Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Armando Magrelli | -- | 3771 | 2023-07-25 11:48:34 | | | |
| 2 | Rita Xu | Meta information modification | 3771 | 2023-07-25 12:01:19 | | | | |
| 3 | Rita Xu | Meta information modification | 3771 | 2023-07-27 10:50:56 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Giorgioni, L.; Ambrosone, A.; Cometa, M.F.; Salvati, A.L.; Magrelli, A. CAR-T Current Challenges. Encyclopedia. Available online: https://encyclopedia.pub/entry/47239 (accessed on 10 August 2026).
Giorgioni L, Ambrosone A, Cometa MF, Salvati AL, Magrelli A. CAR-T Current Challenges. Encyclopedia. Available at: https://encyclopedia.pub/entry/47239. Accessed August 10, 2026.
Giorgioni, Lorenzo, Alessandra Ambrosone, Maria Francesca Cometa, Anna Laura Salvati, Armando Magrelli. "CAR-T Current Challenges" Encyclopedia, https://encyclopedia.pub/entry/47239 (accessed August 10, 2026).
Giorgioni, L., Ambrosone, A., Cometa, M.F., Salvati, A.L., & Magrelli, A. (2023, July 25). CAR-T Current Challenges. In Encyclopedia. https://encyclopedia.pub/entry/47239
Giorgioni, Lorenzo, et al. "CAR-T Current Challenges." Encyclopedia. Web. 25 July, 2023.
Copy Citation
CAR-Ts have started to move past the “ceiling” of third-line treatment with positive results in comparison trials with the Standard of Care (SoC). One such example is the trial Zuma-7 (NCT03391466), which resulted in approval of CAR-T products (Yescarta™) for second-line treatment, a crucial achievement for the field which can increase the use of this type of therapy.
CAR-T
rare diseases
ATMP
1. Introduction
Chimeric Antigen Receptor (CAR)-based therapies represent a significant development in immunotherapy, since they have the potential to be effective in relapsed and refractory (r/r) disease, where efficacy of other therapies is lower. They could also virtually be safer and more effective than traditional chemotherapy, though at the moment they present several unsolved limitations. CAR-T cell therapies are ATMPs and all those currently authorised are orphan drugs. This means they are complex medicinal products, made of biological materials engineered with cutting-edge technologies, currently indicated only for rare diseases, thus being subject to a composite regulatory framework.
Looking at the European pharmaceutical regulatory landscape, CAR-engineered medicinal products are specifically regulated through the following: The Advanced Therapies Regulation (EC) No. 1394/2007, which had the objective to discipline Advanced Therapy Medicinal Products (ATMPs) [1] in the EU, as well as the Orphan Medicines Regulation (EC) No. 141/2000 [2], complemented by the (EC) Regulation Number 847/2000 of the European Parliament and Council, which had the objective of rewarding interest in small patient populations neglected by the pharma industry due to unsustainable returns. These Regulations are presently under revision, so the landscape might change in the future [3].
Other pieces of legislation relevant to this research are those respectively establishing the Hospital Exemption (HE) and Compassionate Use (CU) in Europe. The former describes a regulatory option, foreseen by Article 28 of Regulation (EC) No 1394/2007 amending Article 3 of Directive 2001/83/EC, based on which any ATMP can be prepared as an individual medical prescription for an individual patient (named-based) on a non-routine basis, under the exclusive professional responsibility of a medical practitioner for the treatment of severe, disabling, or life-threatening conditions [1]. The latter (CU) was established by Article 83 of Regulation (EC) No 726/2004 and allows the use of investigational medicinal products in groups of patients affected by diseases with no satisfactory therapies who cannot enter clinical trials [4].
In order to provide an updated picture of CAR-Ts state of the art, together with developments most likely to be achieved in the near- and mid-term, as well as insights on what is currently possible and how the regulatory sector is enabling these products to reach the clinical setting, namely: The EU Clinical Trials Register (clinicaltrialsregister.eu), supplemented by the US database of privately and publicly funded clinical studies (ClinicalTrials.gov); the EMA website’s EPAR Repository, to obtain the latest updates on information and indications of EU-authorised CAR-T medicinal products; the lists of medicinal products awarded with the PRIME scheme, as well as relevant examples of HE and CU to look at possible approaches for future Marketing Authorization Applications (MAAs).
From a structural perspective, a CAR is an artificial protein expressed on the surface of an immune cell, encoded by a transgene introduced via a variety of methods, most commonly transfection via viral vectors. The majority of immune cells presently engineered belong to the cellular component of the adaptive immune system (CD4+ and/or CD8+ T cells), but engineering of other families of immune cells, especially those belonging to the innate immune system like Natural-Killer (NK) [5] and Macrophage (M), [6] have also been reported.
All approved CAR-T products bear a chimeric antigen receptor composed of an antigen-binding domain typical of antibodies, a co-stimulatory factor from the T cell’s surface, and a signalling domain found in T-cell receptors (TCR), all deriving from different sources, hence the name ‘chimeric’. In a process of innovation already visible today and reported by other authors [7], this structure will likely become more complex and diverse in the future, with different proteins being used to fill the role of each domain, with the aim to increase safety and efficacy.
Up to the first promising results from clinical trials in 2014, the participation in CAR-T research was scarce, but in recent years, Chimeric Antigen Receptor T cells have demonstrated to be an efficacious means to achieve enhanced survival in haematological malignancies and presented enough benefit to warrant marketing authorisations for several of the investigated medicinal products. These drugs are showing a very positive risk/benefit balance, with benefits continuing to improve as safety is enhanced by new construct designs and protocols to deal with treatment complications.
