 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alfredo Caturano | -- | 2581 | 2023-08-17 11:29:39 | | | |
| 2 | Jessie Wu | + 10 word(s) | 2591 | 2023-08-18 07:21:52 | | |
Video Upload Options
Oxidative stress is a critical factor in the pathogenesis and progression of diabetes and its associated complications. The imbalance between reactive oxygen species (ROS) production and the body’s antioxidant defence mechanisms leads to cellular damage and dysfunction. In diabetes, chronic hyperglycaemia and mitochondrial dysfunction contribute to increased ROS production, further exacerbating oxidative stress. This oxidative burden adversely affects various aspects of diabetes, including impaired beta-cell function and insulin resistance, leading to disrupted glucose regulation. Additionally, oxidative stress-induced damage to blood vessels and impaired endothelial function contribute to the development of diabetic vascular complications such as retinopathy, nephropathy, and cardiovascular diseases. Moreover, organs and tissues throughout the body, including the kidneys, nerves, and eyes, are vulnerable to oxidative stress, resulting in diabetic nephropathy, neuropathy, and retinopathy. Strategies to mitigate oxidative stress in diabetes include antioxidant therapy, lifestyle modifications, and effective management of hyperglycaemia
1. Genesis of Oxidative Stress in Diabetes
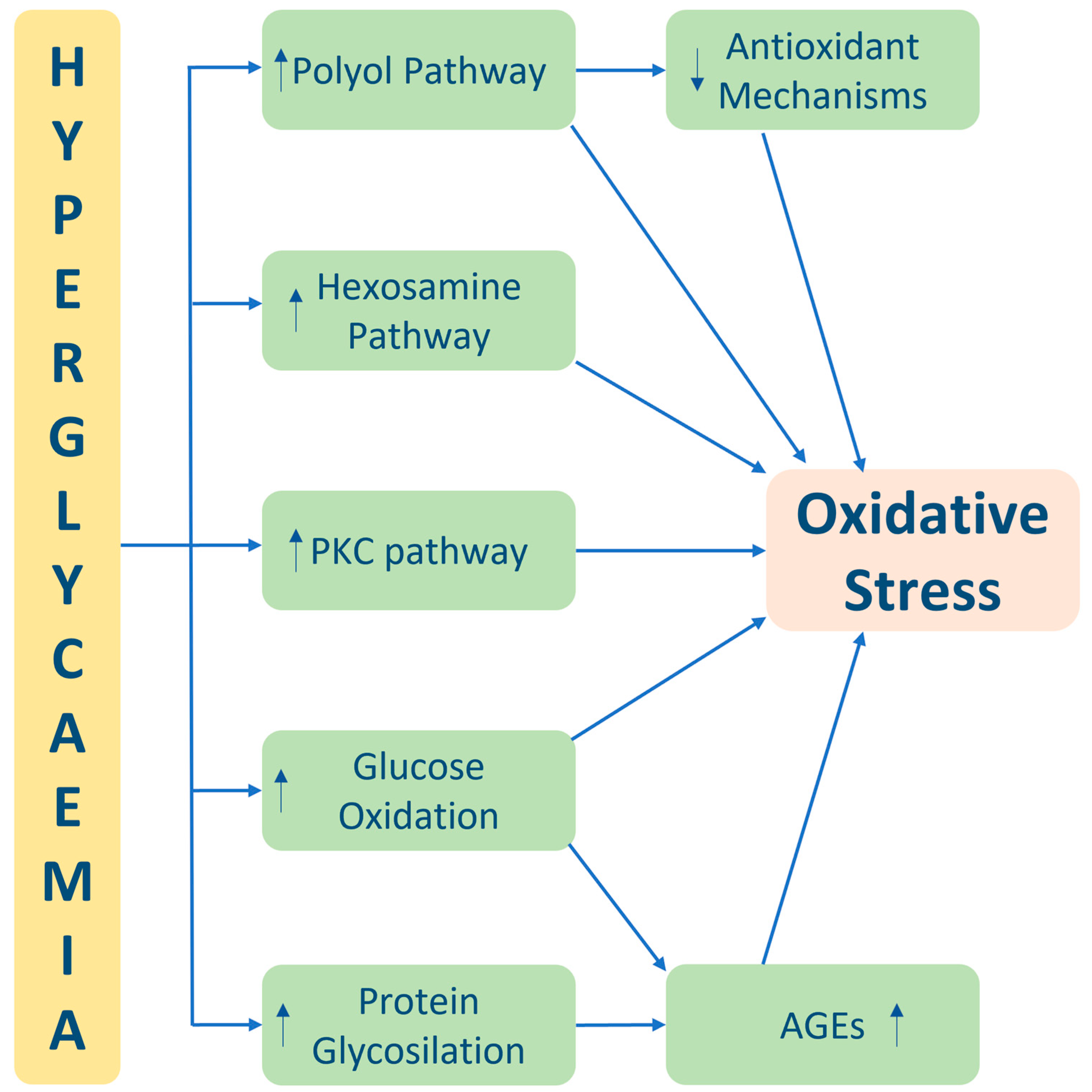
1.1. Pentose Phosphate and Glycolytic Pathways and Oxidative Stress
1.2. Inflammation and Oxidative Stress
2. Oxidative Stress Role in the Development of Type 2 Diabetes Complications
3. Physical Exercise and Oxidative Stress
3.1. Effects of Different Physical Exercise Protocols on Oxidative Stress Markers in Patients with Type 2 Diabetes Mellitus
3.2. Resistance Exercise
3.3. Continuous Moderate-Intensity Exercise
3.4. High-Intensity Interval Exercise
3.5. Concurrent Exercise
References
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63.
