; Zorzano, A. Mechanisms of Modulation of Mitochondrial Architecture. Encyclopedia. Available online: https://encyclopedia.pub/entry/48512 (accessed on 30 June 2026).
Muñoz JP, Basei FL, Rojas ML, Galvis D, Zorzano A. Mechanisms of Modulation of Mitochondrial Architecture. Encyclopedia. Available at: https://encyclopedia.pub/entry/48512. Accessed June 30, 2026.
Muñoz, Juan Pablo, Fernanda Luisa Basei, María Laura Rojas, David Galvis, Antonio Zorzano. "Mechanisms of Modulation of Mitochondrial Architecture" Encyclopedia, https://encyclopedia.pub/entry/48512 (accessed June 30, 2026).
Muñoz, J.P., Basei, F.L., Rojas, M.L., Galvis, D., & Zorzano, A. (2023, August 27). Mechanisms of Modulation of Mitochondrial Architecture. In Encyclopedia. https://encyclopedia.pub/entry/48512
Muñoz, Juan Pablo, et al. "Mechanisms of Modulation of Mitochondrial Architecture." Encyclopedia. Web. 27 August, 2023.
Copy Citation
Mitochondrial architecture is determined by several components, which include the following: mitochondrial distribution in the cytosol, supported by interaction with the cytoskeleton; events of fission and fusion, mediated by mitochondrial dynamics proteins; mitochondrial network contact with other organelles (e.g., endoplasmic reticulum (ER), lipid droplets (LDs), lysosomes, and plasma membrane); and the lipid composition of mitochondrial membranes.
metabolism
mitochondrial dynamics
pharmacology
lipids
metabolic disease
membrane contact sites (MCSs)
tethers
mitochondria
1. Mitochondrial Network Architecture
Cytoskeleton: Recent work evidenced that cytoskeleton elements differentially modulate the mobility and shape of mitochondria [1][2]. Nocodazole, a drug that disrupts microtubule polymerization, reduces mitochondrial network cellular coverage, disrupts mitochondrial alignment with microtubules, and decreases mitochondrial mobility. In contrast, F-actin or intermediate filaments maintain mitochondria confined to the microtubule network, and their disruption alters mitochondrial shape [1][2]. Of note, it has been reported that ARP2/3 and Formin-dependent actin cycling (actin assembly and disassembly) occur through the mitochondrial network [3]. Furthermore, actin polymerization promotes mitochondrial fission, whereas F-actin disruption causes mitochondria fusion [3]. In this regard, F-actin is assembled into the outer mitochondrial membrane (OMM), thereby contributing to mitochondrial fission [3][4].
Mitochondrial dynamics: Continual shifting of mitochondrial network architecture is supported by mitochondrial dynamics proteins. In the last 20 years, several proteins that coordinate mitochondrial fission and fusion events have been defined [5][6][7][8][9]. Moreover, recent studies have indicated that mitochondrial fission and fusion occur in the same site on the mitochondria [10], and also that ER wrapping around mitochondria is required to sustain the dynamics of these organelles [11]. A recent report describes two distinct types of mitochondrial fission. Peripheral fission sustains mitochondrial degradation mediated by mitophagy while midzone mitochondrial fission is required to maintain mitochondrial biogenesis and dynamics [12].
Mitochondrial contacts with other organelles. A growing number of studies have demonstrated the interaction of mitochondria with different organelles. This is an emergent field of research that will permit understanding of how some metabolic and cell signaling mechanisms originate.
Mitochondria–endoplasmic reticulum contacts. Several investigations have reported that mitochondria–endoplasmic reticulum interactions are required to modulate the calcium signal. Close contact between mitochondria and endoplasmic reticulum plays a critical role in mitochondrial calcium uptake. For instance, the complex VDAC1-GRP75-PI3R mediates mitochondria–endoplasmic reticulum tethering and is required to sustain mitochondrial calcium homeostasis [13]. Moreover, mitochondria–endoplasmic reticulum interaction is involved in apoptosis and autophagy activation [14][15][16]. Overnutrition induced via a high fat diet in mice leads to alteration in calcium handling in the liver [17][18]. These studies have demonstrated that mitochondrial–endoplasmic reticulum contacts are disrupted upon ingestion of a high fat diet. On the other hand, the mechanism of phosphatidylethanolamine (PE) synthesis and shuttling requires mitochondrial–endoplasmic reticulum contacts. The enzymes required for phosphatidic acid (PA) conversion to PE are localized in mitochondrial–endoplasmic reticulum contact. Several proteins contribute to mitochondria–endoplasmic reticulum tethering and the maintenance of calcium transfer and lipid synthesis (revised by Wenzel et al., 2022) [19].
Mitochondria–lipid droplets contacts. Contacts between mitochondria and lipid droplets (LDs) are mediated by several proteins including PLIN1, PLIN5, MFN2, and MIGA2 [20]. The highly dynamic interactions between both organelles allow fatty acid migration from LD to mitochondria, where it is oxidized to produce ATP or heat. Furthermore, they are also needed for LD expansion, which stores fatty acids and lipid intermediates to avoid cellular lipotoxicity [21][22].
Mitochondria–peroxisome contacts. Mitochondrial–peroxisome interaction plays a relevant role in fatty acid metabolism and in cellular redox homeostasis; a coordinate function of these organelles is required to sustain mitochondrial activity. Studies have shown that peroxisomal protein (PEX5 or PEX16) ablation in mice livers led to mitochondrial dysfunction and fragmentation [23][24]. Moreover, PEX16 knockout mice showed that peroxisomes are required to sustain mitochondrial homeostasis upon metabolic stress induced by a high fat diet [24]. Two proteins involved in mitochondria–peroxisome tethering have been identified, enoyl-CoA-δ isomerase 2 (ECI2) and the lipid transport protein VPS13D [25][26]. Interestingly, knockdown of VPS13D expression promotes mitochondrial fragmentation, revealing the role of this protein in mitochondrial architecture [27]. Also interesting, a recent study of yeast demonstrated that PEX4 and Fzo1 (the yeast orthologue of MFN1 and MFN2 proteins) are involved in mitochondria–peroxisome tethering [28]. Further investigations are required to elucidate the role of this protein in mammalian cells.
Mitochondria–endosome contacts. A recent report has documented that VDAC2 tethers RAS-PI3K-positive early endosomes with mitochondria [29]. This research also shows that this interaction promotes endosome acidification and maturation [29]. Depletion of the mitochondrial fusion protein MFN1 promotes the association of endosomes and mitochondria through a process that requires Rab5C [30]. In addition, it has been described that endosomes are in close contact with mitochondria in axons of retinal ganglion cells [31]. Rab7a is localized in endosomes, and ribonucleoprotein particles and mitochondria are in contact in axons. In this way, these contacts are required for local translation of mRNA that encodes to mitochondrial proteins. Moreover, Rab7a mutations lead to alteration of mitochondrial morphology, decrease mitochondrial membrane potential, and modify the mitochondrial retrograde and anterograde transport in axons [31].
Mitochondria–lysosome contacts. Lysosome–mitochondrial tethering is mediated by Rab7, is localized in lysosomes, and is modulated by TBC1D15, a Rab7 GTPase activating protein, and Fis1, localized in mitochondria [32]. This research also documented that lysosomes localize in mitochondrial fission sites, suggesting that they modulate mitochondrial dynamics [32]. The functional role of mitochondrial–lysosome tethering on calcium homeostasis has recently been reported [33]. The cation channel TRPML1 localized in lysosomes and late endosomes mediates the direct calcium transfer into mitochondria. Moreover, TRPML1 loss-of-function impairs calcium transfer, and it results in alterations of tethering dynamics [33].