CAR-T Cells Generation
The general principle of CAR-T-cell therapy is summarised in Figure 1.
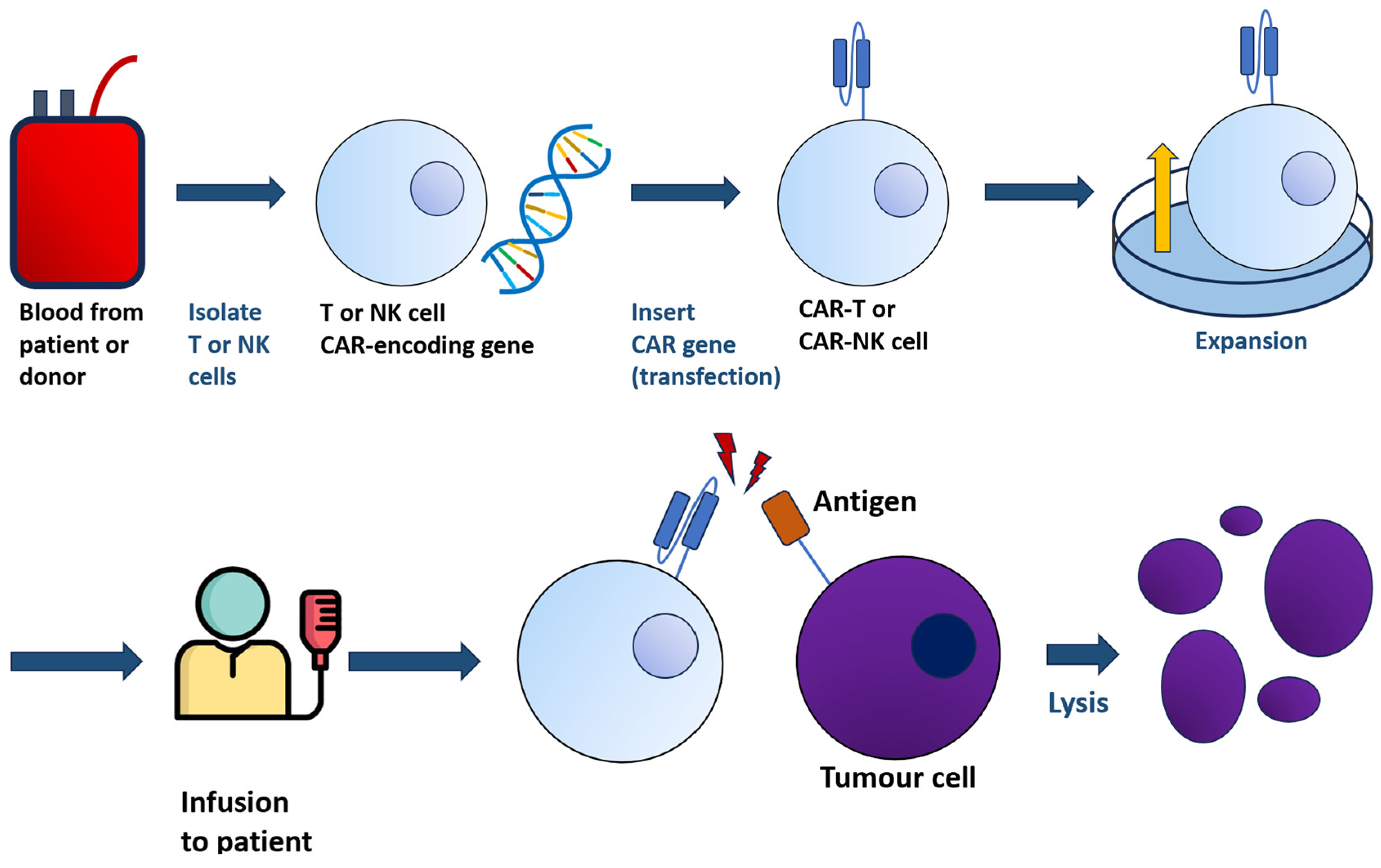
Figure 1. From a sample of blood, either from patients or donors, T or NK cells are harvested and transfected with the gene encoding CAR. The resulting CAR-T/-NK cells, bearing the chimeric receptor, are administered to patients. The interaction between CAR-T/-NK cells with tumour cells will lead to lysis.
In CAR-T design, domains are sometimes referred to as modules [8]. This emphasises the technology’s modular character, with a virtually endless number of possible combinations of different domains making up the construct [9]. This concept of the “structural matrix” is exemplified in Figure 2, which aims to represent the most general possible structure for a CAR construct. Although in a “typical” CAR, the role of antigen-binding domain is filled by an antibody-derived scFv, other options reported in literature are Nanobodies [10], truncated versions of an scFv mainly featuring the heavy chains (VH), or native receptors, or even small peptides like the so-called peptide-centric [11] and SPEAR [12] CAR-Ts designs. Under each domain, parameters which influence its activity are indicated. For instance, the antigen-binding domain activity is affected by the number of “domains”, which can vary for specific constructs (e.g., dual CAR-T share two domains), but it is also influenced by the type of protein chosen to derive the antigen-binding domain, by its affinity and/or avidity for the antigen, or by the interaction of different CAR constructs’ affinities on a single CAR-T-cell surface. Each property originates different behaviours and cytologic phenomena which overall may influence the behaviour of the CAR in vivo, thus impacting its safety and efficacy profile [13].