- Ighodaro, O.M. Molecular pathways associated with oxidative stress in diabetes mellitus. Biomed. Pharmacother. 2018, 108, 656–662.
- Makino, A.; Scott, B.T.; Dillmann, W.H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 2010, 53, 1783–1794.
- Funk, S.D.; Yurdagul, A., Jr.; Orr, A.W. Hyperglycemia and endothelial dysfunction in atherosclerosis: Lessons from type 1 diabetes. Int. J. Vasc. Med. 2012, 2012, 569654.
- Figueroa-Romero, C.; Sadidi, M.; Feldman, E.L. Mechanisms of disease: The oxidative stress theory of diabetic neuropathy. Rev. Endocr. Metab. Disord. 2008, 9, 301–314.
- Garg, S.S.; Gupta, J. Polyol pathway and redox balance in diabetes. Pharmacol. Res. 2022, 182, 106326.
- Twarda-Clapa, A.; Olczak, A.; Białkowska, A.M.; Koziołkiewicz, M. Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells 2022, 11, 1312.
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070.
- Luc, K.; Schramm-Luc, A.; Guzik, T.J.; Mikolajczyk, T.P. Oxidative stress and inflammatory markers in prediabetes and diabetes. J. Physiol. Pharmacol. 2019, 70, 809–824.
- Nie, H.; Yi, W. O-GlcNAcylation, a sweet link to the pathology of diseases. J. Zhejiang. Univ. Sci. B. 2019, 20, 437–448.
- Buse, M.G. Hexosamines, Insulin resistance, and the complications of diabetes: Current status. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1–E8.
- Darenskaya, M.A.; Kolesnikova, L.I.; Kolesnikov, S.I. Oxidative Stress: Pathogenetic Role in Diabetes Mellitus and Its Complications and Therapeutic Approaches to Correction. Bull. Exp. Biol. Med. 2021, 171, 179–189.
- Sreedhar, A.; Zhao, Y. Uncoupling protein 2 and metabolic diseases. Mitochondrion 2017, 34, 135–140.
- Sjöholm, Å.; Corbett, J.A.; Leung, P.S.; Portha, B. Editorial: Understanding the gene expression of β cell dysfunction in diabetes. , Front. Endocrinol. Lausanne 2022, 13, 1069991.
- Badawi, A.; Klip, A.; Haddad, P.; Cole, D.E.; Bailo, B.G.; El-Sohemy, A.; Karmali, M. Type 2 diabetes and inflammation: Prospects for biomarkers of risk and nutritional intervention. Diabetes Metab. Syndr. Obes. 2010, 3, 173–186.
- Navarro, J.F.; Mora, C. Role of inflammation in diabetic complications. Nephrol. Dial. Transplant. 2005, 20, 2601–2604.
- Crook, M. Is type 2 diabetes mellitus a disease of the innate immune system. Diabet. Med. 2004, 21, 203–207.
- Westermann, D.; Van Linthout, S.; Dhayat, S.; Dhayat, N.; Schmidt, A.; Noutsias, M.; Song, X.Y.; Spillmann, F.; Riad, A.; Schultheiss, H.P.; et al. Tumor necrosis factor-alpha antagonism protects from myocardial inflammation and fibrosis in experimental diabetic cardiomyopathy. Basic. Res. Cardiol. 2007, 102, 500–507.