Mitochondrial architecture is altered by environmental stimuli. It has been widely described in cell models that metabolic stress, induced by OXPHOS inhibitors, nutrient starvation, endoplasmic reticulum (ER) stressors, protein and RNA synthesis suppressors, and UV irradiation, induce transient mitochondrial elongation upon acute treatment [34][35][36][37]. Moreover, mitochondrial elongation improves the OXPHOS activity under these stimuli [34][38]. It has been demonstrated that the relationship between mitochondrial morphology and OXPHOS is complex. Given that, maximal mitochondrial respiration is carried out upon mitochondrial uncoupling agents (such as CCCP) treatment and these compounds promote mitochondrial depolarization and fragmentation. A simple relation between mitochondrial elongation and increased OXPHOS activity may not occur under all conditions. Thus, genetic ablation of the mitochondrial fusion protein Mitofusin 1, in which the mitochondrial network is fragmented, showed similar OXPHOS activity in glucose- or galactose-supplemented culture media [39]. In keeping with this, it has been reported that hepatic MFN1 ablation increases mitochondrial mass and OXPHOS activity [40]. Recent evidence has shed new light on the link between the morphology and function of mitochondria. Ngo et al. demonstrated that mitochondrial fragmentation enhances long-chain fatty acid oxidation. Moreover, mitochondrial fragmentation decreases the inhibition of malonyl-CoA-dependent carnitine palmitoyltransferase I. These data reveal that mitochondrial fragmentation upon lipid overload activates mitochondrial β-oxidation [41].
Mitochondrial shape alterations have been reported in conditions such as obesity, diabetes, ischemic–reperfusion, senescence, and cancer [17][42][43][44][45][46][47]. Under metabolic stress induced by nutrient overload, mitochondrial network architecture is fragmented, and this alteration is associated with a metabolic dysfunction that results in a decrease in OXPHOS activity, apoptosis, or mitophagy [48]. These alterations have been evidenced in several cell models, including β-pancreatic cells, skeletal muscle cells, cardiomyocytes, and adipocytes [49][50][51][52][53]. Similarly, disruption of the mitochondrial network architecture and mitochondrial dysfunction have been reported based on in vivo mouse models of diabetes, cardiomyopathy, non-alcoholic fatty liver disease, and obesity [44][54][55][56]. Emergent evidence indicates that mitophagy is impaired in metabolic diseases. The perturbation of mitochondrial homeostasis that causes a decrease in mitochondrial membrane potential induces mitochondrial fragmentation and activates lysosome-dependent mitochondrial degradation [48][57][58]. Recently, Han and colleagues demonstrated the presence of distinct populations of mitochondria in non-small cell lung cancer tumors. They reported a peri-droplet mitochondrial network throughout the cytoplasm in oxidative lung adenocarcinoma (LUAD) cells that was accompanied by an increase in fatty acid oxidation. On the other hand, cells with higher glycolytic activity (lung squamous cell carcinoma) show perinuclear mitochondria. These exciting results support the importance of mitochondrial architecture in sustaining tumor metabolism [47]. Thus, modulation of mitochondrial architecture appears to play a crucial role in cellular responses leading to metabolic diseases.
The discovery of proteins that modulate mitochondrial architecture and the mechanism of action of the same has allowed intense research efforts regarding the link between metabolism and mitochondrial function. These efforts have revealed that changes in the expression or post-translational modification of mitochondrial dynamics proteins are associated with metabolic diseases. As a result of these investigations, new fields of drug discovery aiming to modulate mitochondrial dynamics and thereby redirect cellular metabolism have emerged.
Given the continuous and rapid changes in mitochondrial architecture, sophisticated microscopy approaches are required to elucidate the biological factors that regulate mitochondrial morphology in vivo. Recent studies using super-resolution microscopy have revealed the cellular mechanisms by which mitochondrial network architecture is modulated [10][11][12][59]. In this regard, live-cell imaging has shown that ER–mitochondrial constriction sites prime and coordinate the mitochondrial fission and fusion protein machinery [10]. Remarkably, recent evidence using proximal proteomic and live-cell microscopy demonstrates that ABHD16A, a hydrolase enzyme that mediates the conversion of phosphatidylserine (PS) to lysophosphatidylserine (lysoPS), is localized at ER–mitochondria contacts and is required for mitochondrial fission and fusion [60].
The use of intravital multiphoton microscopy-based technology, which allows collection of cell images in live animals, has revealed that the mitochondrial network is disrupted upon pathological conditions in vivo. This research has demonstrated the transient disruption of the mitochondrial network upon brain ischemia. The mitochondria of dendrites at pyramidal neurons have an elongated shape, as revealed by intravital microscopy and electron microscopy. In contrast, upon transient brain ischemia, mitochondria undergo fragmentation and have a globular morphology, and “mitochondria-on-a-string” (MOAS) structures appear. Ischemic recovery rescues mitochondrial elongation and decreases globular mitochondria, and MOAS morphology is not observed in dendrites [61]. Taken together, these observations indicate that a transient and reversible alteration of mitochondrial morphology occurs under ischemia in vivo. Interestingly, MOAS morphology has also been described in neurons in cellular and mouse models of Alzheimer’s disease, aging, and hypoxia. It has been proposed that energetic collapse upon ischemic injury leads to unfinished mitochondrial fission [62][63]. Intravital microscopy has also been used to study live-cell mitochondrial dynamics and mitophagy in the livers of mice after acute ethanol treatment [63]. This research demonstrated that this treatment disrupts mitochondrial membrane potential; however, only a subpopulation of mitochondria undergoes mitophagy. In addition, ethanol activates selective mitophagy instead of lipophagy in the livers of this model [64]. In this context, a novel state-of-the-art methodological approach will unravel the function and modification of the mitochondrial network under physiological and pathological conditions (Figure 1 and Table 1).
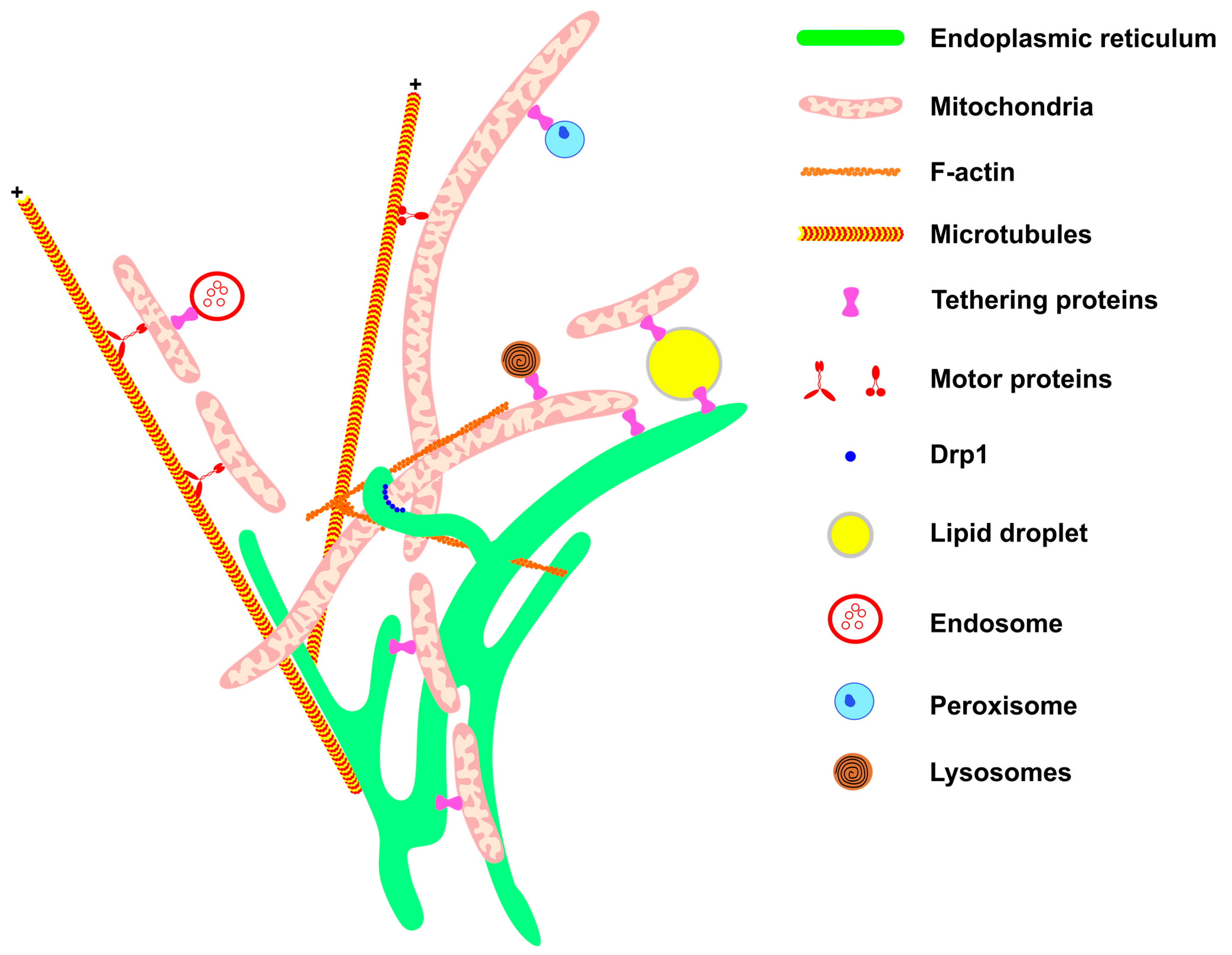
Figure 1. Mitochondrial network architecture. Mitochondria morphology is sustained and affected by interactions with other cellular structures. Communication with the ER (green) provides mitochondrial phospholipids and sustains calcium homeostasis. Interaction with the components of the cytoskeleton (F-actin filaments and microtubules, in orange) moves the mitochondria to specific regions of the cell, such as the appropriate location for cell division, or the periphery of the cell in the case of neurons. Motor protein kinesins and dyneins help to move mitochondria along the cytoskeletons of microtubules (for review, see McElroy et al., 2023) [65]. The endoplasmic reticulum wraps the mitochondria, thereby helping to recruit DRP1 (blue) on the mitochondrial membrane. Through communication with LDs (yellow), mitochondria are provided with fatty acids for β-oxidation and in turn provide energy for LD expansion. The contact between mitochondria and endosome (red) is important for mitochondria quality control through lipid and ion transference. Peroxisome (blue) and mitochondria communication allows the complementation of fatty acid oxidation and anti-oxidant system activation (for review, see Chen et al., 2020) [66]. Mitochondria–lysosome interaction (red and black) facilitates the direct transference of calcium from lysosomes to mitochondria. All these interactions are mediated by specific proteins that connect or tether the membranes.