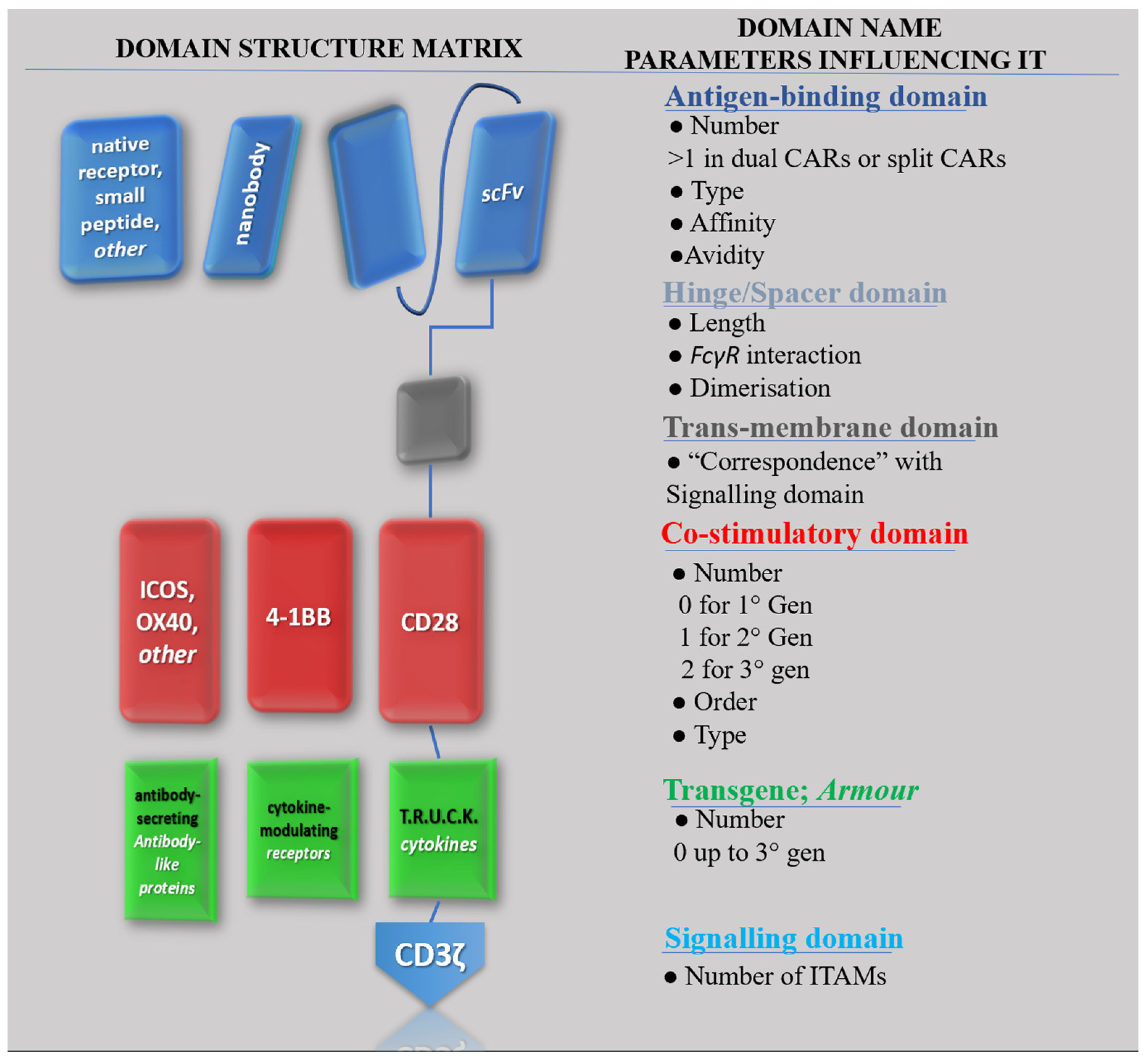
Figure 2. All possible CAR configurations with depictions of possible domains, their name, and the most relevant parameters influencing their activity. Proteins in rows on the left are potential candidates to fill the role of the list of domains on the right.
2. CAR-T Current Challenges
From clinical trials results on liquid or haematologic tumours, it is known that the majority of patients treated with CAR-Ts do have important responses, yet most of these are still not durable.
This is not the case in solid tumours, where current CAR-Ts are not significantly effective, making it an area of unmet need despite being the focus of much of the current research in the field.
CAR-T cells appear to be associated with significant toxicities of inflammatory nature, not only the cited Cytokine Release Syndrome (CRS), but also the Immune effector Cell-Associated Neurotoxicity Syndrome (ICANS), as well as cytopenias which then may lead to opportunistic infections.
On the other hand, the development of allogeneic CAR-engineered cells, including non-T immune cells like Natural Killer (NK) and Macrophage (M), likely represents the largest gap in knowledge in the field today, which will require substantial investments to reach the clinic, although several reports of CAR-NK clinical trials are already available [5].
