Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Teresa Bellón | -- | 2265 | 2023-08-24 10:02:12 | | | |
| 2 | Catherine Yang | Meta information modification | 2265 | 2023-08-25 03:50:07 | | | | |
| 3 | Catherine Yang | Meta information modification | 2265 | 2023-08-28 09:48:33 | | | | |
| 4 | Catherine Yang | Meta information modification | 2265 | 2023-08-30 04:06:56 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rodríguez-Pérez, R.; De Las Vecillas, L.; Cabañas, R.; Bellón, T. Drug Hypersensitivity Reactions. Encyclopedia. Available online: https://encyclopedia.pub/entry/48416 (accessed on 09 August 2026).
Rodríguez-Pérez R, De Las Vecillas L, Cabañas R, Bellón T. Drug Hypersensitivity Reactions. Encyclopedia. Available at: https://encyclopedia.pub/entry/48416. Accessed August 09, 2026.
Rodríguez-Pérez, Rosa, Leticia De Las Vecillas, Rosario Cabañas, Teresa Bellón. "Drug Hypersensitivity Reactions" Encyclopedia, https://encyclopedia.pub/entry/48416 (accessed August 09, 2026).
Rodríguez-Pérez, R., De Las Vecillas, L., Cabañas, R., & Bellón, T. (2023, August 24). Drug Hypersensitivity Reactions. In Encyclopedia. https://encyclopedia.pub/entry/48416
Rodríguez-Pérez, Rosa, et al. "Drug Hypersensitivity Reactions." Encyclopedia. Web. 24 August, 2023.
Copy Citation
Drug hypersensitivity reactions are a serious concern in clinical practice because they can be severe and result in lifelong sequelae. An accurate diagnosis and identification of the culprit drug is essential to prevent future reactions as well as for the identification of safe treatment alternatives.
drug hypersensitivity
in vitro tests
diagnostic
IgE
T cells
1. Introduction
Drug hypersensitivity reactions (DHRs) are adverse effects of pharmaceutical formulations (including active drugs and excipients) that clinically resemble allergies. DHRs can be allergic or non-allergic in nature, with drug allergies being immunologically mediated DHRs. The term non-allergic hypersensitivity is preferred for those reactions in which the underlying pathogenic mechanisms do not involve a B cell-mediated or T cell-mediated drug-specific immune response, which have also been referred to as anaphylactoid or pseudoallergic by several authors [1][2]. However, B and T cell-specific immune mechanisms are not always easily identified, even in allergic reactions.
2. Drug Hypersensitivity Reactions: Clinical Classification and Phenotypes
The classification of drug allergic reactions is challenging. From the clinical point of view, DHRs are usually classified as immediate or non-immediate (delayed) depending on the time of onset during treatment [2]. Immediate reactions typically appear within 1–6 h after the last drug administration; non-immediate reactions take place any time from 1 h after the initial drug administration [2]. They commonly occur after several days of treatment and are often associated with a delayed T cell-dependent type of allergic mechanism [2], although IgG/IgM and immune complex-mediated diseases can also manifest as delayed hypersensitivity. Non-immediate reactions can also occur more rapidly after re-exposure in patients with prior adverse events.
2.1. Immediate Drug Hypersensitivity Reactions
Immediate allergic DHRs are induced by an IgE-mediated mechanism that activates mast cells and basophils in an antigen-specific manner through the high-affinity IgE receptor (FcεRI) expressed on their cell membrane. This activation promotes the release of preformed mediators (mainly tryptase and histamine), inducing symptoms within minutes, and the production of new mediators (such as leukotrienes, prostaglandins, and cytokines) [2].
Symptoms induced by the activation of mast cells and basophils range from mild exanthemas and urticaria/hives, flushing, pruritus, angioedema, gastrointestinal, and respiratory symptoms (rhinorrhea, dyspnea, bronchospasm) to cardiovascular involvement (tachycardia, hypotension) and life-threatening reactions such as severe anaphylaxis [2]. Recognition of anaphylaxis can be difficult, given that different reaction patterns could cause clinical uncertainty. A detailed assessment of clinical reaction features and severity grading might help in diagnosis [3].