Table 1. Cellular components that modulate mitochondrial architecture.
| Organelle | Functional Effect | Ref. |
|---|---|---|
| Endoplasmic reticulum | Mitochondrial dynamics, mitochondrial fusion and fission, endoplasmic reticulum marks fusion and fission sites, calcium transfer, autophagy, iron homeostasis, lipid transport | [10][11][13][16][67][68] |
| Lipid droplets | Lipid droplet expansion, fatty acid oxidation | [69] |
| Peroxisomes | Coordinate fatty acid metabolism, ROS scavenging | [26] |
| Endosomes | Endosome maturation, acidification of early endosomes | [29] |
| Iron transfer | [70] | |
| Mitochondrial mRNA translation in axon | [31] | |
| Lysosomes | Mitochondrial calcium transfer, lipid transport | [33] |
| Actin cytoskeleton | Mitochondrial dynamic, mitochondrial shape, mitochondrial positioning | [1][3] |
| Intermediate filaments | Mitochondrial shape | [1] |
| Tubulin | Mitochondrial transport, mitochondrial shape, mitochondrial positioning | [1][59][71] |
2. Pharmacological Approaches to Modulate Mitochondrial Architecture
Mitochondrial dynamics mediated by several fission and fusion events might be a strategic target with which to modify cellular metabolism. Finding new drugs to modulate mitochondrial architecture is an emergent field. In recent years, there have been reports of some compounds that modulate mitochondrial shape and metabolism.
Imeglimin is an antidiabetic drug structurally related to metformin that improves insulin sensitivity and mitochondrial function and decreases hepatic steatosis of high-fat, high-sucrose diet-treated mice [72][73][74]. This compound increases insulin release in obese db/db diabetic mice [75]. Moreover, this compound also modifies the mitochondrial morphology in pancreatic β-cells of db/db mice [75]. Studies in hepatic HepG2 cell lines show that like metformin, Imeglimin activates similar cellular signals, including AMPK activation and a decrease in mitochondrial respiration [76]. Interestingly, Imeglimin, as opposed to metformin, promotes the expression of Complex I and Complex III mitochondrial subunits [76]. Further investigations are required to elucidate the molecular mechanism of action of Imeglimin in mitochondria.
Mdiv-1 is a quinazoline-derived allosteric DRP1 inhibitor [77]. This compound promotes mitochondrial fusion, and it has been suggested that it protects from mitochondrial dysfunction caused by metabolic diseases and ischemic stroke [78][79][80][81]. Despite these reports, this compound shows Complex I off-target inhibition, and it impairs mitochondrial and cytosolic calcium homeostasis [82]. Also, several studies describe toxic cellular effects in some cells [83]. A recent report describes a novel DRP1 inhibitor, DRP1i27, that interacts with DRP1 and decreases its GTPase activity. This compound promotes mitochondrial elongation and protects from ischemic and reperfusion injury [84].
P110 is a seven-amino acid peptide that decreases the DRP1/FIS1 interaction and increases mitochondrial elongation [85][86]. In addition, this compound reduces oxidative stress and preserves mitochondrial respiration in LPS-treated cardiomyocytes [87]. In vivo studies demonstrated that p110 treatment for 10 or 24 days improves the motor and locomotion function of amyotrophic lateral sclerosis (ALS) mice. These mice carry the G39A SOD1 mutation that promotes oxidative stress, muscle atrophy, mitochondrial dysfunction, and cristae alterations. Overall these parameters were improved after P110 treatment [88].
Recent investigations, using a screening of molecules that can interact with the switch I-adjacent grove (SWAG) on DRP1 have led to the discovery of a novel DRP1 inhibitor, SC9. This molecule increases mitochondrial elongation under a cellular stress condition, such as LPS stimulus [86].
A recent study screened the mitochondrial morphology effect of 10,275 compounds using a high-content live-cell imaging approach [89]. Using this approach, PFK15 (6-phosphofructo-2-kinase (PFKFB3) inhibitor) was identified as a mitochondrial fission inhibitor. Based on the structure of this pharmacophore, a novel mitochondrial fission inhibitor, named MIDI, was synthesized. This compound increases mitochondrial elongation in cells treated with different stressors, such as hydrogen peroxide or mitochondrial function disruptors. Furthermore, MIDI promotes mitochondrial elongation of MFN1 KO cells. Mechanistically, this compound covalently interacts with cysteine C367 in the stalk domain on DRP1 [89].
Furthermore, antioxidant molecules can increase mitochondrial elongation. SS-31 (elamipretide) is a mitochondrial-target tetrapeptide with antioxidant capacity, and it improves mitochondrial elongation and mitochondrial fusion and promotes mitophagy [90][91]. SS-31 has been used in several models of cardiovascular disease. In addition, it is being tested in several clinical trials focused on heart failure, mitochondrial myopathy, renal disease, and aging [92][93][94][95].
The repurposing of FDA-approved drugs has opened new perspectives for drug indications. Leflunomide, a drug used for the treatment of rheumatoid arthritis, increases mitochondrial fusion by activating MFN1 and MFN2 expression [96]. Studies propose that a reversion of fragmented mitochondria in cancer might be a potential therapy. In this regard, leflunomide promotes mitochondrial elongation and increases the survival of pancreatic cancer patients [97].
A recent approach demonstrates that direct modulation of mitochondrial fusion using the TAT-367-384Gly fusogenic peptide creates a potential drug to treat mitochondrial disease. This peptide decreases HR1–HR2 interaction of MFN1 or MFN2 and results in the exposure of the MFN1/MFN2 HR2 domain to the cytoplasm. The open conformation of the HR2 domain might interact with another HR2 domain in other mitochondria and promote mitochondrial fusion. This peptide reverses the mitochondrial defect observed in CMT2A pathology [98]. Moreover, pharmacophore studies focused on HR1–HR2 MFN2 domain interaction and HR1 S378 phosphorylation revealed that a small chimeric molecule (Chimera B-A/l) promotes MFN2-dependent mitochondrial fusion [99]. Based on this molecule, MiM111 was developed. This compound improves mitochondrial transport and promotes axonal regeneration in models of CMT2A pathology [100][101][102].
A recent report describes a small molecule MASM7, which promotes MFN1 and/or MFN2 tethering through HR2 domain interaction. MASM7 increases mitochondrial fusion and enhances mitochondrial respiration, mitochondrial potential, and ATP production [103]. On the other hand, another molecule, MFI8, suppresses Mitofusin tethering, promotes mitochondrial fragmentation, and decreases mitochondrial function [103]. Remarkably, the design of these molecules has been based on the classical Mitofusin topology. Studies that describe Mitofusins as single-spanning membrane proteins may be also used to design novel pharmacological approximations, through the cysteine (Cys 684 and Cys 700) targeting of Mitofusins or the HR2–HR2 interaction region localized in IMS [104]. These studies may open new chemical approaches to modulate mitochondrial architecture. Direct activation or inhibition of the MFN1/2 tethering mechanism emerges as a promising strategy to directly modulate mitochondrial function. Table 6.