The present limitations of CAR-T-cell therapy are summarised in Figure 3:
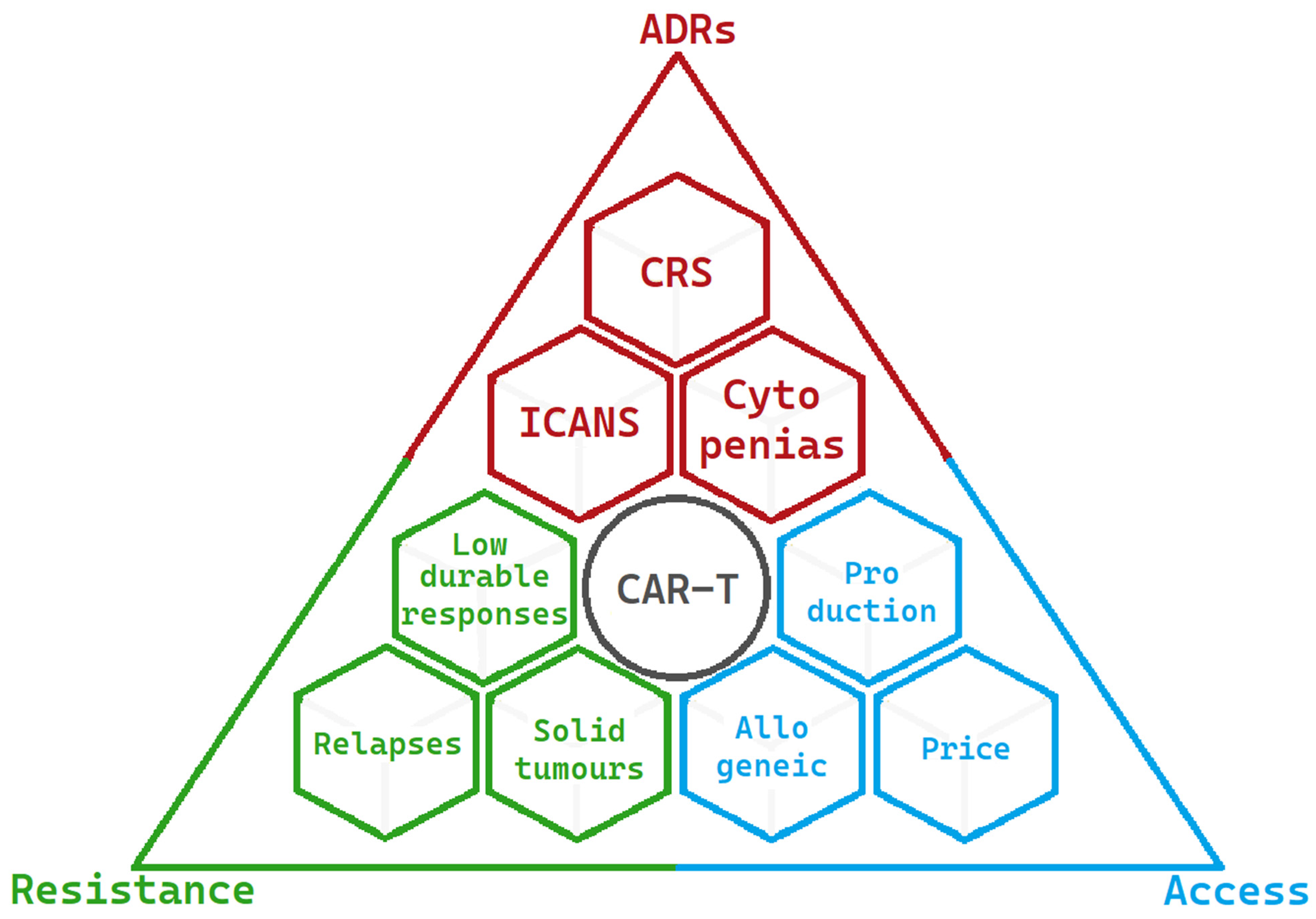
Figure 3. Limitations and challenges of CAR-Ts at present are summarised into three main areas. The most significant adverse drug reactions are in red, the factors resulting in resistance to CAR-T activity are in green, and the factors limiting access to CAR-T-cell therapy are in blue.
2.1. CAR-T Target Choice Impacts Efficacy and Safety
Target choice can be considered a key element in the development of CARs: by choosing an ideal target, the safety of these products can be raised to high standards. The importance of a CAR’s target cannot be overstated, as it ties in directly to both efficacy and safety of these treatments.
Even if there is a vast and growing body of literature and expanding research, both preclinical and clinical, dealing with the discovery and testing of novel targets for CAR constructs [14][15], “target mining” or the search for more and better targets for a CAR’s application remains a significant gap in knowledge.
2.2. Adverse Drug Reactions
While some of the toxicities associated with CAR-T-cell therapy were anticipated (like the B-cell aplasia in anti-CD19 CARs due to its presence on healthy B cells), the two adverse events which have become distinctive of CAR-T-cell therapy were not anticipated from the early murine studies, i.e., CRS and ICANS [16].
2.2.1. Cytokine Release Syndrome (CRS)
CRS usually develops 3–5 days after infusion, while ‘Immune effector-Cell Associated Neurotoxicity Syndrome’ (ICANS) starts 5–7 days after infusion and is likely connected to CRS development.
CRS is caused by extremely high levels of cytokines due to a strong immune response. Typical CRS symptoms range from sustained fevers, hypotension, and in general, the need for airway protection, low blood pressure, high fevers, and circulatory failure, which might require the patient’s admission to the Intensive Care Unit (ICU) and the use of ventilators.
This implies that a small clinical centre, not equipped with advanced facilities, will not be authorised for the use of CAR-Ts, thus contributing to a reduced access to this therapeutic opportunity.
Recent studies in cytokines involvement in CAR-T therapy’s CRS show that many cytokines derive from the CAR-T treatment. This occurs as engineered cells proliferate in vivo when activated by coming in contact with tumour cells, and produce dramatic increase in cytokines. Moreover, some key cytokines mediating CRS, like IL-6, are produced by Tumour-Associated Macrophages (TAMs), myeloid-type cells found in the patient’s tumour [17][18].