Recently, immediate reactions induced by specific IgG anti-drug antibodies (ADAs) have been described [4], called ADA-mediated reactions [5]. The cytokine release induced by some specific drugs, such as chemotherapeutics and biologics, is also related to the development of immediate DHRs, termed cytokine release reactions, with symptoms differing from those triggered by mast cell/basophil activation, including fever, chills, pain (head, back, chest), rigors, and desaturation [5]. When symptoms of both ADA-mediated reactions and cytokine release reactions concur, the reactions are called mixed reactions [5].
2.2. Non-Immediate/Delayed Allergic Drug Hypersensitivity Reactions
Non-immediate or delayed allergic DHRs can manifest with variable cutaneous symptoms, such as delayed urticaria, maculopapular eruptions, fixed drug eruptions (FDEs), vasculitis, Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN), generalized bullous fixed drug eruptions, drug reaction with eosinophilia and systemic symptoms (DRESS), acute generalized exanthematous pustulosis (AGEP), and symmetrical drug-related intertriginous and flexural exanthemas (SDRIFE). Internal organs can be affected either alone or with cutaneous symptoms (hypersensitivity syndrome (HSS)/DRESS/drug-induced hypersensitivity, vasculitis, SJS/TEN) and can include hepatitis, renal failure, pneumonitis, anemia, neutropenia, and thrombocytopenia [2].
DRESS, SJS/TEN, and AGEP are considered severe cutaneous adverse reactions (SCARs) to drugs due to their high morbidity and mortality [6], with SJS/TEN being the most severe form of SCAR [7], with an overall mortality of 34% at one year post-reaction [8].
Accurate diagnosis of specific clinical entities is key for identification of potential culprits. An exhaustive description of criteria for differential diagnosis is beyond the scope of this research. Details on this topic can be found in several previous publications [7][9][10][11][12].
3. Pathogenesis and Pathophysiology
Adverse drug reactions (ADRs) are due to various mechanisms. Rawlings and Thompson proposed a first classification including two groups of ADRs: type A and type B [13]. Type A reactions are due to the pharmacological activity of the drug; they can occur in every individual and are predictable to some extent. Type B reactions are less well defined and comprise approximately 15% of all adverse reactions. Allergic DHRs are a subgroup of type B reactions. In recent classifications, Type A reactions are labeled as “on-target” reactions, whereas type B are referred to as “off-target” adverse reactions [14].
As previously mentioned, the most accepted clinical classification of DHRs takes into account the time between the start of treatment and the onset of symptoms.
The classification of hypersensitivity reactions proposed by Gell and Coombs links the clinical phenotype to the immune mechanism involved. The immediate symptoms (urticaria, anaphylaxis) are categorized as type I hypersensitivity and are caused by allergen-specific IgE and mast cell degranulation. Delayed symptoms (e.g., exanthemas, hepatitis, Stevens-Johnson Syndrome/toxic epidermal necrolysis, DRESS) that are dependent on the activation of drug-specific T cells are classified as type IV hypersensitivity reactions. Some delayed reactions, such as hemolysis, thrombocytopenia, arthralgia, and vasculitis, could be due to drug-induced IgG/IgM antibodies (type II and type III hypersensitivity) and are less frequent [15][16] (Figure 1).
