Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mario Šestan | -- | 4049 | 2023-08-10 07:46:58 | | | |
| 2 | Rita Xu | Meta information modification | 4049 | 2023-08-10 08:12:52 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sestan, M.; Kifer, N.; Arsov, T.; Cook, M.; Ellyard, J.; Vinuesa, C.G.; Jelusic, M. Childhood-Onset Systemic Lupus Erythematosus. Encyclopedia. Available online: https://encyclopedia.pub/entry/47876 (accessed on 26 July 2026).
Sestan M, Kifer N, Arsov T, Cook M, Ellyard J, Vinuesa CG, et al. Childhood-Onset Systemic Lupus Erythematosus. Encyclopedia. Available at: https://encyclopedia.pub/entry/47876. Accessed July 26, 2026.
Sestan, Mario, Nastasia Kifer, Todor Arsov, Matthew Cook, Julia Ellyard, Carola G. Vinuesa, Marija Jelusic. "Childhood-Onset Systemic Lupus Erythematosus" Encyclopedia, https://encyclopedia.pub/entry/47876 (accessed July 26, 2026).
Sestan, M., Kifer, N., Arsov, T., Cook, M., Ellyard, J., Vinuesa, C.G., & Jelusic, M. (2023, August 10). Childhood-Onset Systemic Lupus Erythematosus. In Encyclopedia. https://encyclopedia.pub/entry/47876
Sestan, Mario, et al. "Childhood-Onset Systemic Lupus Erythematosus." Encyclopedia. Web. 10 August, 2023.
Copy Citation
The pathogenesis of childhood-onset systemic lupus erythematosus (cSLE) is complex and not fully understood. It involves three key factors: genetic risk factors, epigenetic mechanisms, and environmental triggers. Genetic factors play a significant role in the development of the disease, particularly in younger individuals. While cSLE has traditionally been considered a polygenic disease, it is now recognized that in rare cases, a single gene mutation can lead to the disease.
childhood-onset systemic lupus erythematosus
monogenic systemic lupus erythematosus
genetics
1. Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune condition with the potential to affect any organ system. It is characterized by the presence of antibodies that specifically target nuclear and cytoplasmic antigens, leading to the widespread inflammation of blood vessels and connective tissue [1]. This immune response also triggers complement activation and the deposition of immune complexes. Consequently, SLE is commonly referred to as “the disease with countless manifestations” due to its capacity to involve multiple organs and exhibit a wide range of clinical symptoms, varying from mild to life-threatening. The inflammatory process typically impacts the skin, kidneys, brain, lungs, and heart [2].
Around 15–20% of individuals with SLE experience the onset of the disease during childhood and receive a diagnosis before the age of 18 [3][4]. This specific form is commonly referred to as cSLE. Although there are similarities in the clinical presentation and immunological markers between children and adults with SLE, it is crucial to recognize cSLE as a distinct clinical entity due to several unique characteristics.
Firstly, in childhood, the clinical picture at the time of diagnosis tends to be more severe, with symptoms such as proteinuria, hemolytic anemia, leukopenia, and a rash in the zygomatic region [3][5][6]. Additionally, cSLE often affects multiple organs and systems, with a predilection for kidney involvement. Renal complications occur in approximately 60–80% of children and 35–50% of adults, as suggested by the literature [5]. Furthermore, there is a notable disparity in central nervous system involvement, affecting 20–50% of children and 10–25% of adults [5]. Conversely, lung issues (20–90% in adults compared to 15–40% in children) and joint problems (80–95% in adults compared to 60–70% in children) are more frequently observed in adult patients [7][8].
In cSLE, procedures like renal biopsies, dialysis, and transplantation are more common, while convulsions occur more frequently and the risk of myocardial infarction is elevated. Generally, cSLE follows a more aggressive clinical course, increasing the likelihood of permanent organ and system damage over time. Considering these factors, children require more intensive treatment, often involving the use of glucocorticoids and immunosuppressants. Consequently, compared to adults, children face a significantly higher risk of corticosteroid-related complications, such as cataracts and avascular bone necrosis [9].
Unlike SLE in adults, where the disease is approximately nine times more prevalent in females, the gender difference in children with cSLE is significantly less pronounced, with a ratio of approximately 4–5 girls to 1 boy. Additionally, it is important to note that cSLE is associated with primary immunodeficiencies, particularly deficiencies in complement components, which contribute to a higher disease activity index. Moreover, when considering early-onset SLE in children, genetic factors may play a more significant role compared to environmental and hormonal factors, which differs from adult-onset SLE [6]. Consequently, rare monogenic forms of SLE resulting from mutations in specific genes occur more frequently in childhood SLE. These monogenic forms follow Mendelian inheritance patterns and have fundamentally challenged the previously established notion of SLE as a solely polygenic disease [10]. Additionally, certain variants exhibited distinguishable differences between cSLE and SLE [11].
2. Polygenic SLE
The precise causes of cSLE are complex and not yet fully understood. The etiology of cSLE involves three primary factors: genetic risk factors, epigenetic mechanisms, and environmental triggers [12].
Genetic factors play a significant role in the development of the disease. In the general population, the risk of developing SLE is approximately 0.1%, while for females, it is around 0.2%. On average, about 7% of SLE patients have first-degree relatives with the same disease [13]. The risk for first-degree relatives ranges from 4% to 8% [14], but in some cases, it can be higher, with sisters of SLE patients having a risk of up to 10% [15]. In countries where consanguineous marriages are more prevalent, the risk can be significantly higher. Siblings of SLE patients have an 8 to 20 times higher risk of developing the disease compared to the general population [16][17][18]. The strong influence of genetics is evident from the fact that monozygotic twins have a 10-fold increased risk compared to dizygotic twins [17][19]. The estimated heritability of SLE ranges from 44% to 66%, with a concordance rate of approximately 24% to 56% among monozygotic twins, whereas in dizygotic twins, it is only 2% to 5% [20][21][22][23][24].
The presence of autoimmune diseases within the family poses a risk factor for the development of SLE, and this risk escalates with the number of relatives affected by autoimmune conditions. Genome-wide association studies (GWAS) have identified over 100 gene loci associated with susceptibility to SLE, although these loci may also contribute to the development of other autoimmune diseases [25][26]. Consequently, having a family history of autoimmune disease increases the risk of SLE by a factor of 4.1, and this risk further rises with the number of relatives affected by autoimmune disease, reaching up to 11.3 times higher [27].