A direct proportionality between the tumour burden, the scale of T-cell activation and action, and the severity of the syndrome has been observed. As a result, the most fragile segment of patients, those with more severe disease conditions, tend to suffer the direst CRSs.
Despite incremental improvement in the ability to recognise Cytokine Release Syndrome and the early treatment with monoclonal antibodies that might induce IL-6 blockade (i.e., Tocilizumab), deaths linked to this side effect continue to be observed. Thus, a deeper understanding of CRS, its causes and mechanism of actions, is needed to control, treat, or reduce occurrence in future CAR-T medicinal products.
2.2.2. CAR-T-Mediated Neurotoxicity (ICANS)
ICANS appears to be due to the expression of CD19 on some neurons and blood–brain barrier lining. ICANS main symptoms are confusion, aphasia, seizures, and encephalopathy.
Even if, according to MRI observations during neurotoxicity, these symptoms tend to be transient, ICANS-induced encephalopathy can be deadly if associated with brain edema, and high-grade ICANS can result in the destruction of the blood–brain barrier (BBB) [19], with potential infiltration of T cells, B cells, and myeloid cells into the Cerebral Spinal Fluid (CSF) and the Central Nervous System (CNS).
GM-CSF seems to play a central role in CAR-T-mediated neurotoxicity, as demonstrated by the analysis of some of the 2017 pivotal clinical trials [20].
In addition, the infiltration into the CSF of CD14-positive cells and monocytes, like Tumour-Associated Macrophages (TAMs), has been associated with Grade ≥ 3 neurotoxicity, thus suggesting a role for these cells as a target for preventing ICANS. Currently, the treatment of ICANS includes solely the administration of corticosteroids and supportive care. In addition, since ICANS often occurs with CRS, the resolution of the former is needed for a proper CRS treatment; however, the spontaneous resolution can only happen if ICANS is low-grade, while severe ICANS (grade ≥ 3) remains a critical condition, thus highlighting a gap in knowledge and the need of finding new and resolutive treatments. ICANS and neurotoxicity are obviously crucial concerns to the application of CAR-Ts in the neurological field. Through the observation of haematologic patients with known CNS involvement, presence of MRI changes or baseline neurological disorders have been linked to increased risk of ICANS [21]. In recent years, several antigens were identified as targets for CAR-T-cell therapy against primary brain tumours (e.g., GBM) which are currently in early clinical development in the US and China [22][23].
2.3. Resistance to CAR-T Activity
Resistance to CAR-T activity is a multifactorial outcome due to different aspects, mainly related to patients’ cells used to generate the CAR-Ts, T-cell exhaustion and tumour microenvironment (TME).
Regarding the influence of starting materials in autologous treatments, patients T cells’ functioning could be defective even before the transduction of T cells with CARs. This seems to be the case in heavily pre-treated patients, who approach CAR-Ts as a third line of treatment (e.g., patients who could benefit from CAR-T treatment may not be eligible because of low WBC counts and/or activity levels), and it is one of the reasons for the interest in allogeneic CAR-Ts. For similar reasons, these therapies have been moved to earlier lines of treatment, like in the case of the Zuma-7 trial (NCT03391466), a comparison trial which allowed Yescarta© to be the first CAR-T to ever be available as second-line treatment, for HGBL and DLBCL.
Defective T-cell function may also appear as a result of genetic modification. This specific form of decline in functions is known as ‘T-cell exhaustion’, a poorly understood continuous differentiation process, in which T cells are transformed from precursors to terminally differentiated T cells, thus losing cellular functionality.
The tumour microenvironment (TME) plays a critical role in the response to tumour treatments. Suppression exerted by the tumour’s TME can down-regulate activity of both CAR-Ts and physiological T cells. CAR-T inhibition mediated by the TME in solid tumours is a major area of study [24]. Currently, remissions in solid tumour indications after CAR-T infusion remain few and short-lived. This may be due to the difficult trafficking to the tumour. In several studies, CAR-Ts were able to get inside the tumour mass, although they were reported to be inhibited at the site. This inhibition is mediated by the immunosuppressive TME, and solving such inhibition could increase the number of possible new medicinal products for new and critical indications in the solid tumour space.
Among the approaches to TME-mediated resistance, dual-targeting CAR-Ts may be used to stop Cancer-Associated Fibroblasts (CAFs) from inhibiting the CAR-Ts. CAFs are a cellular component of the TME, well documented, for example, in multiple myeloma, which can inhibit BCMA CAR T cells [25] such as Abecma (Ide-Cel). A dual CAR targeting BCMA as well as specific CAF antigens such as FAB may allow CAR-Ts to deplete this source of inhibition while carrying out their anti-tumour activity.
Another approach to overcome TME lies in the CAR design called TRUCK CAR Ts, used against inhibitory extracellular vesicles. Such vesicles are part of the milieu of the TME and have been documented [26] expressing PD-1L, thus blocking CAR-Ts immune checkpoints, or even containing inhibitory-micro-RNAs (miRNAs), which can target T-cell pathways resulting in T-cell exhaustion. A CAR able to allow its T cell to secrete FAS-L2 to target such miRNAs, or even PD-1L inhibitor-like moieties may be able to overcome inhibition from the TME’s extra-cellular vesicles.
Lastly, another component of the TME for which potential solutions have been investigated is the cytokine Transforming Growth Factor-β (TGF-β), which has a prominent immunosuppressive role in the TME [27]. An approach that has already been clinically investigated is the development of a CAR armoured with a TGF-β Receptor Dominant-Negative (RDN) [28] that would keep expanding despite TGF-β’s immunosuppressive signal.