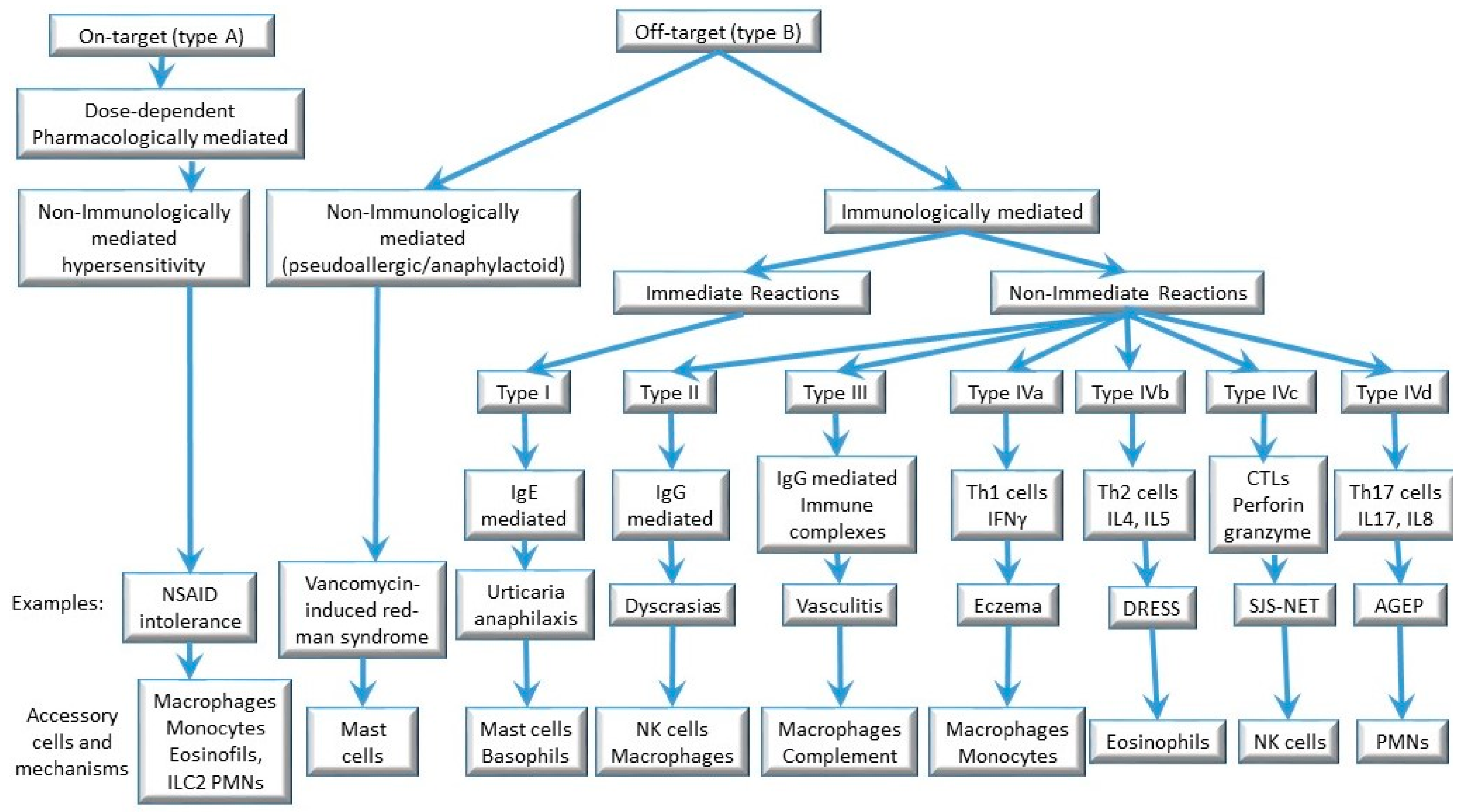
Figure 1. Mechanisms involved in drug hypersensitivity reactions.
Most medications are small molecular weight compounds with simple chemical structures that are not easily recognized by the immune system so as to elicit a primary immune response involving specific T or B cells. Thus, most drugs are not effective immunogens. However, some drugs (or their metabolites) can become immunogenic after covalent binding to host proteins (e.g., albumin). The drug is then referred to as a “hapten”, the host protein as a “carrier”, and the complex or conjugate as “hapten–carrier”. Hapten–carrier conjugates are immunogenic for B cells (antibody responses) and for T cells. Drugs that cannot act directly as haptens but give rise to reactive metabolites are referred to as prohaptens.