The initial gene association identified in SLE was the major histocompatibility complex (MHC) located on chromosome 6, which encompasses human lymphocyte antigens (HLA) [28]. Current knowledge categorizes SLE susceptibility genes into four groups [25] (Figure 1).
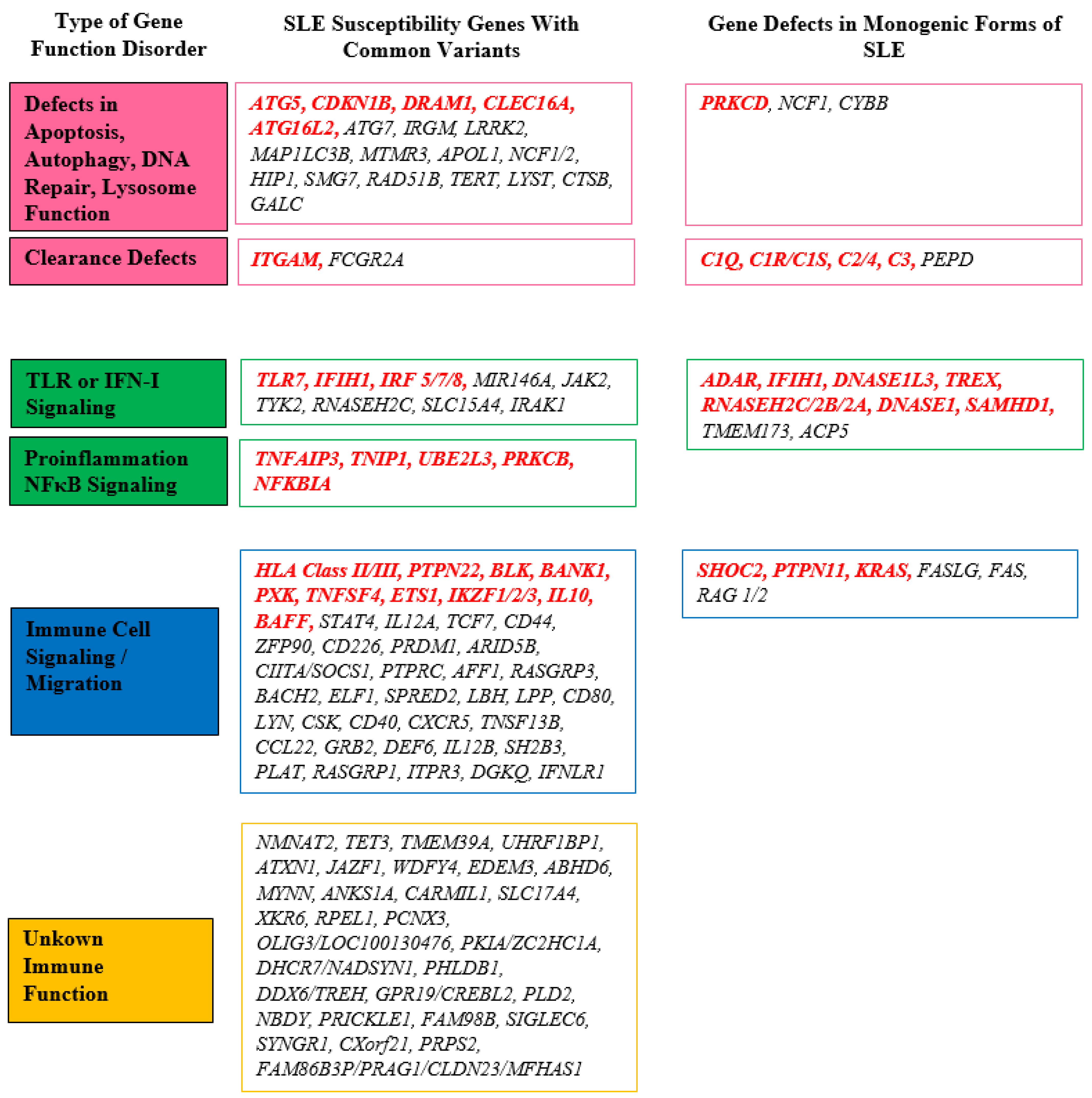
Figure 1. Overview of the important genes involved in SLE pathogenesis. The most important genes are marked in red. DNA: deoxyribonucleic acid; IFN-I: type I interferon; NFκB: nuclear factor κB; SLE: systemic lupus erythematosus; TLR: Toll-like receptor.
2.1. Genes Related to Apoptosis, Autophagy, DNA Repair, Lysosome Function, and Clearance of Immune Complexes
The initial group consists of genes involved in various processes such as apoptosis, autophagy, DNA repair, lysosome function, and immune complex clearance. These genes are categorized together because they are associated with the dysfunctional mechanisms mentioned earlier, which can result in the increased exposure of nuclear autoantigens to the immune system and the deposition of immune complexes. These processes are crucial in initiating and sustaining the autoimmune response in lupus. Autophagy, for example, is a cellular process known as “self-digestion” responsible for breaking down long-lived proteins and cytoplasmic organelles [29]. Autophagy-related mechanisms play a role in regulating multiple immune responses, including antigen delivery to major histocompatibility complex (MHC) compartments, lymphocyte survival and homeostasis, and cytokine production. Through GWAS, several autophagy-related genes, namely ATG5, CDKN1B, DRAM1, CLEC16A, and ATG16L2, have been identified as potentially associated with SLE susceptibility [29]. Additionally, other susceptibility genes such as ATG7, IRGM, LRRK2, MAP1LC3B, MTMR3, and APOL1 play significant roles within this signaling pathway [25][29].
Autophagy-related 5 (Atg5), encoded by the ATG5 gene in humans, is a key protein involved in the formation of autophagic vesicles and is central to autophagy. However, Atg5 also has diverse functions, including mitochondrial quality control after oxidative damage, negative regulation of the innate antiviral immune response, lymphocyte development and proliferation, MHC II antigen presentation, adipocyte differentiation, and apoptosis [30]. While it is known that both common and rare variants of ATG5 are associated with SLE susceptibility, the precise mechanism by which ATG5 contributes to lupus is not yet fully understood [29]. There are indications that ATG5 may initiate the development of SLE by promoting cytokine imbalance or disrupting antigen presentation.