Table 2. Agents that target mitochondria and mitochondrial diseases.
| Drug | Effect on Mitochondria Function |
Disease | Ex Vivo and In Vivo Study Models | Clinical Trial | Ref. | |
|---|---|---|---|---|---|---|
| Dose | Study Models | |||||
| Imeglimin | Modulates OXPHOS, modulates mitochondrial morphology, antioxidant |
Diabetes mellitus type 2 | 0.1; 0.25, 1; 3; 10 mM | HepG2 cells Mouse primary hepatocytes |
EudraCT N. 2006-000909-29; 2011-004086-32; 2010-018580-42; 2010-023915-33 | [75][76][105][106][107] |
| 250 mg/Kg 150 mg/Kg BID 200 mg/Kg BID |
C57BLKS/J Iar-+Leprdb/+Leprdb mice (db/db); pancreatic islets KK-Ay/TaJcl (KK-Ay) mice, BKS; Cg-m +/+ Lepr db/Jcl (db/m) mice, BKS; Cg- + Lepr db/ + Lepr db/Jcl (db/db) mice |
|||||
| SS-31 (elamipretide) |
Improves mitochondrial respiratory capacity, increases mitochondrial supercomplex organization, stabilizes cardiolipin, modulates mitochondrial morphology, antioxidant, promotes mitophagy |
Barth syndrome, dilated cardiomyopathy with ataxia syndrome (DCMA), heart failure, age-related macular degeneration, LHON, skeletal-muscle mitochondrial dysfunction in the elderly, diabetes |
10 nM 100 nM 10 μM 25–125 μM |
Human fibroblast strains from patients with biochemically and/or genetically confirmed DCMA, HK-2 (Human kidney epithelial cells), ARPE-19 (retinal pigment epithelia cell line), INS1 β-cells (rat insulinoma cell line) |
NCT03323749;NCT03891875;NCT02976038;NCT05168774;NCT02814097;NCT03098797;NCT02914665;NCT02848313;NCT03323749;NCT02693119;NCT02245620;NCT02788747 | [90][91][108][109] |
| 0.25–60 mg/kg | TazKD mice CB6F1 mice |
|||||
| Quercetin | Enhances mitochondrial membrane potential, ATP levels, mtDNA PARP-1 inhibition; upregulates Nrf2 expression; modulates mitochondrial morphology; antioxidant |
Osteoarthritis, cardiac hypertrophy, traumatic brain injury, hepatotoxicity |
100 mg/Kg 20 mg/Kg |
Wistar rats, Sprague-Dawley rats, SHRs, Wistar-Kyoto rats, ICR mice |
None | [110][111][112][113][114] |
| 10–200 μM 50 μM |
CHO cells U937 cells THP-1 cells HL-60 cells NB4 cells |
|||||
| Mdiv-1 | Modulates ROS, inhibits DRP1, reduces cytosolic Ca2+ overload, modulates mitochondrial morphology |
Stroke, myocardial infarction, neurodegenerative diseases |
1, 3, 10, 30 μM 5 μM 25 μM 50 μM |
C57BL6/J mice, HL-1 atrial cells, GH3 cells, Primary ventricular myocytes, Neuronal primary culture, Sprague-Dawley rats |
None | [115][116][117][118] |
| DRP1i27 | Inhibits DRP1, modulates mitochondrial morphology |
Ischemia–reperfusion injury |
5, 10, 50 μM | HL-1 cells, human foreskin fibroblasts, iPSCs, CERA007c6 cell line |
None | [84][119] |
| P110 | Inhibits DRP1, blocks DRP1/FIS1 interaction and prevents/reverses excessive mitochondrial fragmentation, modulates mitochondrial morphology |
Neurodegenerative diseases, Friedreich ataxia |
1 μM | SH-5YSY cells, primary skin fibroblasts |
None | [85][120][121] |
| 0.5, 1.0, 1.5 mg/Kg | C57BL/6 mice | |||||
| Idebenone (Coenzyme Q10 Analog) |
Modulates OXPHOS, upregulates Lin28A, inhibits p52Shc PPARα/γ agonist |
Friedreich ataxia (FA), MELAS, LHON, DMD, multiple sclerosis, Parkinson’s disease, Alzheimer’s disease, Huntington’s disease |
5, 15 or 45 mg/kg 150–2250 mg/day |
Cell culture, animal, human |
NCT03891875;NCT02976038;NCT05162768;NCT02814097;NCT03098797;NCT02914665;NCT02848313;NCT03323749;NCT04689360;NCT02693119;NCT02245620;NCT02788747;NCT02805790;NCT01755858;NCT02367014;NCT01572909 | [105][122][123][124][125] |
| MiM111 | Activates mitofusin | Charcot–Marie–Tooth Disease, cardiomyopathy |
100 nM 1 μM |
Dermal fibroblasts (MFN2 T105M, MFN2 H361Y, MFN2 R274W), dermal fibroblasts, control patients, dorsal root ganglion (DRG) neurons were isolated from 8-week-old MFN2 T105M flox-stop transgenic mice |
None | [101][102] |
| 30 mg/kg 50 mg/kg |
MFN2 T105M mice: C57BL/6 Gt(ROSA)26 Sortm1 (CAG-MFN2*T105M)Dple/J) | |||||
* BID: twice a day; DCM: MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes) syndrome; LHON: Leber Hereditary Optic Neuropathy; SHRs: spontaneously hypertensive rats; TazKD: tafazzin knockdown mice; DMD: Duchenne muscular dystrophy.
References
- Fernández Casafuz, A.B.; De Rossi, M.C.; Bruno, L. Mitochondrial Cellular Organization and Shape Fluctuations Are Differentially Modulated by Cytoskeletal Networks. Sci. Rep. 2023, 13, 4065.
- Moore, A.S.; Coscia, S.M.; Simpson, C.L.; Ortega, F.E.; Wait, E.C.; Heddleston, J.M.; Nirschl, J.J.; Obara, C.J.; Guedes-Dias, P.; Boecker, C.A.; et al. Actin Cables and Comet Tails Organize Mitochondrial Networks in Mitosis. Nature 2021, 591, 659–664.
- Moore, A.S.; Wong, Y.C.; Simpson, C.L.; Holzbaur, E.L.F. Dynamic Actin Cycling through Mitochondrial Subpopulations Locally Regulates the Fission-Fusion Balance within Mitochondrial Networks. Nat. Commun. 2016, 7, 12886.
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An Actin-Dependent Step in Mitochondrial Fission Mediated by the ER-Associated Formin INF2. Science 2013, 339, 464–467.
- Smirnova, E.; Shurland, D.L.; Ryazantsev, S.N.; van der Bliek, A.M. A Human Dynamin-Related Protein Controls the Distribution of Mitochondria. J. Cell Biol. 1998, 143, 351–358.
- Santel, A.; Fuller, M.T. Control of Mitochondrial Morphology by a Human Mitofusin. J. Cell Sci. 2001, 114, 867–874.
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.-C. HFis1, a Novel Component of the Mammalian Mitochondrial Fission Machinery. J. Biol. Chem. 2003, 278, 36373–36379.
- Olichon, A.; Emorine, L.J.; Descoins, E.; Pelloquin, L.; Brichese, L.; Gas, N.; Guillou, E.; Delettre, C.; Valette, A.; Hamel, C.P.; et al. The Human Dynamin-Related Protein OPA1 Is Anchored to the Mitochondrial Inner Membrane Facing the Inter-Membrane Space. FEBS Lett. 2002, 523, 171–176.
- Gandre-Babbe, S.; van der Bliek, A.M. The Novel Tail-Anchored Membrane Protein Mff Controls Mitochondrial and Peroxisomal Fission in Mammalian Cells. Mol. Biol. Cell 2008, 19, 2402–2412.
- Abrisch, R.G.; Gumbin, S.C.; Wisniewski, B.T.; Lackner, L.L.; Voeltz, G.K. Fission and Fusion Machineries Converge at ER Contact Sites to Regulate Mitochondrial Morphology. J. Cell Biol. 2020, 219, e201911122.
