Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tamer Fandy | -- | 2432 | 2023-07-23 08:59:54 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Gallimore, F.; Fandy, T.E. Applications of Azanucleoside Analogs as DNA Demethylating Agents. Encyclopedia. Available online: https://encyclopedia.pub/entry/47141 (accessed on 29 July 2026).
Gallimore F, Fandy TE. Applications of Azanucleoside Analogs as DNA Demethylating Agents. Encyclopedia. Available at: https://encyclopedia.pub/entry/47141. Accessed July 29, 2026.
Gallimore, Fallon, Tamer E. Fandy. "Applications of Azanucleoside Analogs as DNA Demethylating Agents" Encyclopedia, https://encyclopedia.pub/entry/47141 (accessed July 29, 2026).
Gallimore, F., & Fandy, T.E. (2023, July 23). Applications of Azanucleoside Analogs as DNA Demethylating Agents. In Encyclopedia. https://encyclopedia.pub/entry/47141
Gallimore, Fallon and Tamer E. Fandy. "Applications of Azanucleoside Analogs as DNA Demethylating Agents." Encyclopedia. Web. 23 July, 2023.
Copy Citation
Azanucleosides, such as 5-azacytidine and decitabine, are DNA demethylating agents used in the treatment of acute myeloid leukemia and myelodysplastic syndromes.
epigenetics
DNA methylation
DNA demethylating agents
leukemia
5-azanucleosides
1. Introduction
Epigenetic modifications are the main mechanism underlying normal and abnormal human development. There are numerous modifications found within the human genome at any given time and DNA methylation is among the most common and well-known of these modifications. DNA methylation regulates many important cellular processes, including gene expression and repression, chromatin remodeling, genomic imprinting, and X-chromosome inactivation. It is essential for normal development and aberrant DNA methylation in the form of hypomethylation or hypermethylation can contribute to the development of disease [1]. The azanucleosides 5-azacytidine, decitabine, and guadecitabine interfere with the DNA methylation machinery and demonstrated efficacy in the treatment of myelodysplastic syndromes, blood dyscrasias, and other malignancies resistant to standard chemotherapeutic agents.
2. DNA Methylation and Demethylation
Histone posttranslational modifications in the mammalian genome include sumoylation, ubiquitylation, acetylation, phosphorylation, citrullination, crotonylation, and methylation. These modifications are regulated by specific enzymes that add or remove the chemical moiety and epigenetically regulate gene expression. Alterations in these epigenetic modifications are associated with aging as well as the development of various cancers and degenerative diseases [1][2]. Among these modifications, DNA methylation and histone acetylation are the most studied, where DNA methylation is involved in the normal development of mammals and key biologic processes, such as X-chromosome inactivation and genomic imprinting [3][4][5][6].
The process of DNA methylation is catalyzed by the DNA methyltransferase (DNMT) family of enzymes. These enzymes facilitate the transfer of a labile methyl group from the methyl donor S-adenosyl-L-methionine (SAM) cofactor to the C5 position of the cytosine ring, forming 5-methylcytosine (5 mC). Mammalian DNMTs include DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L. DNMT1 is a large protein that forms a complex with Ubiquitin-like, containing PHD and RING finger domains 1 (UHRF1) to maintain DNA methylation during the process of DNA replication. DNMT3A and DNMT3B are responsible for establishing methylation patterns during embryonic development. DNMT3L is catalytically inactive but required for gene imprinting and for regulation of activities by DNMT3A and DNMT3B. Lastly, DNMT2 lacks DNMT activity but instead acts as an RNA methyltransferase. In somatic cells, almost all of the DNA methylation occurs in a CpG dinucleotide context. The regions of DNA that contain a large number of CpG dinucleotide repeats are referred to as CpG islands [7].
Methylated DNA is recognized by methyl-CpG-binding domain (MBD) proteins, UHRF proteins, zinc finger-containing proteins, basic leucine zipper-containing transcription factors, and homeodomain-containing transcription factors. MBD-containing proteins, such as MeCP2, MBD1, MBD2, MBD3, and MBD4, are involved in the maintenance and spread of DNA methylation, recruitment of DNMT1 to hemi-methylated DNA, and repression of gene transcription. The MBD domain of MeCP2 is highly specific to methylated CpG sites adjacent to A/T-rich sequences. UHRF proteins, including UHRF1 and UHRF2, maintain DNA methylation by targeting DNMT1 to hemi-methylated DNA during the process of DNA replication. Zinc finger domain proteins, including ZBTB33 (Kaiso), ZBTB4, and ZBTB38, bind to methylated DNA and repress transcription in a DNA methylation-dependent manner [7][8][9].