Cyclin-dependent kinase inhibitor 1b (Cdkn1b) is an enzyme inhibitor encoded by the CDKN1B gene. It functions as an unconventional tumor suppressor and plays various roles in regulating the cell cycle, cell proliferation, and differentiation [31]. Its importance in T lymphocyte development is particularly notable, as it is crucial for inducing T-cell tolerance and anergy. Mice deficient in the cyclin-dependent kinase inhibitor p27 exhibit mild lupus-like abnormalities characterized by a decreased number and activity of regulatory T-cells (Treg cells) [31].
The DNA damage-regulated autophagy modulator 1 (DRAM1) gene encodes a lysosomal membrane protein that is essential for initiating autophagy [29]. DRAM1 expression is induced following DNA damage caused by UV irradiation, which provides a possible explanation for its involvement in the development of SLE [29][32]. It potentially serves as a connection between genetic factors associated with autophagy and environmental triggers.
The C-type lectin domain family 16 member a (Clec16a) protein regulates the selective degradation of mitochondria through autophagy and influences T-cell selection and reactivity in the thymic epithelium [29]. CLEC16A has been genetically linked to multiple autoimmune disorders, including multiple sclerosis, rheumatoid arthritis, Crohn’s disease, and SLE. The exact mechanism by which CLEC16A contributes to the development of SLE is not yet understood. However, the observed reduced expression of CLEC16A isoforms in SLE may lead to increased autophagic activities [33].
ATG16L2 (autophagy-related 16 like 2) is a gene that participates in autophagy and has been suggested as a genetic locus associated with an increased risk of SLE. It has also been linked to multiple sclerosis and Crohn’s disease. While ATG16L2 is believed to have a significant role in autophagy, especially in T-cells, the specific nature of its involvement is still unknown [34].
Among the genes related to SLE susceptibility, particularly in terms of immune complex clearance, the ITGAM gene stands out. The Integrin alpha M (ITGAM) gene encodes the integrin alpha M chain, which combines with the beta 2 chain to form either macrophage receptor 1 (Mac-1) or complement receptor 3 (CR3). These receptors play a crucial role in facilitating the adherence of neutrophils and monocytes to stimulate endothelium. Additionally, they are involved in the phagocytosis of complement-coated particles and immune complexes, as well as the regulation of leukocyte apoptosis [35]. Studies have demonstrated that missense variants in ITGAM impair the phagocytic function of monocytes, neutrophils, and macrophages. This impairment leads to the disrupted clearance of immune complexes, resulting in their deposition, tissue damage, and elevated levels of type I interferon (IFN-I) [36].
2.2. Genes of Innate Immunity
The second category encompasses genes involved in innate immunity and the associated signaling pathways, including IFN-I, Toll-like receptors (TLR), and nuclear factor κB (NFκB) [25]. These genes are grouped together due to their participation in innate immune responses.
IFN-I plays a vital role in the development of SLE, as demonstrated by the increased expression of IFN-I-inducible genes in the peripheral blood cells of the majority of SLE patients [37]. The significance of IFN-I-related genes in SLE susceptibility cannot be overstated, as more than half of the identified SLE susceptibility genes encode proteins directly or indirectly linked to IFN-I production or responses [25]. IFN-I exerts various functions and immune effects, including promoting the differentiation of monocytes and plasmacytoid dendritic cells, activating autoreactive T/B cells, stimulating autoantibody production, and inducing pro-inflammatory cytokines and chemokines [25]. Multiple triggers can induce IFN-I production in SLE, such as increased exposure of nucleic acids within immune complexes, necrotic debris, endosomal receptors (e.g., TLR7), or cytosolic sensors (e.g., IFIH1) [38].
TLR7 is located within intracellular endosomes and plays a central role in antiviral defense by recognizing single-stranded RNA. Activation of TLR7 can lead to both IFN-I production and NFκB activation in various cell populations, including dendritic cells, monocytes, macrophages, and B cells. IFN-I can contribute to the development of SLE [39]. It has been observed that sera from SLE patients contain TLR7 ligands in the form of immune complexes, which can activate plasmacytoid dendritic cells and induce IFN-I secretion. Furthermore, SLE sera can induce TLR7 expression in neutrophils, priming them for NETosis, which is also increased in SLE [40]. NETosis is a form of neutrophil cell death in which neutrophils release neutrophil extracellular traps (NETs) to capture and neutralize pathogens, thereby preventing their spread.
The IFIH1 gene, also known as interferon-induced helicase-1, encodes the protein Mda5 (melanoma differentiation-associated protein 5), an intracellular receptor involved in recognizing double-stranded RNA. During viral replication, double-stranded RNA molecules bind to Mda5, initiating a cascade of events that result in the production of type I and III interferons (IFN-I and IFN-III) [41]. Gain-of-function mutations in IFIH1 lead to the activation of dendritic cells and macrophages, triggering the production of IFN-α in response to nucleic acids. This activation subsequently leads to T-cell activation and the production of autoantibodies [42].
The IRF (interferon regulatory factor) family of genes encodes proteins that regulate interferon transcription [43]. Three IRF genes, namely IRF5, IRF7, and IRF8, have been associated with SLE susceptibility. IRF5 and IRF7 are downstream proteins that interact with the MyD88 adaptor protein upon engagement of Toll-like receptors (TLRs), leading to the transcription of IFN-α mRNA. On the other hand, IRF8, which does not interact with MyD88, appears to be involved in the production of inflammatory cytokines in dendritic cells in response to TLR9 ligands [25][43]. Genetic variants in IRF5 and IRF7 associated with SLE susceptibility are considered gain-of-function variants and are related to increased serum IFN-α levels in SLE patients with specific autoantibodies [25][43]. However, no correlation was found between IRF5 and/or IRF7 and serum IFN-α levels in SLE patients without these autoantibodies [25][43].
IRAK1, situated on the Xq28 chromosome, encodes a serine-threonine protein kinase called IL-1 receptor-associated kinase 1, which plays a regulatory role in various pathways involved in both innate and adaptive immune responses. Its involvement in the regulation of NFκB and TLR activation, as well as the induction of IFN-α and IFN-γ, positions IRAK1 as a promising candidate for thorough genetic and functional analysis in relation to SLE [44]. Jacob et al. propose that IRAK1 may contribute to at least three immune cell functions that have been found to be abnormal in SLE: the induction of IFN-α and IFN-γ, regulation of the NFκB pathway, and TLR activation [44]. The identification of an X chromosome gene as a susceptibility factor in human SLE suggests that gender disparities in SLE might be influenced, at least in part, by sex chromosome genes.