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362.
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct Fission Signatures Predict Mitochondrial Degradation or Biogenesis. Nature 2021, 593, 435–439.
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-Mediated Coupling of Endoplasmic Reticulum and Mitochondrial Ca2+ Channels. J. Cell Biol. 2006, 175, 901–911.
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER-Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610.
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria Supply Membranes for Autophagosome Biogenesis during Starvation. Cell 2010, 141, 656–667.
- Xu, L.; Qiu, Y.; Wang, X.; Shang, W.; Bai, J.; Shi, K.; Liu, H.; Liu, J.-P.; Wang, L.; Tong, C. ER-Mitochondrial Contact Protein Miga Regulates Autophagy through Atg14 and Uvrag. Cell Rep. 2022, 41, 111583.
- Arruda, A.P.; Pers, B.M.; Parlakgül, G.; Güney, E.; Inouye, K.; Hotamisligil, G.S. Chronic Enrichment of Hepatic Endoplasmic Reticulum-Mitochondria Contact Leads to Mitochondrial Dysfunction in Obesity. Nat. Med. 2014, 20, 1427–1435.
- Beaulant, A.; Dia, M.; Pillot, B.; Chauvin, M.-A.; Ji-Cao, J.; Durand, C.; Bendridi, N.; Chanon, S.; Vieille-Marchiset, A.; Da Silva, C.C.; et al. Endoplasmic Reticulum-Mitochondria Miscommunication Is an Early and Causal Trigger of Hepatic Insulin Resistance and Steatosis. J. Hepatol. 2022, 77, 710–722.
- Wenzel, E.M.; Elfmark, L.A.; Stenmark, H.; Raiborg, C. ER as Master Regulator of Membrane Trafficking and Organelle Function. J. Cell Biol. 2022, 221, e202205135.
- Thiam, A.R.; Farese, R.V.; Walther, T.C. The Biophysics and Cell Biology of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2013, 14, 775–786.
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acín-Pérez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics That Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869–885.e6.
- Talari, N.K.; Mattam, U.; Meher, N.K.; Paripati, A.K.; Mahadev, K.; Krishnamoorthy, T.; Sepuri, N.B.V. Lipid-Droplet Associated Mitochondria Promote Fatty-Acid Oxidation through a Distinct Bioenergetic Pattern in Male Wistar Rats. Nat. Commun. 2023, 14, 766.
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18, 1126.
- Chi, L.; Lee, D.; Leung, S.; Hu, G.; Wen, B.; Delgado-Olguin, P.; Vissa, M.; Li, R.; Brumell, J.H.; Kim, P.K.; et al. Loss of Functional Peroxisomes Leads to Increased Mitochondrial Biogenesis and Reduced Autophagy That Preserve Mitochondrial Function. Cell. Mol. Life Sci. 2023, 80, 183.
- Fan, J.; Li, X.; Issop, L.; Culty, M.; Papadopoulos, V. ACBD2/ECI2-Mediated Peroxisome-Mitochondria Interactions in Leydig Cell Steroid Biosynthesis. Mol. Endocrinol. 2016, 30, 763–782.
- Guillén-Samander, A.; Leonzino, M.; Hanna, M.G.; Tang, N.; Shen, H.; De Camilli, P. VPS13D Bridges the ER to Mitochondria and Peroxisomes via Miro. J. Cell Biol. 2021, 220, e202010004.
- Baldwin, H.A.; Wang, C.; Kanfer, G.; Shah, H.V.; Velayos-Baeza, A.; Dulovic-Mahlow, M.; Brüggemann, N.; Anding, A.; Baehrecke, E.H.; Maric, D.; et al. VPS13D Promotes Peroxisome Biogenesis. J. Cell Biol. 2021, 220, e202001188.
- Shai, N.; Yifrach, E.; Van Roermund, C.W.T.; Cohen, N.; Bibi, C.; IJlst, L.; Cavellini, L.; Meurisse, J.; Schuster, R.; Zada, L.; et al. Systematic Mapping of Contact Sites Reveals Tethers and a Function for the Peroxisome-Mitochondria Contact. Nat. Commun. 2018, 9, 1761.
- Satoh, A.O.; Fujioka, Y.; Kashiwagi, S.; Yoshida, A.; Fujioka, M.; Sasajima, H.; Nanbo, A.; Amano, M.; Ohba, Y. Interaction between PI3K and the VDAC2 Channel Tethers Ras-PI3K-Positive Endosomes to Mitochondria and Promotes Endosome Maturation. Cell Rep. 2023, 42, 112229.
- Irazoki, A.; Gordaliza-Alaguero, I.; Frank, E.; Giakoumakis, N.N.; Seco, J.; Palacín, M.; Gumà, A.; Sylow, L.; Sebastián, D.; Zorzano, A. Disruption of Mitochondrial Dynamics Triggers Muscle Inflammation through Interorganellar Contacts and Mitochondrial DNA Mislocation. Nat. Commun. 2023, 14, 108.
- Cioni, J.-M.; Lin, J.Q.; Holtermann, A.V.; Koppers, M.; Jakobs, M.A.H.; Azizi, A.; Turner-Bridger, B.; Shigeoka, T.; Franze, K.; Harris, W.A.; et al. Late Endosomes Act as MRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 2019, 176, 56–72.e15.
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-Lysosome Contacts Regulate Mitochondrial Fission via RAB7 GTP Hydrolysis. Nature 2018, 554, 382–386.
- Peng, W.; Wong, Y.C.; Krainc, D. Mitochondria-Lysosome Contacts Regulate Mitochondrial Ca2+ Dynamics via Lysosomal TRPML1. Proc. Natl. Acad. Sci. USA 2020, 117, 19266–19275.
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep. 2018, 22, 2827–2836.
- Park, Y.-Y.; Nguyen, O.T.K.; Kang, H.; Cho, H. MARCH5-Mediated Quality Control on Acetylated Mfn1 Facilitates Mitochondrial Homeostasis and Cell Survival. Cell Death Dis. 2014, 5, e1172.
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 Is Required for Stress-Induced Mitochondrial Hyperfusion. EMBO J. 2009, 28, 1589–1600.
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular Network Formation Protects Mitochondria from Autophagosomal Degradation during Nutrient Starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195.
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During Autophagy Mitochondria Elongate, Are Spared from Degradation and Sustain Cell Viability. Nat. Cell Biol. 2011, 13, 589–598.
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic Cleavage of Opa1 Stimulates Mitochondrial Inner Membrane Fusion and Couples Fusion to Oxidative Phosphorylation. Cell Metab. 2014, 19, 630–641.
- Kulkarni, S.S.; Joffraud, M.; Boutant, M.; Ratajczak, J.; Gao, A.W.; Maclachlan, C.; Hernandez-Alvarez, M.I.; Raymond, F.; Metairon, S.; Descombes, P.; et al. Mfn1 Deficiency in the Liver Protects against Diet-Induced Insulin Resistance and Enhances the Hypoglycemic Effect of Metformin. Diabetes 2016, 65, 3552–3560.
- Ngo, J.; Choi, D.W.; Stanley, I.A.; Stiles, L.; Molina, A.J.A.; Chen, P.-H.; Lako, A.; Sung, I.C.H.; Goswami, R.; Kim, M.-Y.; et al. Mitochondrial Morphology Controls Fatty Acid Utilization by Changing CPT1 Sensitivity to Malonyl-CoA. EMBO J. 2023, 42, e111901.
- Correia-Melo, C.; Marques, F.D.M.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria Are Required for Pro-Ageing Features of the Senescent Phenotype. EMBO J. 2016, 35, 724–742.
- Boudina, S.; Sena, S.; Theobald, H.; Sheng, X.; Wright, J.J.; Hu, X.X.; Aziz, S.; Johnson, J.I.; Bugger, H.; Zaha, V.G.; et al. Mitochondrial Energetics in the Heart in Obesity-Related Diabetes: Direct Evidence for Increased Uncoupled Respiration and Activation of Uncoupling Proteins. Diabetes 2007, 56, 2457–2466.