Drugs with the capability to haptenate host proteins are penicillins and other beta-lactam antibiotics (cephalosporins, carbapenems, and monobactams), sulfonamides, metamizole, quinolones, radiocontrast media, and muscle relaxants (rocuronium, succinylcholine), although the list is not exhaustive. Drugs that can generate reactive metabolites (prohaptens) are sulfonamide antimicrobials (sulfamethoxazole), phenacetin, or halothane, phenytoin, carbamazepine, and lamotrigine (although aromatic anticonvulsants are not common elicitors of IgE response). Also, several drugs can behave as a hapten, and their metabolites also have this capacity (e.g., metamizole). Recombinant proteins such as monoclonal antibodies, solubilized receptors and cytokines, insulin and other hormones, enzymes, protamine, antisera, and vaccines are pharmaceutical products that can elicit an antibody response by themselves because they are peptides or proteins (true allergens or immunogens) [17]. Hapten–carrier complexes are processed by antigen-presenting cells, and the haptenated peptides are then presented to T cells via human leukocyte antigen (HLA) molecules, which are recognized by T cell receptors.
3.1. Immediate IgE-Mediated Allergic Reactions (Type I Reactions)
Various mechanisms and pathways can be involved in mast cell activation that lead to immediate hypersensitivity symptoms, including immunoglobulin-mediated and direct mast cell activation. Symptoms are similar, independent of the underlying mechanism involved and caused by the release of mediators such as histamine, tryptase, platelet-activating factor (PAF), and cysteinyl leukotrienes. Histamine induces smooth muscle constriction and increases vascular permeability, leading to flushing, pruritus, rhinorrhea, tachycardia, and bronchospasm. Tryptase activates the complement cascade, the coagulation pathway, and the kallikrein–kinin system, contributing to the development of hypotension and angioedema. PAF and cysteinyl leukotrienes also enhance vascular permeability, leading to hypotension [18]. Type I reactions require the presence of drug-specific immunoglobulin IgE.
The formation of drug-specific IgE starts with the sensitization stage, including the activation of Th2 and Tfh cells by the drug. B and T cell interactions allow the production of primary response of antibodies (IgM). Upon antigen encounter and B and T cell activation, CD40-CD40L interactions, in conjunction with interleukin (IL)-21 and Th2 cytokines, enable further immunoglobulin class-switching [19][20]. The B cells then proliferate, mature, and differentiate into IgE-secreting plasma cells. Drug-specific IgE molecules diffuse through the circulation and attach to the high-affinity IgE receptor (FcεRI) on the surface of mast cells and basophils throughout the body.
When an individual is re-exposed to the medication, the effector stage is induced. During the effector stage, the drug–carrier complex binds to drug-specific IgE on the surface of mast cells and/or basophils. Consequently, the cross-linking of two or more high-affinity IgE receptors results in a sudden and widespread activation and release of an array of vasoactive mediators (e.g., histamine, prostaglandins, leukotrienes) [21].
Sensitization and effector stages can also occur during the same treatment course if it lasts long enough.
Beta-lactam antibiotics are considered the primary hapten triggers of IgE-mediated anaphylaxis induced by drugs [22]. An additional mechanism by which IgE-mediated reactions arise is previous sensitization to a cross-reacting agent, e.g., sensitization to quaternary ammonium compounds in cosmetic and personal care products, leading to type I reactions to neuromuscular-blocking agents used in anesthesia induction, which contain tertiary and quaternary substituted ammonium structures [17].