Adenine bases in the mammalian genome can also undergo SAM-dependent methylation similar to cytosine. Adenine methylation has recently been discovered through the use of deep sequencing technology. This modification is catalyzed by N6-adenine-specific DNMT1 (N6AMT1) and commonly takes place on the amino group at the sixth position of the purine ring. The result is the formation of N6-methyldeoxyadenosine. Demethylation of adenine is accomplished by alkylated DNA repair protein B homolog (ALKBH1). Studies have shown that decreased levels of genomic adenine methylation are associated with tumorigenesis and poor prognosis for cancer patients. Studies have also reported that there is a connection between adenine methylation and mitochondrial DNA damage [7].
DNA demethylation is mediated by either an active or passive mechanism. Active DNA demethylation is catalyzed by the ten-eleven tanslocation (TET) proteins. These are Fe(II) and α-ketoglutarate-dependent dioxygenases that utilize molecular oxygen and α-ketoglutarate as co-substrates to generate sequentially oxidized 5 mC derivatives, such as 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxycytosine. These entities are recognized and excised by thymine DNA glycosylase, which generates apyrimidinic sites that are subsequently corrected with base excision repair. On the other hand, passive DNA demethylation occurs when 5 mC is lost due to DNMT1 loss of function and successive rounds of DNA replication; this ultimately results in the dilution of the methylated DNA. Figure 1 summarizes the mechanism of DNA methylation and demethylation by passive and active ways.
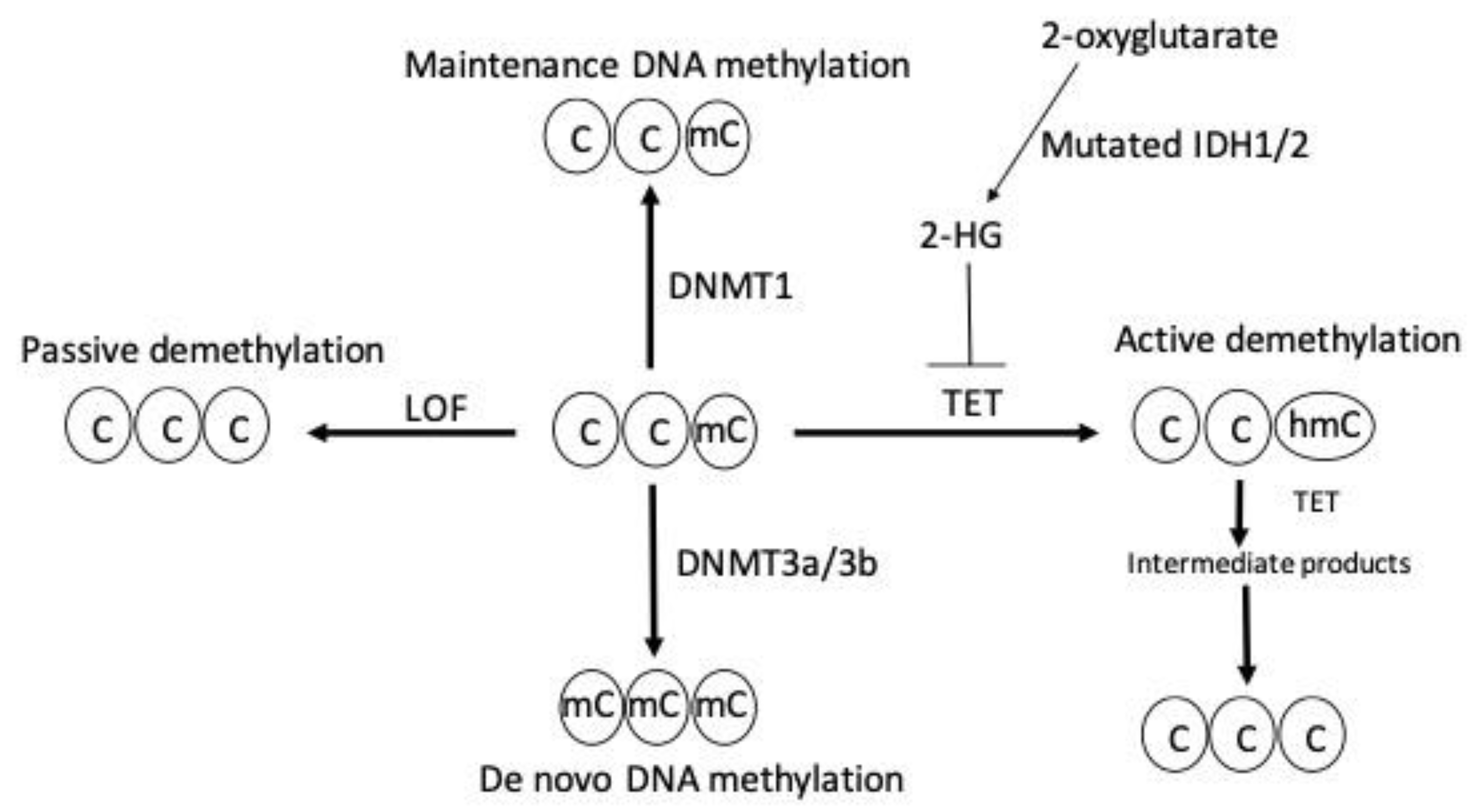
Figure 1. DNA methylation and demethylation. Maintenance and de novo DNA methylation are catalyzed by DNMT1 and DNMT3a/3b, respectively. Loss of function (LOF) mutation of DNMT enzymes induces passive demethylation. TET enzymes catalyze active demethylation by sequential oxidation reactions that leads to the formation of sequential intermediate products, such as 5 hydroxylmethylcytosine (5 hmc), 5-formylcytosine, and, finally, 5-carboxylcytosine, which is replaced by unmodified cytosine through base excision repair. TET enzymes are inhibited by the oncometabolite 2-hydroxyglutarate (2-HG) generated by neomorphic mutations in isocitrate dehydrogenase 1 (IDH1) and IDH2 enzymes resulting in active demethylation inhibition. C indicates cytosine, mC indicates methylcytosine, and hmC indicates 5-hydroxylmethylcytosine.
3. Normal and Aberrant DNA Methylation