The TYK2 gene is situated on chromosome 19p13.2 and is responsible for encoding non-receptor tyrosine-protein kinase 2. TYK2 belongs to the Janus kinase (JAK) family and plays a crucial role in the signaling pathways of IFN-I, IL-6, IL-10, IL-12, and IL-23, particularly in the transmission of signals from IFN-α and β. Recent research indicates that TYK2 variants are associated with various autoimmune disorders, such as type 1 diabetes, psoriasis, multiple sclerosis, and increased susceptibility to SLE [45][46]. Interestingly, certain polymorphisms were found to have a protective effect in some autoimmune diseases while posing a risk factor for others, suggesting diverse underlying pathogenic mechanisms. In a study by Contreras-Cubas et al., genetic variants with a protective effect were identified for both childhood-onset and adult-onset SLE in the Mexican population [47].
The second group of SLE susceptibility genes includes genes associated with the NFκB pathway, such as TNFAIP3, TNIP1, UBE2L3, PRKCB, and NFKBIA [25]. The NFκB pathway regulates the activation of various cytokines, and NFκB target genes are involved in diverse immune functions, including lymphocyte development, activation, and differentiation, as well as the maturation and inflammatory functions of innate immune cells [48]. Abnormal NFκB signaling can lead to the production of autoreactive T-cells, which play a significant role in SLE, and promote plasma cell development.
The TNFAIP3 (tumor necrosis factor alpha-induced protein 3) gene, which encodes the enzyme A20, has been demonstrated to inhibit NFκB activation, TNF-mediated apoptosis, and NLRP3 inflammasome [49]. Risk alleles of TNFAIP3 are linked to reduced expression of A20 in SLE patients, resulting in heightened NFκB signaling.
TNIP1, also known as tumor necrosis factor alpha-induced protein 3-interacting protein 1, is a protein encoded by the TNIP1 gene. It interacts with A20 and functions as a physiological inhibitor of NFκB. Variants of TNIP1 that impair its inhibitory function contribute to the development of SLE by promoting increased NFκB activation [50].
Ube2l3, a ubiquitin-conjugating enzyme E2 L3, participates in the ubiquitination of NFκB precursor proteins to facilitate targeted degradation [48]. It may also play a role in B-cell proliferation and differentiation. The risk allele of Ube2l3 is associated with enhanced expression, leading to increased NFκB activation and elevated numbers of circulating plasma cells in SLE patients [51].
Prkcb, also known as protein kinase C beta type, is an enzyme involved in NFκB activation mediated by the B-cell receptor [48]. Variants of PRKCB have been linked to SLE, characterized by heightened NFκB activation and B-cell hyperactivity [52].
NFKBIA, or NFκB inhibitor alpha, is a transcription factor gene that participates in the activation of genes involved in immune responses [53]. Its association with SLE susceptibility is likely due to increased NFκB activation.
2.3. Genes of Adaptive Immunity
The third group of genes consists of those involved in adaptive immunity, specifically in the signaling and migration of immune cells. This group can be further divided into HLA genes and genes outside the HLA system. It encompasses various kinases, cytokines, and transcription factors associated with signal transduction within lymphocytes, as well as immune cell activation, proliferation, and interaction [25]. Variants in these genes may lead to the loss of immune cell tolerance and sustained production of autoantibodies.
The chromosomal region 6p21.3, referred to as the major histocompatibility complex (MHC) or human leukocyte antigen (HLA) region in humans, contains more than 200 genes. These genes encode leukocyte antigens, complement factors, and other molecules related to the immune system [31]. The MHC region is divided into three regions: class I, class II, and class III. Class I and class II regions consist of genes that encode glycoproteins responsible for processing and presenting peptides to T-cells. Class I molecules present peptides from within the cell to trigger CD8+ cytotoxic immune responses, while class II molecules present peptides from outside the cell to elicit helper T and B-cell antibody responses. The class III region encodes complement components, TNF, and other immune-related genes. Through meta-analysis of genome-wide association studies (GWAS), the HLA region has been identified as the most significant genetic risk factor for SLE [54][55]. However, the specific variants within this region that contribute to susceptibility are not yet fully understood due to its complexity. In white populations, the most consistent associations with SLE involve class II alleles HLA-DR3 (DRB1*0301) and HLA-DR2 (DRB1*1501) [56]. Large-scale GWAS studies have identified a combination of HLA alleles in class I (B08:01 and B18:01), class II (DQB1*02:01, DRB3*02:00, and DQA*01:02), and an SNP (rs74290525) in SLC44A4 in the class III region as the strongest associations [57]. Various mechanisms have been proposed to explain the association of DR and DQ alleles with autoimmunity, including variation in peptide binding regions, the selection of autoreactive T cells, and misfolded class II genes [58][59][60].
Apart from the HLA system, genes outside the HLA system, such as PTPN22, BLK, BANK1, PXK, TNFSF4, ETS1, IKZF1, IKZF2, IKZF3, IL10, and BAFF, also play roles in T- and B-cell signaling, transcription factors, and cytokines, and have been implicated in SLE susceptibility [25][61].
The gene PTPN22 encodes the enzyme tyrosine phosphatase nonreceptor type 22, which is predominantly expressed in lymphoid tissues. This enzyme is involved in multiple signaling pathways associated with the immune response. Its main function is to inhibit T-cell activation and contribute to the central and peripheral tolerance of B-cells at various developmental stages [25][61][62]. A gain-of-function variant of PTPN22 leads to the production of a more active phosphatase, resulting in lower thresholds for T-cell receptor signaling. This alteration affects B-cell receptor signaling, leading to increased autoreactivity and influencing the elimination of self-reactive B-cells during development. Ultimately, it contributes to both central and peripheral B-cell tolerance, thereby promoting autoimmunity [63].
The BLK gene encodes B-lymphoid tyrosine kinase (Blk), which plays various roles in intracellular signaling and regulates B-cell proliferation, differentiation, and tolerance [25]. Variants of the BLK gene associated with SLE susceptibility result in decreased expression of Blk, potentially affecting B-cell development and functional responses.
BANK1, encoded by the BANK1 gene, is an adaptor protein known as the B-cell scaffold protein with ankyrin repeats 1. It facilitates the release of intracellular calcium and modifies the activation threshold of B cells [25]. Variants of the BANK1 gene, linked to SLE, lead to reduced B-cell signaling and increased expansion of memory B-cells [64]. Rare variants found in patients impair the suppression of IFN-I in human B-cell lines and contribute to an increase in pathogenic lymphocytes in lupus-prone mice [65].