- Zhang, P.; Zhang, X.; Brown, J.; Vistisen, D.; Sicree, R.; Shaw, J.; Nichols, G. Global healthcare expenditure on diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010, 87, 293–301.
- Derosa, G.; D’Angelo, A.; Bonaventura, A.; Bianchi, L.; Romano, D.; Maffioli, P. Effects of berberine on lipid profile in subjects with low cardiovascular risk. Expert. Opin. Biol. Ther. 2013, 13, 475–482.
- Butkowski, E.G.; Jelinek, H.F. Hyperglycemia, oxidative stress and inflammation markers. Redox Rep. 2017, 22, 257–264.
- Collier, B.; Dosssett, L.A.; May, A.K.; Diaz, J.J. Glucose control and the inflammatory response. Nutr. Clin. Pract. 2008, 23, 3–15.
- Palmiero, G.; Cesaro, A.; Vetrano, E.; Pafundi, P.C.; Galiero, R.; Caturano, A.; Moscarella, E.; Gragnano, F.; Salvatore, T.; Rinaldi, L.; et al. Impact of SGLT2 Inhibitors on Heart Failure: From Pathophysiology to Clinical Effects. Int. J. Mol. Sci. 2021, 22, 5863.
- Falco, L.; Tessitore, V.; Ciccarelli, G.; Malvezzi, M.; D’Andrea, A.; Imbalzano, E.; Golino, P.; Russo, V. Antioxidant Properties of Oral Antithrombotic Therapies in Atherosclerotic Disease and Atrial Fibrillation. Antioxidants 2023, 12, 1185.
- Russo, V.; Fabiani, D. Put out the fire: The pleiotropic anti-inflammatory action of non-vitamin K oral anticoagulants. Pharmacol. Res. 2022, 182, 106335.
- Salvatore, T.; Galiero, R.; Caturano, A.; Vetrano, E.; Rinaldi, L.; Coviello, F.; Di Martino, A.; Albanese, G.; Marfella, R.; Sardu, C.; et al. Effects of Metformin in Heart Failure: From Pathophysiological Rationale to Clinical Evidence. Biomolecules 2021, 11, 1834.
- Tousoulis, D.; Papageorgiou, N.; Androulakis, E.; Siasos, G.; Latsios, G.; Tentolouris, K.; Stefanadis, C. Diabetes mellitus-associated vascular impairment: Novel circulating biomarkers and therapeutic approaches. J. Am. Coll. Cardiol. 2013, 62, 667–676.
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820.
- Esposito, K.; Nappo, F.; Marfella, R. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: Role of oxidative stress. Circulation 2002, 106, 2067–2072.
- Monnier, L.; Lapinski, H.; Colette, C. Contributions of fasting and postprandial plasma glucose increments to the overall diurnal hyperglycemia of type 2 diabetic patients: Variations within increasing levels of HbA(1c). Diabetes Care 2003, 26, 881–885.
- Huang, C.; Kim, Y.; Caramori, M.L.; Fish, A.J.; Rich, S.S.; Miller, M.E.; Russell, G.B.; Mauer, M. Cellular basis of diabetic nephropathy: II. The transforming growth factor-beta system and diabetic nephropathy lesions in type 1 diabetes. Diabetes 2002, 51, 3577–3581.
- Halliwell, B. The wanderings of a free radical. Free Radic. Biol. Med. 2009, 46, 531–542.
- Yadav, P.; Sarkar, S.; Bhatnagar, D. Lipid peroxidation and antioxidant enzymes in erythrocytes and tissues in aged diabetic rats. Indian. J. Exp. Biol. 1997, 35, 389–392.
- Hamden, K.; Carreau, S.; Jamoussi, K.; Miladi, S.; Lajmi, S.; Aloulou, D.; Ayadi, F.; Elfeki, A. 1Alpha, 25 dihydroxyvitamin D3: Therapeutic and preventive effects against oxidative stress, hepatic, pancreatic and renal injury in alloxan-induced diabetes in rats. J. Nutr. Sci. Vitaminol. 2009, 55, 215–222.
- Fialkow, L.; Wang, Y.; Downey, G.P. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic. Biol. Med. 2007, 42, 153–164.
- González, R.G.; Barnett, P.; Aguayo, J.; Cheng, H.M.; Chylack, L.T., Jr. Direct measurement of polyol pathway activity in the ocular lens. Diabetes. 1984, 33, 196–199.