Novel designs, such as peptide-centric (PC) CAR-Ts [29], are possible solutions to this type of roadblock.
2.4. Access to CAR-T-cell therapy
The number of patients having access to CAR-T-cell treatment is currently quite limited, despite the high and increasing level of interest, investments, and research in the field. According to the EBMT registry [30], 2500 patients were infused with a CAR-T medicinal product in 2021. When comparing this figure to the total number of patients for diseases where at least one CAR-T product has that indication [31], they only represent about 1% of the total number of cases. Crucial roadblocks to the increase of CAR-T-cell therapy access to patients are the long and complex manufacturing processes and high costs.
Although the actual CAR-T-based treatment price can vary significantly among EU member states, it may indicatively be considered at around EUR 350.000, with possible variations due to different agreements stipulated between the Marketing Authorization Holders (MAHs) and member states’ NHSs.
Despite this very high price tag, in principle making CAR-Ts profitable products, it crushes their ability to fully become part of the mainstream cancer treatment options.
Two potential solutions have been discussed in the available literature, consisting of (i) increasing production outputs to lower costs (e.g., increased levels of automation and miniaturisation of GMP-grade production facilities), and (ii) manufacturing of allogeneic products to lower costs (e.g., development of non-T CAR-engineered cells, like CAR NK and CAR M and/or the use of novel sources of biologic materials, ranging from healthy donors to iPSCs, generating so-called iCAR-Ts). The first is reasonably the near-term solution that can be expected in the coming years, while the second can constitute instead a long-term objective. Allogeneic products will allow a virtually unlimited quantity of medicinal products, with lower costs and the possibility to increase the complexity, and thus hopefully the effectiveness of CARs, by introducing multiple transgenes and/or mutations to the engineered cell.
2.4.1. GMP Cell Manufacturing: Cost and Complexity of Production
When analysing and comparing the efforts to address the cost and complexity of CAR-T-cell therapy, a recurrent theme across both literature and scientific institutions developing this technology is the need to pass from a centralised process to a decentralised production process [32][33], both shown in Figure 4:
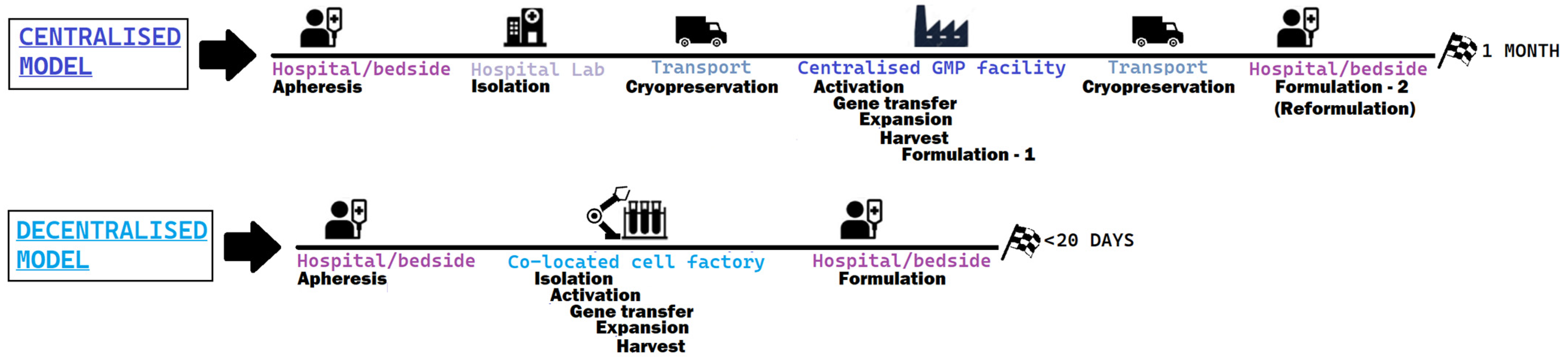
Figure 4. Steps of the prototypical centralised model, the one adopted by authorised CAR-T products versus a decentralised model, much more compact and less variable.
In a centralised model, the key to production is the company and its GMP facilities. Current challenges in access to therapy partly stem from the complex nature of the process, that can take between 3–4 to 6–8 weeks from the time that leukapheresis is performed and patient’s cells are shipped to the company, and the product is back to the hospital for being reinfused. The extremely expensive nature of this kind of medicinal product, due to this complex production process as well as the low scale of production, translates to the fact that only major academic centres can afford this therapeutic option, further decreasing access, similarly to the established third-line treatment alternative to CAR-Ts (bone marrow transplantation), which is not available in small clinical centres. In addition, by looking at the clinical trial results with the centralised model, it was observed that some patients were invariably lost because the time to acquire the medicinal product was too long for the patient to wait, and the bridging therapy was not sufficient.
On the other side, the decentralised production model aims at significantly reducing the overall time to reach the patient, by cutting storage and transportation times through the use of mobile cell factories. The possibility of doing CAR-T infusions as an outpatient therapy, an increase in the number of centres that can adopt CAR-T-cell therapies, shortening the time for potentially life-saving innovations to reach the clinical setting, are some of the potential advantages of this approach. A nice example of a decentralised and flexible approach was tested in St. James’s Hospital and the Trinity College Dublin Clinical Research Facility in Dublin (July 2022), moving a cell factory closer to patients using a mobile GMP facility for CAR-T [34].