3.2. Non-Immediate T Cell-Mediated Allergic Reactions (Type IV Reactions)
Type IV hypersensitivity reactions are induced by activated T cells, with participation of both CD4+ and CD8+ T cells. Advances in the understanding of T cell functions led to a sub-classification of T cell-mediated hypersensitivity reactions into three subgroups (types IVa, IVb, IVc) [23]. A fourth subgroup, type IVd, was later added to sub-categorize type IV reactions into types IVa–IVd according to the dominant cytokines and to the contribution of certain subpopulations of leukocytes to skin inflammation and tissue damage [24] (Figure 1).
In this classification, type IVa reactions involve the actions of CD4+ T helper (Th)1 cells. Type IVb reactions correspond to Th2 responses with production of IL-4, IL-13, and IL-5, which facilitate eosinophilic inflammation. In type IVc reactions, cytotoxic CD8+ T cells are the main effectors of tissue injury, with contribution of natural killer cells. Lastly, in type IVd reactions, T cells secreting IL-8/CXCL8 promote neutrophilia as well as neutrophil recruitment to affected tissue [24]. Traditionally, DRESS is considered a type IVb Th2-driven reaction, SJS/TEN is a type IVc cytotoxic reaction, and AGEP is a type IVd reaction [25][26] (Figure 1). Although this classification might be useful, there is overlap between the subtypes, which are not mutually exclusive. For example, high interferon (IFN)-γ levels have been identified in serum and blister fluid from patients with SJS/TEN [27], and drug-specific CD8+ cytotoxic T cells can also be involved in DRESS and AGEP [28]. Of note, any of these reactions can occur in response to any drug, meaning that similar immune mechanisms are triggered in response to different chemical compounds.
T cells recognize the antigen through their antigen-specific T cell receptors (TCRs). Somatic recombination of the TCR genes can lead to a unique, randomly generated repertoire of TCR specificities in each individual [29]. To be stimulated, TCRs must bind a bimolecular complex displayed at the surface of the antigen-presenting (or target) cells, consisting of a fragment of a protein antigen (peptide) bound in the peptide-binding cleft of an HLA molecule. The genes coding HLA molecules are clustered together in the major histocompatibility complex (MHC) in chromosome 6 in humans. Given that the MHC is the most polymorphic region in the human genome, the available HLA haplotypes tend to be specific for each individual. The repertoire of available TCRs, together with the available HLA haplotype in each individual, limits the possibilities for developing specific adaptive immune cell responses to encountered antigens. The occurrence of a DHR needs the conjunction of several risk factors, including the availability of adequate HLA and TCR variants (Jackpot theory) [30].
Various models have been proposed for xenobiotic drug recognition by specific TCRs. The first model considers the fact that some drugs behave as haptens, as explained above. Hapten–protein conjugates can elicit hapten-specific adaptive immune responses because the hapten can be recognized by specific TCRs after covalent binding to protein-derived peptides presented by HLA molecules. However, in vitro development of drug-specific T cell clones has led to the finding that some lymphocytes can be stimulated in the presence of specific drugs and HLA molecules in the absence of antigen-presenting cells’ active metabolism. This led to the hypothesis of non-covalent interaction between TCR, the HLA–peptide complex, and the drug (no need for haptenization), and the development of the so-called “pharmacological interaction (p-i)” model [31]. Any drug can stimulate specific T cells through this mechanism. More recently, it has been demonstrated that the anti-retroviral drug, abacavir, can accommodate itself within one of the pockets of the peptide-binding cleft, specifically in HLA-B*57:01, changing its conformation and allowing the binding of a new repertoire of peptides. Given that this new peptide repertoire is not found in the thymus at the moment of thymic selection, T cells autoreactive to these new peptides presented by HLA-B*57:01 “filled” with abacavir can be found in the periphery and can behave as alloreactive T cells, leading to the development of hypersensitivity reactions to abacavir in carriers of the risk allele HLA-B*57:01 [32][33][34]. This is known as the altered peptide model (Reviewed in [35]).