There are several biological functions of DNA methylation. These include the transcriptional repression of retrotransposons, monoallelic expression of imprinted genes, X-chromosome inactivation in female cells, and regulation of tissue-specific gene expression [8][9][10]. DNA methylation’s repressive effect on gene expression is observed across different regions of the genome. For instance, intergenic regions, including transposable and viral elements, compose nearly 45% of the mammalian genome. DNA methylation plays a repressive role in these regions to avoid their expression and consequent genetic mutations. Likewise, CpG islands in or around promoter regions could be methylated to impair the binding of transcription factors, recruit repressive methyl-binding proteins, and silence gene expression. This is important because the majority of gene promoters, especially for housekeeping genes, reside within CpG islands [9].
Aberrant DNA methylation is characterized by a genome that is generally hypomethylated or hypermethylated. Mechanisms for hypermethylation and hypomethylation include changes in the activity of DNMTs and TET enzymes, structure of chromatin, and selective recruitment of DNA modifying enzymes. These aberrant patterns are known to produce genomic instability and promote pathological conditions, including neurological diseases, immunological diseases, atherosclerosis, osteoporosis, and cancer [11][12][13][14].
Abnormal gene-specific methylation and global hypomethylation contribute to disease in humans. The effects of global DNA hypomethylation are thought to be due to the activation of normally dormant transposable elements within the genome. Activation events could result in changes in transcription factor levels, especially those involved in growth regulation, as well as the accumulation of genomic mutations and abnormal chromosomal recombination. As a result, there is an increase in DNA damage, decondensation of chromatin, and chromosomal instability [13].
Cancers typically display widespread hypomethylation of DNA repetitive elements as well as focal DNA hypermethylation. Hypermethylation of an active CpG-rich promoter often corresponds to repression of the gene’s expression except in cases where there is an alternative unmethylated promoter. Tumor suppressor genes, miRNAs, and other regulatory elements are often targets for this modification in disease states.
4. DNA Demethylating Agents
DNA demethylating agents, also known as DNMT inhibitors (DNMTi), are a class of drugs used for a reversal of DNA methylation. There are two main classes of DNA demethylating agents: nucleoside DNMTi and non-nucleoside DNMTi. Nucleoside DNMTi incorporate into DNA, trap DNMTs, facilitate their degradation by cellular proteosomes, and consequently lead to DNA demethylation. Examples of nucleoside DNMTi include 5-azacytidine, decitabine, zebularine, and guadecitabine. On the other hand, the non-nucleoside DNMTi induce DNA demethylation by mechanisms that do require incorporation into DNA. Examples of non-nucleoside DNMTIs include curcumin, procaine, hydralazine, and isooxazoline [15][16].