- Morré, D.M.; Lenaz, G.; Morré, D.J. Surface oxidase and oxidative stress propagation in aging. J. Exp. Biol. 2000, 203, 1513–1521.
- Ishill, N.; Patel, K.P.; Lane, P.H. Nitric oxide synthesis and oxidative stress in the renal cortex of rats with diabetes mellitus. J. Am. Soc. Nephrol. 2001, 12, 1630–1639.
- Etoh, T.; Inoguchi, T.; Kakimoto, M.; Sonoda, N.; Kobayashi, K.; Kuroda, J.; Sumimoto, H.; Nawata, H. Increased expression of NAD(P)H oxidase subunits, NOX4 and p22phox, in the kidney of streptozotocin-induced diabetic rats and its reversibility by interventive insulin treatment. Diabetologia 2003, 46, 1428–1437.
- Wu, L.J.; Wu, G.; Akhavan Sharif, M.R.; Baker, A.; Jia, Y.; Fahey, F.H.; Luo, H.R.; Feener, E.P.; Clapham, D.E. The voltage-gated proton channel, Hv1, enhances brain damage from ischemic stroke. Nat. Neurosci. 2012, 15, 565–573.
- Thallas-Bonke, V.; Thorpe, S.R.; Coughlan, M.T.; Fukami, K.; Yap, F.Y.; Sourris, K.C.; Penfold, S.A.; Bach, L.A.; Cooper, M.E.; Forbes, J.M. Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha-dependent pathway. Diabetes 2008, 57, 460–469.
- Noh, H.; King, G.L. The role of protein kinase C activation in diabetic nephropathy. Kidney Int. 2007, 72, S49–S53.
- Way, K.J.; Katai, N.; King, G.L. Protein kinase C and the development of diabetic vascular complications. Diabet. Med. 2001, 18, 945–959.
- Harani, H.; Otmane, A.; Makrelouf, M.; Ouadahi, N.; Abdi, A.; Berrah, A.; Zenati, A.; Alamir, B.; Koceir, E.A. The relationship between inflammation, oxidative stress and metabolic risk factors in type 2 diabetic patients. Ann. Biol. Clin. 2012, 70, 669–677.
- Salvatore, T.; Galiero, R.; Caturano, A.; Vetrano, E.; Loffredo, G.; Rinaldi, L.; Catalini, C.; Gjeloshi, K.; Albanese, G.; Di Martino, A.; et al. Coronary Microvascular Dysfunction in Diabetes Mellitus: Pathogenetic Mechanisms and Potential Therapeutic Options. Biomedicines 2022, 10, 2274.
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome, and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150.
- Elmarakby, A.A.; Sullivan, J.C. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc. Ther. 2012, 30, 49–59.
- Al Hannan, F.; Culligan, K.G. Human resistin and the RELM of inflammation in diabesity. Diab Metab. Syndr. 2015, 7, 54.
- Cozzolino, D.; Sessa, G.; Salvatore, T.; Sasso, F.C.; Giugliano, D.; Lefebvre, P.J.; Torella, R. The involvement of the opioid system in human obesity: A study in normal weight relatives of obese people. J. Clin. Endocrinol. Metab. 1996, 81, 713–718.
- Du, Y.; Miller, C.M.; Kern, T.S. Hyperglycemia increases mitochondrial superoxide in the retina and retinal cells. Free Rad. Biol. Med. 2003, 35, 1491–1499.
- Kafle, D.; Singh, N.; Singh, S.K.; Islam, N. Relationship between hyperglycemia, inflammation, and oxidative stress in type 2 diabetic nephropathy subjects. Int. J. Pharm. Biol. Arch. 2012, 3, 1203–1206.
- Sasso, F.C.; Carbonara, O.; Persico, M.; Iafusco, D.; Salvatore, T.; D’Ambrosio, R.; Torella, R.; Cozzolino, D. Irbesartan reduces the albumin excretion rate in microalbuminuric type 2 diabetic patients independently of hypertension: A randomised double-blind placebo-controlled crossover study. Diabetes Care 2002, 25, 1909–1913.
- Russell, J.W.; Sullivan, K.A.; Windebank, A.J.; Herrmann, D.N.; Feldman, E.L. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol. Dis. 1999, 6, 347–363.
- Gómez-Cabrera, M.; Domenech, E.; Viña, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free Radic. Biol. Med. 2008, 44, 126–131.
- Gacitua, T.; Karachon, L.; Romero, E.; Parra, P.; Poblete, C.; Russell, J.; Rodrigo, R. Effects of resistance training on oxidative stress-related biomarkers in metabolic diseases: A review. Sport. Sci. Health 2017, 14, 1–7.