Although primary immune responses are well demonstrated for xenobiotics behaving as haptens, the tendency for some drug reactions to occur with short latency periods while maintaining long-lasting memory T cell responses suggests the absence of a sensitization or priming phase. It also suggests that cross-reactive memory T cells could be important in individuals exposed to a drug for the first time. As an example, abacavir-reacting memory CD8+ T cells have been identified in unexposed individuals, suggesting priming by earlier exposure to another antigen [36]. This phenomenon is known as heterologous immunity [37], and it is not mutually exclusive to other models of T cell activation in drug hypersensitivity reactions [38].
References
- Johansson, S.G.; Bieber, T.; Dahl, R.; Friedmann, P.S.; Lanier, B.Q.; Lockey, R.F.; Motala, C.; Ortega Martell, J.A.; Platts-Mills, T.A.; Ring, J.; et al. Revised Nomenclature for Allergy for Global Use: Report of the Nomenclature Review Committee of the World Allergy Organization, October 2003. J. Allergy Clin. Immunol. 2004, 113, 832–836.
- Demoly, P.; Adkinson, N.F.; Brockow, K.; Castells, M.; Chiriac, A.M.; Greenberger, P.A.; Khan, D.A.; Lang, D.M.; Park, H.S.; Pichler, W.; et al. International Consensus on Drug Allergy. Allergy Eur. J. Allergy Clin. Immunol. 2014, 69, 420–437.
- Brown, S.G.A. Clinical Features and Severity Grading of Anaphylaxis. J. Allergy Clin. Immunol. 2004, 114, 371–376.
- Jönsson, F.; De Chaisemartin, L.; Granger, V.; Gouel-Chéron, A.; Gillis, C.M.; Zhu, Q.; Dib, F.; Nicaise-Roland, P.; Ganneau, C.; Hurtado-Nedelec, M.; et al. An IgG-Induced Neutrophil Activation Pathway Contributes to Human Drug-Induced Anaphylaxis. Sci. Transl. Med. 2019, 11, eaat1479.
- Bavbek, S.; Pagani, M.; Alvarez-Cuesta, E.; Castells, M.; Dursun, A.B.; Hamadi, S.; Madrigal-Burgaleta, R.; Sanchez-Sanchez, S.; Vultaggio, A. Hypersensitivity Reactions to Biologicals: An EAACI Position Paper. Allergy Eur. J. Allergy Clin. Immunol. 2022, 77, 39–54.
- Mockenhaupt, M. Epidemiology and Causes of Severe Cutaneous Adverse Reactions to Drugs. In Drug Hypersensitivity; Karger Publishers: Basel, Karger, 2007.
- Paulmann, M.; Mockenhaupt, M. Severe Drug-Induced Skin Reactions: Clinical Features, Diagnosis, Etiology, and Therapy. J. Dtsch. Dermatol. Ges. 2015, 13, 625–645.
- Sekula, P.; Dunant, A.; Mockenhaupt, M.; Naldi, L.; Bouwes Bavinck, J.N.; Halevy, S.; Kardaun, S.; Sidoroff, A.; Liss, Y.; Schumacher, M.; et al. Comprehensive Survival Analysis of a Cohort of Patients with Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis. J. Investig. Dermatol. 2013, 133, 1197–1204.
- Guvenir, H.; Arikoglu, T.; Vezir, E.; Misirlioglu, E.D. Clinical Phenotypes of Severe Cutaneous Drug Hypersensitivity Reactions. Curr. Pharm. Des. 2019, 25, 3840–3854.
- Mockenhaupt, M. Bullous Drug Reactions. Acta Derm. Venereol. 2020, 100, 122–134.
- Lipowicz, S.; Sekula, P.; Ingen-Housz-Oro, S.; Liss, Y.; Sassolas, B.; Dunant, A.; Roujeau, J.-C.; Mockenhaupt, M. Prognosis of Generalized Bullous Fixed Drug Eruption: Comparison with Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis. Br. J. Dermatol. 2013, 168, 726–732.