Further investigations into the use of more automated manufacturing platforms and co-located GMP-compliant facilities are needed. Examples of currently ongoing innovative developments in the field are carried out by several players, such as Lonza pharmaceuticals® [35], Mylteni therapeutics® [36], and aCGT Vector [37].
The establishment of GMP manufacturing quality standards is considered an important challenge with regulatory implications. Specific guidelines do not exist yet, and coordination between regulators and manufacturers is needed to bridge this gap in the near future.
2.4.2. Allogeneic CAR-T
Another sprawling field of interest which addresses the limitations of CAR-T-cell therapy are “Off-the-shelf” CAR-Ts, obtained from healthy donors (allogeneic T cells), which can provide high amounts of fully functional cells and allow multiple CAR-T-cell products to generate. Such an approach may increase patients’ access to therapy, also reducing the delivery time of products that can be stored like conventional biologics.
Their major limitation is the need to undergo an additional editing step to prevent Graft-versus-Host (GvHD) disease-type rejection [38]. Though results are inferior to the autologous CAR-Ts at the moment, the presence of therapeutic action proves the potential of this platform. Along with T cells engineered to be allogeneic CAR-Ts, Natural Killer (NK) cells have been successfully used as a separate cell type to produce allogeneic CAR-engineered medicinal products, with no relevant toxic effect [39].
Another promising approach for the generation of allogeneic CAR-T products foresees the use of in vitro-induced pluripotent stem cells (iPSCs) [40], instead of patients’ blood cells from healthy donors.
The promise of these engineered stem cells is fascinating: with a potentially perpetual supply of virtually unlimitedly editable cells, the versatility of iPSC-derived CAR-T cells (iCAR-Ts) has garnered increasing investments and interest. iPSC technology is currently in its “first wave” and suffers from several limitations, like the absence of a clear regulation for the product’s quality assessment and release for clinical use, the lacking acceptance of key stakeholders towards iPSC technology, especially patients, and the non-availability of robust and scalable manufacturing protocols for clinical-grade iCAR-T cells.
Other gaps in knowledge that are likely to impact in the long term the access to iCAR-T therapies are the suboptimal function and developmental maturity of iPSC-derived CAR-Ts in comparison to ‘primary’, autologous CAR-Ts. So far, studies conducted on T cells derived from iPSCs (TiPSCs) only managed to produce CD8αα CAR-T cells with a low activity profile, similar to innate T cells [41]. This phenomenon is most probably attributable to the β-selection stage of T-cell development, which seemed to be skipped. This can deprive CD8+ cells of important moieties, such as CD28 and CCR7, respectively, a co-stimulatory receptor which allows significant activation and survival of T cells, and a memory marker which characterises the central memory phenotype, hence leading to significant persistence in vivo [42].
Currently, to use TiPSCs as raw material to engineer CAR-Ts, a master iPSC cell bank must be created via gene editing and subcloning, specifically dedicated to the disease in study. This is a long and technically challenging, substantially expensive process, which can generate genotoxic products, and it prevents the introduction of more complex CAR designs (armoured or dual CARs). Surpassing this technological challenge appears to be crucial to the ability to scale up quantities of CAR-T produced starting from iPSCs, and the potential of iPSCs to compete with cells from healthy donors.
References
- Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on Advanced Therapy Medicinal Products and Amending Directive 2001/83/EC and Regulation (EC) No 726/2004 25th of February. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2007:324:0121:0137:en:PDF (accessed on 23 May 2023).
- Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999 on Orphan Medicinal Products. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32000R0141&from=EN (accessed on 23 May 2023).
- European Commission Proposal on Pharmaceutical Legislation. Available online: https://health.ec.europa.eu/medicinal-products/pharmaceutical-strategy-europe/reform-eu-pharmaceutical-legislation_en (accessed on 23 May 2023).
- Compassionate Use of Medicinal Products, Pursuant to Article 83 of Regulation (EC) No 726/2004 Guideline on Compassionate Use of Medicinal Products. Available online: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/guideline-compassionate-use-medicinal-products-pursuant-article-83-regulation-ec-no-726/2004_en.pdf (accessed on 23 May 2023).
- Berrien-Elliott, M.M.; Jacobs, M.T.; Fehniger, T.A. Allogeneic natural killer cell therapy. Blood 2023, 141, 856–868.
- Chen, Y.; Yu, Z.; Tan, X.; Jiang, H.; Xu, Z.; Fang, Y.; Han, D.; Hong, W.; Wei, W.; Tu, J. CAR-macrophage: A new immunotherapy candidate against solid tumors. Biomed. Pharmacother. 2021, 139, 111605.
- De Marco, R.C.; Monzo, H.J.; Ojala, P.M. CAR T Cell Therapy: A Versatile Living Drug. Int. J. Mol. Sci. 2023, 24, 6300.
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. EBioMedicine 2020, 58, 102931.
- Tokarew, N.; Ogonek, J.; Endres, S.; Von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2018, 120, 26–37.
- Bao, C.; Gao, Q.; Li, L.-L.; Han, L.; Zhang, B.; Ding, Y.; Song, Z.; Zhang, R.; Zhang, J.; Wu, X.-H. The Application of Nanobody in CAR-T Therapy. Biomolecules 2021, 11, 238.
- Ma, Q.; Garber, H.R.; Lu, S.; He, H.; Tallis, E.; Ding, X.; Sergeeva, A.; Wood, M.S.; Dotti, G.; Salvado, B.; et al. A novel TCR-like CAR with specificity for PR1/HLA-A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy 2016, 18, 985–994.