Azanucleosides (AZN) were first synthesized in 1964, and clinical trials began to demonstrate their anticancer activity shortly after this. The mechanism of action of AZN drugs starts with their cellular uptake, intracellular metabolism, and incorporation into nucleic acids. Incorporation of the 5-aza-dCTP metabolite impairs methylation of DNA by irreversible inhibition of DNMTs. DNMT1 is responsible for the maintenance of methylation patterns during DNA replication; therefore, the inhibition and eventual degradation of this enzyme means that methylation patterns are lost during DNA replication [15][16][17][18].
Azacitidine (5-azacytidine) was the first DNA hypomethylating agent to receive regulatory approval by the FDA in 2004 for the treatment of myelodysplastic syndrome (MDS). Since then, it has also received approval for treatment of acute myeloid leukemia (AML) with 20–30% bone marrow blasts. Azacitidine is mostly incorporated into RNA, and the remaining 10–20% of the drug is incorporated into DNA after a multistep conversion by the enzyme ribonucleotide reductase. The incorporation of azacytidine into DNA and RNA results in DNA damage, growth inhibition, G2 cell cycle arrest, apoptosis, and inhibition of DNA synthesis and repair [15].
Decitabine (5-aza-2′-deoxycytidine) was the second DNA hypomethylating agent to receive regulatory approval by the FDA in 2006 for the treatment of high-risk MDS. Decitabine has a similar chemical structure and mechanism to azacitidine; however, it only incorporates into the DNA, induces apoptosis in a p53-dependent manner, and has greater demethylation potential than azacitidine [16]. Guadecitabine (SGI-110) is a second-generation DNMTi that was designed by chemically linking decitabine to deoxyguanosine by a phosphodiester bond. This design makes the drug less susceptible to degradation by the enzyme cytidine deaminase and is expected to improve the exposure time and degree of marrow penetration by the drug. Early studies with guadecitabine have demonstrated greater DNA hypomethylation activity compared to azacitidine and decitabine [19].
5. Therapeutic Applications of AZN
Azacitidine, decitabine, and guadecitabine are primarily used to manage hematologic malignancies, such as MDS and AML [20][21]. The anticancer activity of these drugs is hypothesized to be mediated through inhibiting the activity of DNMTs and consequent reactivation of epigenetically silenced tumor suppressor genes. The reactivation of these genes may lead to various events, including cell differentiation, death, inhibition of proliferation, and sensitization to chemotherapy and immunotherapy. In the tumor microenvironment, AZNs induce an increase in immune responses and decrease in angiogenesis [22][23]. Furthermore, it has been demonstrated that decitabine augments the responsiveness of natural killer cells, which are a part of the body’s innate response against malignant cells [24].
MDS refers to a collection of bone marrow diseases characterized by cytopenia, dysplastic morphology, and genetic evidence of clonality. Cases of MDS are classified based on whether there is single-lineage or multilineage dysplasia, ring sideroblasts, excess blasts, or an isolated del(5q) abnormality [25]. AML is characterized by abnormal proliferation and differentiation of clonal myeloid stem cells. Cases of AML are classified based on whether there are recurrent genetic abnormalities, myelodysplasia-related changes, therapy-related myeloid neoplasms, myeloid sarcomas, and myeloid proliferations related to Down syndrome [26].
6. AZN Combination Therapies
Combination therapy using AZN has demonstrated the following effects: enhanced radiation sensitivity, increased sensitivity to anticancer drugs, cancer cell reprogramming, and induction of an immune response against cancer. Most of the combinations that have been assessed so far involve the combination of decitabine and platinum-based drugs (e.g., cisplatin and carboplatin). It is important to note that these findings are mostly for the treatment of solid tumors [27]. The rationale behind using combination therapy is to allow for hitting multiple targets and, consequently, sensitize resistant tumors [16].