The PXK gene encodes a phox domain-containing protein involved in regulating synaptic transmission [25]. Risk variants in PXK associated with SLE lead to a decrease in B-cell receptor internalization. Although the genetic mechanism underlying this alteration is not fully understood, it may impact the regulation of B-cell signaling [66].
Tnfsf4, also known as tumor necrosis factor ligand superfamily member 4, is an inflammatory factor that has been associated with various inflammatory diseases and cancers. It is primarily expressed on activated CD4+ T cells. Increased expression of TNFSF4 is believed to predispose individuals to SLE by promoting interactions between T-cells and antigen-presenting cells or by disrupting peripheral tolerance through the inhibition of IL-10-producing CD4+ type 1 regulatory T-cells [67].
ETS1, encoded by the ETS1 gene, produces the protein C-ets-1, which belongs to the Erythroblast Transformation Specific family of transcription factors (ETS). Ets1 is primarily expressed in lymphocytes and is found at reduced levels in peripheral blood mononuclear cells from SLE patients [68]. It plays a crucial role in maintaining B-cell tolerance.
Ikzf1, Ikzf2, and Ikzf3 belong to the Ikaros family of zinc finger proteins. These proteins function as transcription factors and play a crucial role in regulating the differentiation, proliferation, and self-tolerance of lymphocytes. They are involved in controlling the signaling processes of B-cells, T-cells, and dendritic cells [69]. However, the specific mechanism by which causative variants in IKZF1, IKZF2, and IKZF3 are associated with SLE is still unknown [25].
Interleukin 10 (IL-10) is an immunoregulatory cytokine with both immunosuppressive and immunostimulatory properties. It is primarily produced by B cells, which utilize it for proliferation, and myeloid cells, which employ it to suppress proinflammatory responses. In SLE patients, risk alleles of IL10 lead to the increased production of IL-10 by peripheral blood B cells and monocytes, and elevated levels of IL-10 in the serum are correlated with disease activity [70]. Elevated levels of IL-10 contribute to SLE susceptibility and severity by promoting B-cell proliferation [71].
B cell-activating factor (BAFF) is a cytokine encoded by the TNFSF13B gene. It plays a significant role in the survival, proliferation, and maturation of B lymphocytes. Risk variants associated with BAFF increase its expression and are linked to active disease as well as renal and hematological involvement [72]. Excessive expression of BAFF is associated with enhanced survival and expansion of autoreactive B cells.
2.4. Genes with Unknown Immune Function
The fourth group comprises genes involved in immune functions, but their specific roles have not yet been fully elucidated. Some of these genes encode membrane proteins (e.g., C3orf21, DHCR7, PLD2), while others produce gene products with unknown immune functions [25].
The identified genetic variants or single nucleotide polymorphisms (SNPs) within the designated loci are common but have a relatively small effect on disease susceptibility, carrying a low relative risk of developing SLE. These variants explain approximately 30–50% of SLE heritability, indicating that other factors, such as rare genetic variants, epigenetic effects, and gene interactions (epistasis), play more significant roles in SLE susceptibility [73][74].
In addition to SNPs, copy number variations (CNVs) involving the deletion, insertion, or duplication of genomic regions also contribute to SLE susceptibility [75]. For example, the NCF1 gene, which encodes neutrophil cytosol factor 1, is affected by certain SNPs that result in reduced oxidative burst and lower production of reactive oxygen species. This leads to increased expression of IFN-I-regulated genes and association with SLE [76]. Decreased CNVs (0 and 1 copy) of NCF1 predispose individuals to SLE, while increased CNVs (≥3 copies) have a protective effect [77].
Complement components C1q, C4A, C4B, and C2B are the gene loci with the highest risk for developing SLE, followed by genes involved in the IFN-I signaling pathway (IRF5, ITGAM) and genes related to B lymphocyte signaling (BANK1 and BLK).
Although SLE and other autoimmune diseases share many susceptibility loci, the role of a particular locus in predisposition is not always consistent across different diseases. Sometimes the same variant may have an opposite effect, or the degree of effect may vary. For example, certain PTPN22 variants predispose to SLE but confer protection against inflammatory bowel diseases. Similarly, some NCF1 variants show a strong association with SLE but only a mild association with rheumatoid arthritis and Sjögren’s syndrome [25].
SLE, a complex disease, is influenced by both genetic predisposition and environmental factors, highlighting its polygenic nature. These genetic variations often reside outside the coding segments of genes. To uncover common variants that may not reach genome-wide significance, it is crucial to focus on multiple independent variants within each locus, perform meta-analyses using available data, and promote international collaborations to strengthen association studies. Furthermore, integrating information from gene expression profiles, protein complexes, signal transduction pathways, and regulatory networks can offer additional insights into the disease [31].
References
- Klein-Gitelman, M.S.; Beresford, M.W. Systemic Lupus Erythematosus, Mixed Connective Tissue Disease, and Undifferentiated Connective Tissue Disease. In Textbook of Pediatric Rheumatology, 8th ed.; Mellins, E.D., Petty, R.E., Laxer, R.M., Lindsley, C.B., Wedderburn, L., Fuhlbrigge, R.C., Eds.; Elsevier: Philadelphia, PA, USA, 2021; pp. 295–329.
- Edworthy, S.M. Clinical manifestations of systemic lupus erythematosus. In Kelley’s Textbook of Rheumatology, 7th ed.; Harris, E.D., Budd, R.C., Firestein, G.S., Eds.; WB Saunders: Philadelphia, PA, USA, 2005; pp. 1201–1224.
- Hoffman, I.E.; Lauwerys, B.R.; De Keyser, F.; Huizinga, T.W.; Isenberg, D.; Cebecauer, L.; Dehoorne, J.; Joos, R.; Hendrickx, G.; Houssiau, F.; et al. Juvenile-onset systemic lupus erythematosus: Different clinical and serological pattern than adult-onset systemic lupus erythematosus. Ann. Rheum. Dis. 2009, 68, 412–415.
- Silva, C.A.; Avcin, T.; Brunner, H.I. Taxonomy for systemic lupus erythematosus with onset before adulthood. Arthritis Care Res. 2012, 64, 1787–1793.