- Rajab, B.S.; Kassab, S.; Stonall, C.D.; Daghistani, H.; Gibbons, S.; Mamas, M.; Smith, D.; Mironov, A.; AlBalawi, Z.; Zhang, Y.H.; et al. Differential Remodelling of Mitochondrial Subpopulations and Mitochondrial Dysfunction Are a Feature of Early Stage Diabetes. Sci. Rep. 2022, 12, 978.
- Bugger, H.; Chen, D.; Riehle, C.; Soto, J.; Theobald, H.A.; Hu, X.X.; Ganesan, B.; Weimer, B.C.; Abel, E.D. Tissue-Specific Remodeling of the Mitochondrial Proteome in Type 1 Diabetic Akita Mice. Diabetes 2009, 58, 1986–1997.
- Bagur, R.; Tanguy, S.; Foriel, S.; Grichine, A.; Sanchez, C.; Pernet-Gallay, K.; Kaambre, T.; Kuznetsov, A.V.; Usson, Y.; Boucher, F.; et al. The Impact of Cardiac Ischemia/Reperfusion on the Mitochondria-Cytoskeleton Interactions. Biochim. Biophys. Acta 2016, 1862, 1159–1171.
- Han, M.; Bushong, E.A.; Segawa, M.; Tiard, A.; Wong, A.; Brady, M.R.; Momcilovic, M.; Wolf, D.M.; Zhang, R.; Petcherski, A.; et al. Spatial Mapping of Mitochondrial Networks and Bioenergetics in Lung Cancer. Nature 2023, 615, 712–719.
- Nisr, R.B.; Shah, D.S.; Ganley, I.G.; Hundal, H.S. Proinflammatory NFkB Signalling Promotes Mitochondrial Dysfunction in Skeletal Muscle in Response to Cellular Fuel Overloading. Cell. Mol. Life Sci. 2019, 76, 4887–4904.
- Molina, A.J.A.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial Networking Protects Beta-Cells from Nutrient-Induced Apoptosis. Diabetes 2009, 58, 2303–2315.
- Stiles, L.; Shirihai, O.S. Mitochondrial Dynamics and Morphology in Beta-Cells. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 725–738.
- Nisr, R.B.; Shah, D.S.; Hundal, H.S. Mono- and Polyunsaturated Fatty Acids Counter Palmitate-Induced Mitochondrial Dysfunction in Rat Skeletal Muscle Cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2020, 54, 975–993.
- Parra, V.; Verdejo, H.E.; Iglewski, M.; Del Campo, A.; Troncoso, R.; Jones, D.; Zhu, Y.; Kuzmicic, J.; Pennanen, C.; Lopez-Crisosto, C.; et al. Insulin Stimulates Mitochondrial Fusion and Function in Cardiomyocytes via the Akt-MTOR-NFκB-Opa-1 Signaling Pathway. Diabetes 2014, 63, 75–88.
- Zhou, Z.; Torres, M.; Sha, H.; Halbrook, C.J.; Van den Bergh, F.; Reinert, R.B.; Yamada, T.; Wang, S.; Luo, Y.; Hunter, A.H.; et al. Endoplasmic Reticulum-Associated Degradation Regulates Mitochondrial Dynamics in Brown Adipocytes. Science 2020, 368, 54–60.
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease With Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131.
- Shami, G.J.; Cheng, D.; Verhaegh, P.; Koek, G.; Wisse, E.; Braet, F. Three-Dimensional Ultrastructure of Giant Mitochondria in Human Non-Alcoholic Fatty Liver Disease. Sci. Rep. 2021, 11, 3319.
- Hammerschmidt, P.; Ostkotte, D.; Nolte, H.; Gerl, M.J.; Jais, A.; Brunner, H.L.; Sprenger, H.-G.; Awazawa, M.; Nicholls, H.T.; Turpin-Nolan, S.M.; et al. CerS6-Derived Sphingolipids Interact with Mff and Promote Mitochondrial Fragmentation in Obesity. Cell 2019, 177, 1536–1552.e23.
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and Selective Fusion Govern Mitochondrial Segregation and Elimination by Autophagy. EMBO J. 2008, 27, 433–446.
- Wikstrom, J.D.; Mahdaviani, K.; Liesa, M.; Sereda, S.B.; Si, Y.; Las, G.; Twig, G.; Petrovic, N.; Zingaretti, C.; Graham, A.; et al. Hormone-Induced Mitochondrial Fission Is Utilized by Brown Adipocytes as an Amplification Pathway for Energy Expenditure. EMBO J. 2014, 33, 418–436.
- Friedman, J.R.; Webster, B.M.; Mastronarde, D.N.; Verhey, K.J.; Voeltz, G.K. ER Sliding Dynamics and ER-Mitochondrial Contacts Occur on Acetylated Microtubules. J. Cell Biol. 2010, 190, 363–375.
- Nguyen, T.T.; Voeltz, G.K. An ER Phospholipid Hydrolase Drives ER-Associated Mitochondrial Constriction for Fission and Fusion. eLife 2022, 11, e84279.
- Kirov, S.A.; Fomitcheva, I.V.; Sword, J. Rapid Neuronal Ultrastructure Disruption and Recovery during Spreading Depolarization-Induced Cytotoxic Edema. Cereb. Cortex 2020, 30, 5517–5531.
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered Brain Energetics Induces Mitochondrial Fission Arrest in Alzheimer’s Disease. Sci. Rep. 2016, 6, 18725.
- Morozov, Y.M.; Datta, D.; Paspalas, C.D.; Arnsten, A.F.T. Ultrastructural Evidence for Impaired Mitochondrial Fission in the Aged Rhesus Monkey Dorsolateral Prefrontal Cortex. Neurobiol. Aging 2017, 51, 9–18.
- Samuvel, D.J.; Li, L.; Krishnasamy, Y.; Gooz, M.; Takemoto, K.; Woster, P.M.; Lemasters, J.J.; Zhong, Z. Mitochondrial Depolarization after Acute Ethanol Treatment Drives Mitophagy in Living Mice. Autophagy 2022, 18, 2671–2685.
- McElroy, T.; Zeidan, R.S.; Rathor, L.; Han, S.M.; Xiao, R. The Role of Mitochondria in the Recovery of Neurons after Injury. Neural Regen. Res. 2023, 18, 317–318.
- Chen, C.; Li, J.; Qin, X.; Wang, W. Peroxisomal Membrane Contact Sites in Mammalian Cells. Front. Cell Dev. Biol. 2020, 8, 512.
- Vance, J.E. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic 2015, 16, 1–18.
- Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernández, V.; Vasconcelos, N.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895.e17.
- Hong, Z.; Adlakha, J.; Wan, N.; Guinn, E.; Giska, F.; Gupta, K.; Melia, T.J.; Reinisch, K.M. Mitoguardin-2-Mediated Lipid Transfer Preserves Mitochondrial Morphology and Lipid Droplet Formation. J. Cell Biol. 2022, 221, e202207022.
- Barra, J.; Crosbourne, I.; Wang, L.; Nelson, I.; Goldwag, J.; Jourd’heuil, F.; Jourd’heuil, D.; Barroso, M. DMT1-mediated Endosome-mitochondria Interactions Regulates Iron Homeostasis and Mitochondrial Metabolism. FASEB J. 2022, 36, fasebj.2022.36.S1.R5276.
- Wang, X.; Schwarz, T.L. The Mechanism of Ca2+ -Dependent Regulation of Kinesin-Mediated Mitochondrial Motility. Cell 2009, 136, 163–174.
- Perry, R.J.; Cardone, R.L.; Petersen, M.C.; Zhang, D.; Fouqueray, P.; Hallakou-Bozec, S.; Bolze, S.; Shulman, G.I.; Petersen, K.F.; Kibbey, R.G. Imeglimin Lowers Glucose Primarily by Amplifying Glucose-Stimulated Insulin Secretion in High-Fat-Fed Rodents. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E461–E470.
- Vial, G.; Chauvin, M.-A.; Bendridi, N.; Durand, A.; Meugnier, E.; Madec, A.-M.; Bernoud-Hubac, N.; Pais de Barros, J.-P.; Fontaine, É.; Acquaviva, C.; et al. Imeglimin Normalizes Glucose Tolerance and Insulin Sensitivity and Improves Mitochondrial Function in Liver of a High-Fat, High-Sucrose Diet Mice Model. Diabetes 2015, 64, 2254–2264.