- Mitranun, W.; Deerochanawong, C.; Tanaka, H.; Suksom, D. Continuous vs interval training on glycemic control and macro- and microvascular reactivity in type 2 diabetic patients. Scand. J. Med. Sci. Sports 2014, 24, 69–76.
- Vinetti, G.; Mozzini, C.; Desenzani, P.; Boni, E.; Bulla, L.; Lorenzetti, I.; Romano, C.; Pasini, A.; Cominacini, L.; Assanelli, D. Supervised exercise training reduces oxidative stress and cardiometabolic risk in adults with type 2 diabetes: A randomised controlled trial. Sci. Rep. 2015, 5, 9238.
- Gordon, B.A.; Benson, A.C.; Bird, S.R.; Fraser, S.F. Resistance training improves metabolic health in type 2 diabetes: A systematic review. Diabetes Res. Clin. Pract. 2009, 83, 157–175.
- de Oliveira, V.N.; Bessa, A.; Jorge, M.L.; Oliveira, R.J.; de Mello, M.T.; De Agostini, G.G.; Jorge, P.T.; Espindola, F.S. The effect of different training programs on antioxidant status, oxidative stress, and metabolic control in type 2 diabetes. Appl. Physiol. Nutr. Metab. 2012, 37, 334–344.
- Azizbeigi, K.; Azarbayjani, M.A.; Atashak, S.; Stannard, S.R. Effect of moderate and high resistance training intensity on indices of inflammatory and oxidative stress. Res. Sports Med. 2015, 23, 73–87.
- Cakir-Atabek, H.; Demir, S.; PinarbaŞili, R.D.; Gündüz, N. Effects of different resistance training intensity on indices of oxidative stress. J. Strength. Cond. Res. 2010, 24, 2491–2497.
- Quílez Llopiz, P.; Reig García-Galbis, M. Glycemic control through physical exercise in type 2 diabetes systematic review. Nutr. Hosp. 2015, 31, 1465–1472.
- Kurban, S.; Mehmetoglu, I.; Yerlikaya, H.F.; Gonen, S.; Erdem, S. Effect of chronic regular exercise on serum ischemia-modified albumina levels and oxidative stress in type 2 diabetes mellitus. Endocr. Res. 2011, 36, 116–123.
- Moghaddam, D.A.; Heber, A.; Capin, D.; Kreutz, T.; Opitz, D.; Lenzen, E.; Bloch, W.; Brixius, K.; Brinkmann, C. Training increases peroxiredoxin 2 contents in the erythrocytes of overweight/obese men suffering from type 2 diabetes. Wien. Med. Wochenschr. 2011, 161, 511–518.
- Krause, M.; Rodrigues-Krause, J.; O’Hagan, C.; Medlow, P.; Davison, G.; Susta, D.; Boreham, C.; Newsholme, P.; O’Donnell, M.; Murphy, C.; et al. The effects of aerobic exercise training at two different intensities in obesity and type 2 diabetes: Implications for oxidative stress, low-grade inflammation and nitric oxide production. Eur. J. Appl. Physiol. 2014, 114, 251–260.
- Nojima, H.; Watanabe, H.; Yamane, K.; Kitahara, Y.; Sekikawa, K.; Yamamoto, H.; Yokoyama, A.; Inamizu, T.; Asahara, T.; Kohno, N.; et al. Effect of aerobic exercise training on oxidative stress in patients with type 2 diabetes mellitus. Metabolism 2008, 57, 170–176.
- Poblete-Aro, C.; Russell-Guzmán, J.; Parra, P.; Díaz, E.; Reyes, D.; Saavedra, M.; Lanas, F. Efecto del ejercicio físico sobre marcadores de estrés oxidativo en pacientes con diabetes mellitus tipo 2 . Rev. Med. Chil. 2018, 146, 362–372.
- Buchheit, M.; Laursen, P.B. High-intensity interval training, solutions to the programming puzzle: Part I: Cardiopulmonary emphasis. Sports Med. 2013, 43, 313–338.
- Coffey, V.G.; Hawley, J.A. Concurrent exercise training: Do opposites distract? J. Physiol. 2016, 595, 2883–2896.
- Oliveira, C.; Simões, M.; Carvalho, J.; Ribeiro, J. Combined exercise for people with type 2 diabetes mellitus: A systematic review. Diabetes Res. Clin. Pract. 2012, 98, 187–198.