- Morgan, T.A.; Watson, L.; McCann, L.J.; Beresford, M.W. Children and adolescents with SLE: Not just little adults. Lupus 2013, 22, 1309–1319.
- Webb, R.; Kelly, J.A.; Somers, E.C.; Hughes, T.; Kaufman, K.M.; Sanchez, E.; Nath, S.K.; Bruner, G.; Alarcón-Riquelme, M.E.; Gilkeson, G.S.; et al. Early disease onset is predicted by a higher genetic risk for lupus and is associated with a more severe phenotype in lupus patients. Ann. Rheum. Dis. 2011, 70, 151–156.
- Hiraki, L.T.; Benseler, S.M.; Tyrrell, P.N.; Hebert, D.; Harvey, E.; Silverman, E.D. Clinical and laboratory characteristics and long-term outcome of pediatric systemic lupus erythematosus: A longitudinal study. J. Pediatr. 2008, 15, 550–556.
- Brunner, H.I.; Gladman, D.D.; Ibanez, D.; Urowitz, M.D.; Silverman, E.D. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008, 15, 556–562.
- Lo, M.S. Monogenic lupus. Curr. Rheumatol. Rep. 2016, 18, 71.
- Charras, A.; Smith, E.; Hedrich, C.M. Systemic Lupus Erythematosus in Children and Young People. Curr. Rheumatol. Rep. 2021, 23, 20.
- Hersh, A.O.; von Scheven, E.; Yazdany, J.; Panopalis, P.; Trupin, L.; Julian, L.; Katz, P.; Criswell, L.A.; Yelin, E. Differences in long-term disease activity and treatment of adult patients with childhood- and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2009, 15, 13–20.
- Harry, O.; Yasin, S.; Brunner, H. Childhood-Onset Systemic Lupus Erythematosus: A Review and Update. J. Pediatr. 2018, 196, 22–30.e2.
- Estes, D.; Christian, C.L. The natural history of systemic lupus erythematosus by prospective analysis. Medicine 1971, 50, 85–95.
- Michel, M.; Johanet, C.; Meyer, O.; Francès, C.; Wittke, F.; Michel, C.; Arfi, S.; Tournier-Lasserve, E.; Piette, J.C.; Group for Research on Auto-Immune Disorders (GRAID). Familial lupus erythematosus. Clinical and immunologic features of 125 multiplex families. Medicine 2001, 80, 153–158.
- Giles, I.; Isenberg, D. Lupus in the family—Analysis of a cohort followed from 1978 to 1999. Lupus 2001, 10, 38–44.
- Foster, H.E.; Brogan, P.A. (Eds.) Paediatric Rheumatology, 2nd ed.; Oxford University Press: Oxford, UK, 2018.
- Wallace, D.J.; Hahn, B.; Dubois, E.L. Dubois’ Lupus Erythematosus and Related Syndromes, 8th ed.; Elsevier/Saunders: Philadelphia, PA, USA, 2013; p. 15.
- Lloyd, P.; Doaty, S.; Hahn, B. Aetiopathogenesis of systemic lupus erythematosus. In Systemic Lupus Erythematosus; Gordon, C., Isenberg, D., Eds.; Oxford Rheumatology Library; Oxford University Press: Oxford, UK, 2016.
- Lewandowski, L.; Scott, C.; Gomez-Martin, D.; Silverman, E.D.; Aksentijevich, I.; Deng, Z.; Siegel, R.M.; Rider, L.G.; Hasni, S.; Kaplan, M.J. GG-10 Imagine SLE: International multi-site assessment of genetics and inflammation in early onset and familial systemic lupus erythematosus. Lupus Sci. Med. 2016, 3 (Suppl. S1), A32.
- Kuo, C.F.; Grainge, M.J.; Valdes, A.M.; See, L.C.; Luo, S.F.; Yu, K.H.; Zhang, W.; Doherty, M. Familial aggregation of systemic lupus erythematosus and coaggregation of autoimmune diseases in affected families. JAMA Intern. Med. 2015, 175, 1518–1526.
- Frangou, E.A.; Bertsias, G.K.; Boumpas, D.T. Gene expression and regulation in systemic lupus erythematosus. Eur. J. Clin. Invest. 2013, 43, 1084–1096.
- Criswell, L.A. The genetic contribution to systemic lupus erythematosus. Bull. NYU Hosp Jt. Dis. 2008, 66, 176–183.
- Crow, Y.J. Lupus: How much “complexity” is really (just) genetic heterogeneity? Arthritis Rheum. 2011, 63, 3661–3664.
- Deng, Y.; Tsao, B.P. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat. Rev. Rheumatol. 2010, 6, 683–692.
- Deng, Y.; Tsao, B.P. Updates in Lupus Genetics. Curr. Rheumatol. Rep. 2017, 19, 68.
- Goulielmos, G.N.; Zervou, M.I.; Vazgiourakis, V.M.; Ghodke-Puranik, Y.; Garyfallos, A.; Niewold, T.B. The genetics and molecular pathogenesis of systemic lupus erythematosus (SLE) in populations of different ancestry. Gene 2018, 668, 59–72.
- Priori, R.; Medda, E.; Conti, F.; Cassara, E.A.; Danieli, M.G.; Gerli, R.; Giacomelli, R.; Franceschini, F.; Manfredi, A.; Pietrogrande, M. Familial autoimmunity as a risk factor for systemic lupus erythematosus and vice versa: A case-control study. Lupus 2003, 12, 735–740.
- Waters, H.; Konrad, P.; Walford, R.L. The distribution of HL-A histocompatibility factors and genes in patients with systemic lupus erythematosus. Tissue Antigens 1971, 1, 68–73.
- Qi, Y.Y.; Zhou, X.J.; Zhang, H. Autophagy and immunological aberrations in systemic lupus erythematosus. Eur. J. Immunol. 2019, 49, 523–533.
- Ye, X.; Zhou, X.J.; Zhang, H. Exploring the Role of Autophagy-Related Gene 5 (ATG5) Yields Important Insights Into Autophagy in Autoimmune/Autoinflammatory Diseases. Front. Immunol. 2018, 9, 2334.
- Yang, W.; Lau, Y.L. Solving the genetic puzzle of systemic lupus erythematosus. Pediatr. Nephrol. 2015, 30, 1735–1748.