- Aster, R.H. Blood Dyscrasias Caused by Hypersensitivity to Drugs. In Drug Hypersensitivity; Karger Publishers: Berlin, Germany, 2007; pp. 306–320.
- Rawlins, M.D.; Thompson, J.W. Pathogenesis of Adverse Drug Reactions; Oxford University Press: New York, NY, USA, 1977; ISBN 0-19-263206-X.
- Phillips, E.J. Classifying ADRs--Does Dose Matter? Br. J. Clin. Pharmacol. 2016, 81, 10–12.
- Coombs, R.R.A.; Gell, P. Classification of Allergic Reactions Responsible for Clinical Hypersensitivity and Disease. In Clinical Aspects of Immunology; Coombs, R.R.A., Gells, P.G.H., Eds.; Oxford University Press: New York, NY, USA, 1976; p. 575.
- Pichler, W.J. Immune Pathomechanism and Classification of Drug Hypersensitivity. Allergy Eur. J. Allergy Clin. Immunol. 2019, 74, 1457–1471.
- Pichler, W.J. Drug Allergy: Pathogenesis-UpToDate. Available online: https://www.uptodate.com/contents/drug-allergy-pathogenesis?search=drug allergy &source=search_result&selectedTitle=4~150&usage_type=default&display_rank=4 (accessed on 26 May 2023).
- Montañez, M.I.; Mayorga, C.; Bogas, G.; Barrionuevo, E.; Fernandez-Santamaria, R.; Martin-Serrano, A.; Laguna, J.J.; Torres, M.J.; Fernandez, T.D.; Doña, I. Epidemiology, Mechanisms, and Diagnosis of Drug-Induced Anaphylaxis. Front. Immunol. 2017, 8, 614.
- Kuchen, S.; Robbins, R.; Sims, G.P.; Sheng, C.; Phillips, T.M.; Lipsky, P.E.; Ettinger, R. Essential Role of IL-21 in B Cell Activation, Expansion, and Plasma Cell Generation during CD4+ T Cell-B Cell Collaboration. J. Immunol. 2007, 179, 5886–5896.
- Kobayashi, T.; Iijima, K.; Dent, A.L.; Kita, H. Follicular Helper T Cells Mediate IgE Antibody Response to Airborne Allergens. J. Allergy Clin. Immunol. 2017, 139, 300–313.e7.
- Metcalfe, D.D.; Peavy, R.D.; Gilfillan, A.M. Mechanisms of Mast Cell Signaling in Anaphylaxis. J. Allergy Clin. Immunol. 2009, 124, 639.
- Torres, M.J.; Blanca, M. The Complex Clinical Picture of Beta-Lactam Hypersensitivity: Penicillins, Cephalosporins, Monobactams, Carbapenems, and Clavams. Med. Clin. N. Am. 2010, 94, 805–820.
- Janeway, C.A.; Travers, P.; Walport, M.S.M. Allergy and Hypersensitivity. In Immunology: The Immune System in Health and Disease; Garland Publisher: New York, NY, USA, 2001.
- Pichler, W.J. Delayed Drug Hypersensitivity Reactions. Ann. Intern. Med. 2003, 139, 683–693.
- Pichler, W.J. Drug Hypersensitivity Reactions: Classification and Relationship to T-Cell Activation. In Drug Hypersensitivity; Pichler, W.J., Ed.; Karger: Basel, Switzerland, 2007; pp. 168–189.
- Phillips, E.J.; Bigliardi, P.; Bircher, A.J.; Broyles, A.; Chang, Y.-S.; Chung, W.-H.; Lehloenya, R.; Mockenhaupt, M.; Peter, J.; Pirmohamed, M.; et al. Controversies in Drug Allergy: Testing for Delayed Reactions. J. Allergy Clin. Immunol. 2019, 143, 66–73.