- Dubourg, J.; Fouqueray, P.; Thang, C.; Grouin, J.-M.; Ueki, K. Efficacy and Safety of Imeglimin Monotherapy Versus Placebo in Japanese Patients With Type 2 Diabetes (TIMES 1): A Double-Blind, Randomized, Placebo-Controlled, Parallel-Group, Multicenter Phase 3 Trial. Diabetes Care 2021, 44, 952–959.
- Sanada, J.; Obata, A.; Fushimi, Y.; Kimura, T.; Shimoda, M.; Ikeda, T.; Nogami, Y.; Obata, Y.; Yamasaki, Y.; Nakanishi, S.; et al. Imeglimin Exerts Favorable Effects on Pancreatic β-Cells by Improving Morphology in Mitochondria and Increasing the Number of Insulin Granules. Sci. Rep. 2022, 12, 13220.
- Hozumi, K.; Sugawara, K.; Ishihara, T.; Ishihara, N.; Ogawa, W. Effects of Imeglimin on Mitochondrial Function, AMPK Activity, and Gene Expression in Hepatocytes. Sci. Rep. 2023, 13, 746.
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204.
- Ong, S.-B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting Mitochondrial Fission Protects the Heart against Ischemia/Reperfusion Injury. Circulation 2010, 121, 2012–2022.
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor Mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor That Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6.
- Maneechote, C.; Palee, S.; Apaijai, N.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. Mitochondrial Dynamic Modulation Exerts Cardiometabolic Protection in Obese Insulin-Resistant Rats. Clin. Sci. 2019, 133, 2431–2447.
- Finocchietto, P.; Perez, H.; Blanco, G.; Miksztowicz, V.; Marotte, C.; Morales, C.; Peralta, J.; Berg, G.; Poderoso, C.; Poderoso, J.J.; et al. Inhibition of Mitochondrial Fission by Drp-1 Blockade by Short-Term Leptin and Mdivi-1 Treatment Improves White Adipose Tissue Abnormalities in Obesity and Diabetes. Pharmacol. Res. 2022, 178, 106028.
- Ruiz, A.; Alberdi, E.; Matute, C. Mitochondrial Division Inhibitor 1 (Mdivi-1) Protects Neurons against Excitotoxicity through the Modulation of Mitochondrial Function and Intracellular Ca2+ Signaling. Front. Mol. Neurosci. 2018, 11, 3.
- Ruiz, A.; Quintela-López, T.; Sánchez-Gómez, M.V.; Gaminde-Blasco, A.; Alberdi, E.; Matute, C. Mitochondrial Division Inhibitor 1 Disrupts Oligodendrocyte Ca2+ Homeostasis and Mitochondrial Function. Glia 2020, 68, 1743–1756.
- Rosdah, A.A.; Abbott, B.M.; Langendorf, C.G.; Deng, Y.; Truong, J.Q.; Waddell, H.M.M.; Ling, N.X.Y.; Smiles, W.J.; Delbridge, L.M.D.; Liu, G.-S.; et al. A Novel Small Molecule Inhibitor of Human Drp1. Sci. Rep. 2022, 12, 21531.
- Qi, X.; Qvit, N.; Su, Y.-C.; Mochly-Rosen, D. A Novel Drp1 Inhibitor Diminishes Aberrant Mitochondrial Fission and Neurotoxicity. J. Cell Sci. 2013, 126, 789–802.
- Rios, L.; Pokhrel, S.; Li, S.-J.; Heo, G.; Haileselassie, B.; Mochly-Rosen, D. Targeting an Allosteric Site in Dynamin-Related Protein 1 to Inhibit Fis1-Mediated Mitochondrial Dysfunction. Nat. Commun. 2023, 14, 4356.
- Haileselassie, B.; Mukherjee, R.; Joshi, A.U.; Napier, B.A.; Massis, L.M.; Ostberg, N.P.; Queliconi, B.B.; Monack, D.; Bernstein, D.; Mochly-Rosen, D. Drp1/Fis1 Interaction Mediates Mitochondrial Dysfunction in Septic Cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 130, 160–169.
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 Interaction Slows Progression of Amyotrophic Lateral Sclerosis. EMBO Mol. Med. 2018, 10, e8166.
- Yang, J.; Chen, P.; Cao, Y.; Liu, S.; Wang, W.; Li, L.; Li, J.; Jiang, Z.; Ma, Y.; Chen, S.; et al. Chemical Inhibition of Mitochondrial Fission via Targeting the DRP1-Receptor Interaction. Cell Chem. Biol. 2023, 30, 278–294.e11.
- Mitchell, W.; Tamucci, J.D.; Ng, E.L.; Liu, S.; Birk, A.V.; Szeto, H.H.; May, E.R.; Alexandrescu, A.T.; Alder, N.N. Structure-Activity Relationships of Mitochondria-Targeted Tetrapeptide Pharmacological Compounds. eLife 2022, 11, e75531.
- Petcherski, A.; Trudeau, K.M.; Wolf, D.M.; Segawa, M.; Lee, J.; Taddeo, E.P.; Deeney, J.T.; Liesa, M. Elamipretide Promotes Mitophagosome Formation and Prevents Its Reduction Induced by Nutrient Excess in INS1 β-Cells. J. Mol. Biol. 2018, 430, 4823–4833.
- Chiao, Y.A.; Zhang, H.; Sweetwyne, M.; Whitson, J.; Ting, Y.S.; Basisty, N.; Pino, L.K.; Quarles, E.; Nguyen, N.-H.; Campbell, M.D.; et al. Late-Life Restoration of Mitochondrial Function Reverses Cardiac Dysfunction in Old Mice. eLife 2020, 9, e55513.
- Miyamoto, S.; Zhang, G.; Hall, D.; Oates, P.J.; Maity, S.; Madesh, M.; Han, X.; Sharma, K. Restoring Mitochondrial Superoxide Levels with Elamipretide (MTP-131) Protects Db/Db Mice against Progression of Diabetic Kidney Disease. J. Biol. Chem. 2020, 295, 7249–7260.
- Siegel, M.P.; Kruse, S.E.; Percival, J.M.; Goh, J.; White, C.C.; Hopkins, H.C.; Kavanagh, T.J.; Szeto, H.H.; Rabinovitch, P.S.; Marcinek, D.J. Mitochondrial-Targeted Peptide Rapidly Improves Mitochondrial Energetics and Skeletal Muscle Performance in Aged Mice. Aging Cell 2013, 12, 763–771.
- Escribano-Lopez, I.; Diaz-Morales, N.; Iannantuoni, F.; Lopez-Domenech, S.; de Marañon, A.M.; Abad-Jimenez, Z.; Bañuls, C.; Rovira-Llopis, S.; Herance, J.R.; Rocha, M.; et al. The Mitochondrial Antioxidant SS-31 Increases SIRT1 Levels and Ameliorates Inflammation, Oxidative Stress and Leukocyte-Endothelium Interactions in Type 2 Diabetes. Sci. Rep. 2018, 8, 15862.
- Miret-Casals, L.; Sebastián, D.; Brea, J.; Rico-Leo, E.M.; Palacín, M.; Fernández-Salguero, P.M.; Loza, M.I.; Albericio, F.; Zorzano, A. Identification of New Activators of Mitochondrial Fusion Reveals a Link between Mitochondrial Morphology and Pyrimidine Metabolism. Cell Chem. Biol. 2018, 25, 268–278.e4.
- Yu, M.; Nguyen, N.D.; Huang, Y.; Lin, D.; Fujimoto, T.N.; Molkentine, J.M.; Deorukhkar, A.; Kang, Y.; San Lucas, F.A.; Fernandes, C.J.; et al. Mitochondrial Fusion Exploits a Therapeutic Vulnerability of Pancreatic Cancer. JCI Insight 2019, 5, e126915.
- Franco, A.; Kitsis, R.N.; Fleischer, J.A.; Gavathiotis, E.; Kornfeld, O.S.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, S.K.; et al. Correcting Mitochondrial Fusion by Manipulating Mitofusin Conformations. Nature 2016, 540, 74–79.
- Larrea, D.; Pera, M.; Gonnelli, A.; Quintana–Cabrera, R.; Akman, H.O.; Guardia-Laguarta, C.; Velasco, K.R.; Area-Gomez, E.; Dal Bello, F.; De Stefani, D.; et al. MFN2 Mutations in Charcot–Marie–Tooth Disease Alter Mitochondria-Associated ER Membrane Function but Do Not Impair Bioenergetics. Hum. Mol. Genet. 2019, 28, 1782–1800.