- Valbuena, A.; Castro-Obregon, S.; Lazo, P.A. Downregulation of VRK1 by p53 in response to DNA damage is mediated by the autophagic pathway. PLoS ONE 2011, 6, e17320.
- Tam, R.C.; Lee, A.L.; Yang, W.; Lau, C.S.; Chan, V.S. Systemic Lupus Erythematosus Patients Exhibit Reduced Expression of CLEC16A Isoforms in Peripheral Leukocytes. Int. J. Mol. Sci. 2015, 16, 14428–14440.
- Yin, L.; Liu, J.; Dong, H.; Xu, E.; Qiao, Y.; Wang, L.; Zhang, L.; Jia, J.; Li, L.; Geng, X. Autophagy-related gene16L2, a potential serum biomarker of multiple sclerosis evaluated by bead-based proteomic technology. Neurosci. Lett. 2014, 562, 34–38.
- Kim, K.; Brown, E.E.; Choi, C.B.; Alarcón-Riquelme, M.E.; BIOLUPUS; Kelly, J.A.; Glenn, S.B.; Ojwang, J.O.; Adler, A.; Lee, H.S.; et al. Variation in the ICAM1-ICAM4-ICAM5 locus is associated with systemic lupus erythematosus susceptibility in multiple ancestries. Ann. Rheum. Dis. 2012, 71, 1809–1814.
- Fossati-Jimack, L.; Ling, G.S.; Cortini, A.; Szajna, M.; Malik, T.H.; McDonald, J.U.; Pickering, M.C.; Cook, H.T.; Taylor, P.R.; Botto, M. Phagocytosis is themain CR3-mediated function affected by the lupus-associated variant of CD11b in human myeloid cells. PLoS ONE 2013, 8, e57082.
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615.
- Jensen, M.A.; Niewold, T.B. Interferon regulatory factors: Critical mediators of human lupus. Transl Res. 2015, 165, 283–295.
- Wolf, S.J.; Theros, J.; Reed, T.J.; Liu, J.; Grigorova, I.L.; Martínez-Colón, G.; Jacob, C.O.; Hodgin, J.B.; Kahlenberg, J.M. TLR7-Mediated Lupus Nephritis Is Independent of Type I IFN Signaling. J. Immunol. 2018, 201, 393–405.
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20.
- Domsgen, E.; Lind, K.; Kong, L.; Hühn, M.H.; Rasool, O.; van Kuppeveld, F.; Korsgren, O.; Lahesmaa, R.; Flodström-Tullberg, M. An IFIH1 gene polymorphism associated with risk for autoimmunity regulates canonical antiviral defence pathways in Coxsackievirus infected human pancreatic islets. Sci. Rep. 2016, 6, 39378.
- Funabiki, M.; Kato, H.; Miyachi, Y.; Toki, H.; Motegi, H.; Inoue, M.; Minowa, O.; Yoshida, A.; Deguchi, K.; Sato, H.; et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 2014, 40, 199–212.
- Salloum, R.; Niewold, T.B. Interferon regulatory factors in human lupus pathogenesis. Transl Res. 2011, 157, 326–331.
- Jacob, C.O.; Zhu, J.; Armstrong, D.L.; Yan, M.; Han, J.; Zhou, X.J.; Thomas, J.A.; Reiff, A.; Myones, B.L.; Ojwang, J.O.; et al. Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 2009, 106, 6256–6261.
- Sigurdsson, S.; Nordmark, G.; Göring, H.H.; Lindroos, K.; Wiman, A.C.; Sturfelt, G.; Jönsen, A.; Rantapää-Dahlqvist, S.; Möller, B.; Kere, J.; et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am. J. Hum. Genet. 2005, 76, 528–537.
- Li, Z.; Rotival, M.; Patin, E.; Michel, F.; Pellegrini, S. Two common disease-associated TYK2 variants impact exon splicing and TYK2 dosage. PLoS ONE 2020, 15, e0225289.
- Contreras-Cubas, C.; García-Ortiz, H.; Velázquez-Cruz, R.; Barajas-Olmos, F.; Baca, P.; Martínez-Hernández, A.; Barbosa-Cobos, R.E.; Ramírez-Bello, J.; López-Hernández, M.A.; Svyryd, Y.; et al. Catalytically Impaired TYK2 Variants are Protective Against Childhood- and Adult-Onset Systemic Lupus Erythematosus in Mexicans. Sci. Rep. 2019, 9, 12165.
- Mishra, R.K. Involvement of NF-κB signaling pathway in the pathogenesis of systemic lupus erythematosus. Nephrol. Open J. 2016, 2, 9–13.
- Catrysse, L.; Vereecke, L.; Beyaert, R.; van Loo, G. A20 in inflammation and autoimmunity. Trends Immunol. 2014, 35, 22–31.
- Brady, M.P.; Korte, E.A.; Caster, D.J.; Powell, D.W. TNIP1/ABIN1 and lupus nephritis: Review. Lupus Sci. Med. 2020, 7, e000437.
- Lewis, M.J.; Vyse, S.; Shields, A.M.; Boeltz, S.; Gordon, P.A.; Spector, T.D.; Lehner, P.J.; Walczak, H.; Vyse, T.J. UBE2L3 polymorphism amplifies NF-kappaB activation and promotes plasma cell development, linking linear ubiquitination to multiple autoimmune diseases. Am. J. Hum. Genet. 2015, 96, 221–234.
- Sheng, Y.J.; Gao, J.P.; Li, J.; Han, J.W.; Xu, Q.; Hu, W.L.; Pan, T.M.; Cheng, Y.L.; Yu, Z.Y.; Ni, C.; et al. Follow-up study identifies two novel susceptibility loci PRKCB and 8p11.21 for systemic lupus erythematosus. Rheumatology 2011, 50, 682–688.
- Romzova, M.; Hohenadel, D.; Kolostova, K.; Pinterova, D.; Fojtikova, M.; Ruzickova, S.; Dostal, C.; Bosak, V.; Rychlik, I.; Cerna, M. NFκB and its inhibitor IκB in relation to type 2 diabetes and its microvascular and atherosclerotic complications. Hum. Immunol. 2006, 67, 706–713.
- Forabosco, P.; Gorman, J.D.; Cleveland, C.; Kelly, J.A.; Fisher, S.A.; Ortmann, W.A.; Johansson, C.; Johanneson, B.; Moser, K.L.; Gaffney, P.M.; et al. Meta-analysis of genome-wide linkage studies of systemic lupus erythematosus. Genes Immun. 2006, 7, 609–614.