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 2019, 7, 276.
- Labanieh, L.; Mackall, C.L. CAR immune cells: Design principles, resistance and the next generation. Nature 2023, 614, 635–648.
- Yan, T.; Zhu, L.; Chen, J. Current advances and challenges in CAR T-Cell therapy for solid tumors: Tumor-associated antigens and the tumor microenvironment. Exp. Hematol. Oncol. 2023, 12, 14.
- Zmievskaya, E.; Valiullina, A.; Ganeeva, I.; Petukhov, A.; Rizvanov, A.; Bulatov, E. Application of CAR-T Cell Therapy beyond Oncology: Autoimmune Diseases and Viral Infections. Biomedicines 2021, 9, 59.
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638.
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55.
- Rooney, C.; Sauer, T. Modeling cytokine release syndrome. Nat. Med. 2018, 24, 705–706.
- Sterner, R.C.; Sterner, R.M. Immune effector cell associated neurotoxicity syndrome in chimeric antigen receptor-T cell therapy. Front. Immunol. 2022, 13, 879608.
- Siegler, E.L.; Kenderian, S.S. Neurotoxicity and Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy: Insights into Mechanisms and Novel Therapies. Front. Immunol. 2020, 11, 1973.
- Tallantyre, E.C.; Evans, N.A.; Parry-Jones, J.; Morgan, M.P.G.; Jones, C.H.; Ingram, W. Neurological updates: Neurological complications of CAR-T therapy. J. Neurol. 2021, 268, 1544–1554.
- Luksik, A.S.; Yazigi, E.; Shah, P.; Jackson, C.M. CAR T Cell Therapy in Glioblastoma: Overcoming Challenges Related to Antigen Expression. Cancers 2023, 15, 1414.
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941.
- Fonkoua, L.A.K.; Sirpilla, O.; Sakemura, R.; Siegler, E.L.; Kenderian, S.S. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol. Ther.-Oncolytics 2022, 25, 69–77.
- Sakemura, R.; Hefazi, M.; Siegler, E.L.; Cox, M.J.; Larson, D.P.; Hansen, M.J.; Roman, C.M.; Schick, K.J.; Can, I.; Tapper, E.E.; et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood 2022, 139, 3708–3721.
- Cox, M.J.; Lucien, F.; Sakemura, R.; Boysen, J.C.; Kim, Y.; Horvei, P.; Roman, C.M.; Hansen, M.J.; Tapper, E.E.; Siegler, E.L.; et al. Leukemic extracellular vesicles induce chimeric antigen receptor T cell dysfunction in chronic lymphocytic leukemia. Mol. Ther. 2021, 29, 1529–1540.
- Liu, M.; Kuo, F.; Capistrano, K.J.; Kang, D.; Nixon, B.G.; Shi, W.; Chou, C.; Do, M.H.; Stamatiades, E.G.; Gao, S.; et al. TGF-β suppresses type 2 immunity to cancer. Nature 2020, 587, 115–120.
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation and Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866.
- Yarmarkovich, M.; Marshall, Q.F.; Warrington, J.M.; Premaratne, R.; Farrel, A.; Groff, D.; Li, W.; di Marco, M.; Runbeck, E.; Truong, H.; et al. Cross-HLA targeting of intracellular oncoproteins with peptide-centric CARs. Nature 2021, 599, 477–484.
- The EMBT Registry. Available online: https://www.ebmt.org/registry/data-collection-car-t-cells (accessed on 8 June 2023).
- Cancer Today—Database by the International Agency for Research on Cancer, World Health Organisation. Available online: https://gco.iarc.fr/today/data/factsheets/populations/908-europe-fact-sheets.pd (accessed on 8 June 2023).
- Gee, A.P. GMP CAR-T cell production. Best Pr. Res. Clin. Haematol. 2018, 31, 126–134.
- Abou-El-Enein, M.; Elsallab, M.; Feldman, S.A.; Fesnak, A.D.; Heslop, H.E.; Marks, P.; Till, B.G.; Bauer, G.; Savoldo, B. Scalable Manufacturing of CAR T Cells for Cancer Immunotherapy. Blood Cancer Discov. 2021, 2, 408–422.
- The Future of the Delivery of Modern Medicine. Available online: https://www.acgtvector.com/the-future-of-the-delivery-of-modern-medicine/ (accessed on 23 May 2023).
- Cocoon® Platform|Cell Therapy Manufacturing|Lonza. Available online: https://www.lonza.com/knowledge-center/cellgene/brief/cocoon-platform-brochure (accessed on 23 May 2023).
- CliniMACS Prodigy®|Automated Cell Processing with the CliniMACS Prodigy|USA. Available online: https://www.miltenyibiotec.com/US-en/products/clinimacs-prodigy.html (accessed on 23 May 2023).
- GMP in a POD. Available online: https://www.acgtvector.com/ (accessed on 23 May 2023).
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: Results of two phase 1 studies. Lancet 2020, 396, 1885–1894.
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553.
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130.
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933.
- Maeda, T.; Nagano, S.; Ichise, H.; Kataoka, K.; Yamada, D.; Ogawa, S.; Koseki, H.; Kitawaki, T.; Kadowaki, N.; Takaori-Kondo, A.; et al. Regeneration of CD8αβ T Cells from T-cell–Derived iPSC Imparts Potent Tumor Antigen-Specific Cytotoxicity. Cancer Res. 2016, 76, 6839–6850.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
626
Revisions:
3 times
(View History)
Update Date:
27 Jul 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No