- Nassif, A.; Moslehi, H.; Le Gouvello, S.; Bagot, M.; Lyonnet, L.; Michel, L.; Boumsell, L.; Bensussan, A.; Roujeau, J.-C. Evaluation of the Potential Role of Cytokines in Toxic Epidermal Necrolysis. J. Investig. Dermatol. 2004, 123, 850–855.
- Neukomm, C.B.; Yawalkar, N.; Helbling, A.P.W. T-Cell Reactions to Drugs in Distinct Clinical Manifestations of Drug Allergy. Le. J. Investig. Allergol. Clin. Immunol. 2001, 11, 275–284.
- Venturi, V.; Price, D.A.; Douek, D.C.; Davenport, M.P. The Molecular Basis for Public T-Cell Responses? Nat. Rev. Immunol. 2008, 8, 231–238.
- Yang, S.C.; Chen, C.B.; Lin, M.Y.; Zhang, Z.Y.; Jia, X.Y.; Huang, M.; Zou, Y.F.; Chung, W.H. Genetics of Severe Cutaneous Adverse Reactions. Front. Med. 2021, 8, 652091.
- Pichler, W.J.; Adam, J.; Watkins, S.; Wuillemin, N.; Yun, J.; Yerly, D. Drug Hypersensitivity: How Drugs Stimulate T Cells via Pharmacological Interaction with Immune Receptors. Int. Arch. Allergy Immunol. 2015, 168, 13–24.
- Illing, P.T.; Vivian, J.P.; Dudek, N.L.; Kostenko, L.; Chen, Z.; Bharadwaj, M.; Miles, J.J.; Kjer-Nielsen, L.; Gras, S.; Williamson, N.A.; et al. Immune Self-Reactivity Triggered by Drug-Modified HLA-Peptide Repertoire. Nature 2012, 486, 554–558.
- Norcross, M.A.; Luo, S.; Lu, L.; Boyne, M.T.; Gomarteli, M.; Rennels, A.D.; Woodcock, J.; Margulies, D.H.; McMurtrey, C.; Vernon, S.; et al. Abacavir Induces Loading of Novel Self-Peptides into HLA-B*57: 01: An Autoimmune Model for HLA-Associated Drug Hypersensitivity. AIDS 2012, 26, F21–F29.
- Ostrov, D.A.; Grant, B.J.; Pompeu, Y.A.; Sidney, J.; Harndahl, M.; Southwood, S.; Oseroff, C.; Lu, S.; Jakoncic, J.; de Oliveira, C.A.F.; et al. Drug Hypersensitivity Caused by Alteration of the MHC-Presented Self-Peptide Repertoire. Proc. Natl. Acad. Sci. USA 2012, 109, 9959–9964.
- Illing, P.T.; Mifsud, N.A.; Purcell, A.W. Allotype Specific Interactions of Drugs and HLA Molecules in Hypersensitivity Reactions. Curr. Opin. Immunol. 2016, 42, 31–40.
- Lucas, A.; Lucas, M.; Strhyn, A.; Keane, N.M.; McKinnon, E.; Pavlos, R.; Moran, E.M.; Meyer-Pannwitt, V.; Gaudieri, S.; D’Orsogna, L.; et al. Abacavir-Reactive Memory T Cells Are Present in Drug Naïve Individuals. PLoS ONE 2015, 10, e0117160.
- Agrawal, B. Heterologous Immunity: Role in Natural and Vaccine-Induced Resistance to Infections. Front. Immunol. 2019, 10, 2631.
- Gibson, A.; Deshpande, P.; Campbell, C.N.; Krantz, M.S.; Mukherjee, E.; Mockenhaupt, M.; Pirmohamed, M.; Palubinsky, A.M.; Phillips, E.J. Updates on the Immunopathology and Genomics of Severe Cutaneous Adverse Drug Reactions. J. Allergy Clin. Immunol. 2023, 151, 289–300.e4.
More
Information
Subjects:
Allergy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
912
Revisions:
4 times
(View History)
Update Date:
30 Aug 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No