- Graham, R.R.; Hom, G.; Ortmann, W.; Behrens, T.W. Review of recent genome-wide association scans in lupus. J. Intern. Med. 2009, 265, 680–688.
- International MHC and Autoimmunity Genetics Network; Rioux, J.D.; Goyette, P.; Vyse, T.J.; Hammarström, L.; Fernando, M.M.; Green, T.; De Jager, P.L.; Foisy, S.; Wang, J.; et al. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc. Natl. Acad. Sci. USA 2009, 106, 18680–18685.
- Bentham, J.; Morris, D.L.; Cunninghame Graham, D.S.; Pinder, C.L.; Tombleson, P.; Behrens, T.W.; Martín, J.; Fairfax, B.P.; Knight, J.C.; Chen, L.; et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 2015, 47, 457–464.
- Gough, S.C.; Simmonds, M.J. The HLA Region and Autoimmune Disease: Associations and Mechanisms of Action. Curr. Genomics. 2007, 8, 453–465.
- Zanelli, E.; Breedveld, F.C.; de Vries, R.R. HLA association with autoimmune disease: A failure to protect? Rheumatology 2000, 39, 1060–1066.
- Gromme, M.; Neefjes, J. Antigen degradation or presentation by MHC class I molecules via classical and non-classical pathways. Mol. Immunol. 2002, 39, 181–202.
- Saeed, M. Lupus pathobiology based on genomics. Immunogenetics 2017, 69, 1–12.
- Menard, L.; Saadoun, D.; Isnardi, I.; Ng, Y.S.; Meyers, G.; Massad, C.; Price, C.; Abraham, C.; Motaghedi, R.; Buckner, J.H.; et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J. Clin. Invest. 2011, 121, 3635–3644.
- Kyogoku, C.; Langefeld, C.D.; Ortmann, W.A.M.; Lee, A.; Selby, S.; Carlton, V.E.; Chang, M.; Ramos, P.; Baechler, E.C.; Batliwalla, F.M.; et al. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am. J. Hum. Genet. 2004, 75, 504–507.
- Kozyrev, S.V.; Abelson, A.K.; Wojcik, J.; Zaghlool, A.; Linga Reddy, M.V.; Sanchez, E.; Gunnarsson, I.; Svenungsson, E.; Sturfelt, G.; Jönsen, A.; et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat. Genet. 2008, 40, 211–216.
- Jiang, S.H.; Athanasopoulos, V.; Ellyard, J.I.; Chuah, A.; Cappello, J.; Cook, A.; Prabhu, S.B.; Cardenas, J.; Gu, J.; Stanley, M.; et al. Functional rare and low frequency variants in BLK and BANK1 contribute to human lupus. Nat. Commun. 2019, 10, 2201.
- Vaughn, S.E.; Foley, C.; Lu, X.; Patel, Z.H.; Zoller, E.E.; Magnusen, A.F.; Williams, A.H.; Ziegler, J.T.; Comeau, M.E.; Marion, M.C.; et al. Lupus risk variants in the PXK locus alter B-cell receptor internalization. Front. Genet. 2015, 5, 450.
- Ito, T.; Wang, Y.H.; Duramad, O.; Hanabuchi, S.; Perng, O.A.; Gilliet, M.; Qin, F.X.; Liu, Y.J. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13138–13143.
- Yang, W.; Shen, N.; Ye, D.Q.; Liu, Q.; Zhang, Y.; Qian, X.X.; Hirankarn, N.; Ying, D.; Pan, H.F.; Mok, C.C.; et al. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 2010, 6, e1000841.
- Chen, L.; Niu, Q.; Huang, Z.; Yang, B.; Wu, Y.; Zhang, J. IKZF1 polymorphisms are associated with susceptibility, cytokine levels, and clinical features in systemic lupus erythematosus. Medicine 2020, 99, e22607.
- Hagiwara, E.; Gourley, M.F.; Lee, S.; Klinman, D.K. Disease severity in patients with systemic lupus erythematosus correlates with an increased ratio of interleukin-10:Interferon-gamma-secreting cells in the peripheral blood. Arthritis Rheum. 1996, 39, 379–385.
- Blenman, K.; Duan, B.; Xu, Z.; Wan, S.; Atkinson, M.A.; Flotte, T.R.; Croker, B.P.; Morel, L. IL-10 regulation of lupus in the NZM2410 murine model. Lab. Invest. 2006, 86, 1136–1148.
- Marín-Rosales, M.; Cruz, A.; Salazar-Camarena, D.C.; Santillán-López, E.; Espinoza-García, N.; Muñoz-Valle, J.F.; Ramírez-Dueñas, M.G.; Oregón-Romero, E.; Orozco-Barocio, G.; Palafox-Sánchez, C.A. High BAFF expression associated with active disease in systemic lupus erythematosus and relationship with rs9514828C>T polymorphism in TNFSF13B gene. Clin. Exp. Med. 2019, 19, 183–190.
- Chen, L.; Morris, D.L.; Vyse, T.J. Genetic advances in systemic lupus erythematosus: An update. Curr. Opin. Rheumatol. 2017, 29, 423–433.
- Cui, Y.; Sheng, Y.; Zhang, X. Genetic susceptibility to SLE: Recent progress from GWAS. J. Autoimmun. 2013, 41, 25–33.
- Ptacek, T.; Li, X.; Kelley, J.M.; Edberg, J.C. Copy number variants in genetic susceptibility and severity of systemic lupus erythematosus. Cytogenet. Genome Res. 2008, 123, 142–147.
- Olsson, L.M.; Johansson, Å.C.; Gullstrand, B.; Jönsen, A.; Saevarsdottir, S.; Rönnblom, L.; Leonard, D.; Wetterö, J.; Sjöwall, C.; Svenungsson, E.; et al. A single nucleotide polymorphism in the NCF1 gene leading to reduced oxidative burst is associated with systemic lupus erythematosus. Ann. Rheum. Dis. 2017, 76, 1607–1613.
- Zhao, J.; Ma, J.; Deng, Y.; Kelly, J.A.; Kim, K.; Bang, S.Y.; Lee, H.S.; Li, Q.Z.; Wakeland, E.K.; Qiu, R.; et al. A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat. Genet. 2017, 49, 433–437.
More
Information
Subjects:
Rheumatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
489
Revisions:
2 times
(View History)
Update Date:
10 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No