- Dang, X.; Zhang, L.; Franco, A.; Li, J.; Rocha, A.G.; Devanathan, S.; Dolle, R.E.; Bernstein, P.R.; Dorn, G.W. Discovery of 6-Phenylhexanamide Derivatives as Potent Stereoselective Mitofusin Activators for the Treatment of Mitochondrial Diseases. J. Med. Chem. 2020, 63, 7033–7051.
- Franco, A.; Dang, X.; Walton, E.K.; Ho, J.N.; Zablocka, B.; Ly, C.; Miller, T.M.; Baloh, R.H.; Shy, M.E.; Yoo, A.S.; et al. Burst Mitofusin Activation Reverses Neuromuscular Dysfunction in Murine CMT2A. eLife 2020, 9, e61119.
- Franco, A.; Dang, X.; Zhang, L.; Molinoff, P.B.; Dorn, G.W. Mitochondrial Dysfunction and Pharmacodynamics of Mitofusin Activation in Murine Charcot-Marie-Tooth Disease Type 2A. J. Pharmacol. Exp. Ther. 2022, 383, 137–148.
- Zacharioudakis, E.; Agianian, B.; Kumar Mv, V.; Biris, N.; Garner, T.P.; Rabinovich-Nikitin, I.; Ouchida, A.T.; Margulets, V.; Nordstrøm, L.U.; Riley, J.S.; et al. Modulating Mitofusins to Control Mitochondrial Function and Signaling. Nat. Commun. 2022, 13, 3775.
- Mattie, S.; Riemer, J.; Wideman, J.G.; McBride, H.M. A New Mitofusin Topology Places the Redox-Regulated C Terminus in the Mitochondrial Intermembrane Space. J. Cell Biol. 2018, 217, 507–515.
- Singh, A.; Faccenda, D.; Campanella, M. Pharmacological Advances in Mitochondrial Therapy. EBioMedicine 2021, 65, 103244.
- Nowak, M.; Grzeszczak, W. Imeglimin: A New Antidiabetic Drug with Potential Future in the Treatment of Patients with Type 2 Diabetes. Endokrynol. Pol. 2022, 73, 361–370.
- Fauzi, M.; Murakami, T.; Fujimoto, H.; Botagarova, A.; Sakaki, K.; Kiyobayashi, S.; Ogura, M.; Inagaki, N. Preservation Effect of Imeglimin on Pancreatic β-Cell Mass: Noninvasive Evaluation Using 111In-Exendin-4 SPECT/CT Imaging and the Perspective of Mitochondrial Involvements. Front. Endocrinol. 2022, 13, 1010825.
- Chavez, J.D.; Tang, X.; Campbell, M.D.; Reyes, G.; Kramer, P.A.; Stuppard, R.; Keller, A.; Zhang, H.; Rabinovitch, P.S.; Marcinek, D.J.; et al. Mitochondrial Protein Interaction Landscape of SS-31. Proc. Natl. Acad. Sci. USA 2020, 117, 15363–15373.
- Russo, S.; De Rasmo, D.; Signorile, A.; Corcelli, A.; Lobasso, S. Beneficial Effects of SS-31 Peptide on Cardiac Mitochondrial Dysfunction in Tafazzin Knockdown Mice. Sci. Rep. 2022, 12, 19847.
- Gibellini, L.; Bianchini, E.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Natural Compounds Modulating Mitochondrial Functions. Evid.-Based Complement. Altern. Med. 2015, 2015, 527209.
- Waseem, M.; Kaushik, P.; Dutta, S.; Chakraborty, R.; Hassan, M.I.; Parvez, S. Modulatory Role of Quercetin in Mitochondrial Dysfunction in Titanium Dioxide Nanoparticle-Induced Hepatotoxicity. ACS Omega 2022, 7, 3192–3202.
- Qiu, L.; Luo, Y.; Chen, X. Quercetin Attenuates Mitochondrial Dysfunction and Biogenesis via Upregulated AMPK/SIRT1 Signaling Pathway in OA Rats. Biomed. Pharmacother. 2018, 103, 1585–1591.
- Chen, W.-J.; Cheng, Y.; Li, W.; Dong, X.-K.; Wei, J.; Yang, C.-H.; Jiang, Y.-H. Quercetin Attenuates Cardiac Hypertrophy by Inhibiting Mitochondrial Dysfunction Through SIRT3/PARP-1 Pathway. Front. Pharmacol. 2021, 12, 739615.
- Li, X.; Wang, H.; Gao, Y.; Li, L.; Tang, C.; Wen, G.; Zhou, Y.; Zhou, M.; Mao, L.; Fan, Y. Protective Effects of Quercetin on Mitochondrial Biogenesis in Experimental Traumatic Brain Injury via the Nrf2 Signaling Pathway. PLoS ONE 2016, 11, e0164237.
- Nhu, N.T.; Li, Q.; Liu, Y.; Xu, J.; Xiao, S.-Y.; Lee, S.-D. Effects of Mdivi-1 on Neural Mitochondrial Dysfunction and Mitochondria-Mediated Apoptosis in Ischemia-Reperfusion Injury After Stroke: A Systematic Review of Preclinical Studies. Front. Mol. Neurosci. 2021, 14, 778569.
- So, E.C.; Hsing, C.-H.; Liang, C.-H.; Wu, S.-N. The Actions of Mdivi-1, an Inhibitor of Mitochondrial Fission, on Rapidly Activating Delayed-Rectifier K+ Current and Membrane Potential in HL-1 Murine Atrial Cardiomyocytes. Eur. J. Pharmacol. 2012, 683, 1–9.
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related Protein 1 (Drp1)-mediated Diastolic Dysfunction in Myocardial Ischemia-reperfusion Injury: Therapeutic Benefits of Drp1 Inhibition to Reduce Mitochondrial Fission. FASEB J. 2014, 28, 316–326.
- Ding, J.; Zhang, Z.; Li, S.; Wang, W.; Du, T.; Fang, Q.; Wang, Y.; Wang, D.W. Mdivi-1 Alleviates Cardiac Fibrosis Post Myocardial Infarction at Infarcted Border Zone, Possibly via Inhibition of Drp1-Activated Mitochondrial Fission and Oxidative Stress. Arch. Biochem. Biophys. 2022, 718, 109147.
- Grohm, J.; Kim, S.-W.; Mamrak, U.; Tobaben, S.; Cassidy-Stone, A.; Nunnari, J.; Plesnila, N.; Culmsee, C. Inhibition of Drp1 Provides Neuroprotection in Vitro and in Vivo. Cell Death Differ. 2012, 19, 1446–1458.
- Tokuyama, T.; Hirai, A.; Shiiba, I.; Ito, N.; Matsuno, K.; Takeda, K.; Saito, K.; Mii, K.; Matsushita, N.; Fukuda, T.; et al. Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients. Biomolecules 2020, 10, 450.
- Filichia, E.; Hoffer, B.; Qi, X.; Luo, Y. Inhibition of Drp1 Mitochondrial Translocation Provides Neural Protection in Dopaminergic System in a Parkinson’s Disease Model Induced by MPTP. Sci. Rep. 2016, 6, 32656.
- Gueven, N.; Ravishankar, P.; Eri, R.; Rybalka, E. Idebenone: When an Antioxidant Is Not an Antioxidant. Redox Biol. 2021, 38, 101812.
- Tinker, R.J.; Lim, A.Z.; Stefanetti, R.J.; McFarland, R. Current and Emerging Clinical Treatment in Mitochondrial Disease. Mol. Diagn. Ther. 2021, 25, 181–206.
- Jaber, S.M.; Ge, S.X.; Milstein, J.L.; VanRyzin, J.W.; Waddell, J.; Polster, B.M. Idebenone Has Distinct Effects on Mitochondrial Respiration in Cortical Astrocytes Compared to Cortical Neurons Due to Differential NQO1 Activity. J. Neurosci. 2020, 40, 4609–4619.
- Heitz, F.D.; Erb, M.; Anklin, C.; Robay, D.; Pernet, V.; Gueven, N. Idebenone Protects against Retinal Damage and Loss of Vision in a Mouse Model of Leber’s Hereditary Optic Neuropathy. PLoS ONE 2012, 7, e45182.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
812
Revisions:
3 times
(View History)
Update Date:
28 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No