The antiapoptotic proteins of the B-cell lymphoma 2 (BCL-2) family are commonly overexpressed in leukemic stem cell subpopulations. This suggests that the use of BCL-2 inhibitors in the treatment of AML and MDS will sensitize the tumor cells [28]. Venetoclax (ABT-199) is an oral BCL-2 inhibitor that has currently received FDA approval for the treatment of chronic lymphocytic leukemia (CLL) and small lymphocytic leukemia [16]. These inhibitors, including venetoclax, are known to sensitize the myeloid malignancies to the actions of AZN, such as 5-azacytidine, decitabine, and guadecitabine [29]. Indeed, the combination of 5-azacytidine and venetoclax was demonstrated to block amino acid metabolism, which is crucial to the survival of the leukemic stem cells [30]. This finding ultimately led to the FDA approval of venetoclax in combination with DNMT inhibitors for the treatment of AML patients who cannot undergo intensive chemotherapy.
Histone deacetylases (HDACs) are enzymes that remove the acetyl groups from lysine residues, controlling the remodeling of chromatin, and gene expression. Different classes of HDAC inhibitors, including short chain fatty acids, benzamides, cyclic peptides, hydroxamic acids, and miscellaneous compounds were developed as antitumor agents [31]. A study looking at a murine mammary tumor model used 5-azacytidine in combination with butyrate was found to reduce the number of cancer stem cells and increase overall survival through inhibition of growth-promoting proteins [32].
The combination therapy of DNMTIs and poly ADP-ribose polymerase inhibitors (PARPIs) was also investigated. PARPs are nuclear proteins that are involved in DNA base-excision and single-strand repairs. PARP1 and PARP2 are the main targets of many anticancer drugs due to their specific roles in DNA damage repair [33]. Examples of PARPIs that have shown potential usefulness in the treatment of various cancers include veliparib and talazoparib. A recent study reported that the combination of guadecitabine- and talazoparib-sensitized breast and ovarian cancers are resistant to PARPIs independent of BRCA mutations, which indicates that this combination may be useful in clinical settings [34].
Recently, a combination therapy involving decitabine and cedazuridine was approved by the FDA. The effectiveness of oral decitabine is limited by its extensive metabolism by the enzyme cytidine deaminase. Cedazuridine is responsible for the inhibition of cytidine deaminase in the gastrointestinal tract and liver and, consequently, increases the systemic exposure time of decitabine following oral administration. A fixed oral dose combination of decitabine and cedazuridine is currently being developed for the treatment of MDS, chronic myelomonocytic leukemia (CMML), AML, glioma, and solid tumors. The combination has approval for the treatment of MDS and CMML at this moment. This includes treated and untreated MDS and CMML with specific subtypes (i.e., refractory anemia, refractory anemia with ringed sideroblasts, and refractory anemia with excess blasts) and scores using the International Prognostic Scoring System (i.e., intermediate-1, intermediate-2, and high-risk). The recommended dosage is 35 mg of decitabine and 100 mg of cedazuridine taken for five days on a 28-day cycle with a minimum of four cycles. Furthermore, other studies have shown that the cedazuridine combination therapy increases the bioavailability of decitabine, produces consistent efficacy similar to AZN single agent treatment, and has a tolerability profile similar to IV decitabine [35][36][37].
Resistance development to AZN therapy was correlated with upregulating the expression of PD-1 and PD-L1 in immune T cells. Persistent expression and engagement of PD-1 results in tumor immune evasion. Rational combination of the PD-1 inhibitor nivolumab with AZN showed a higher overall response rate than AZN therapy alone in older AML patients who were ineligible for intensive chemotherapy [38]. Moreover, AZN treatment enhanced responses to anti-CTLA-4 immunotherapy in a melanoma model by activating the endogenous retroviral pathway (ERV). Stimulation of the ERV pathway culminates in T cell activation through IFN production [39].
References
- Hashimoto, H.; Vertino, P.M.; Cheng, X. Molecular coupling of DNA methylation and histone methylation. Epigenomics 2010, 2, 657–669.
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7.
- Bestor, T.H.; Edwards, J.R.; Boulard, M. Notes on the role of dynamic DNA methylation in mammalian development. Proc. Natl. Acad. Sci. USA 2015, 112, 6796–6799.
- Geiman, T.M.; Muegge, K. DNA methylation in early development. Mol. Reprod. Dev. 2010, 77, 105–113.
- Guibert, S.; Forne, T.; Weber, M. Dynamic regulation of DNA methylation during mammalian development. Epigenomics 2009, 1, 81–98.
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093.
- Sriraman, A.; Debnath, T.K.; Xhemalce, B.; Miller, K.M. Making it or breaking it: DNA methylation and genome integrity. Essays Biochem. 2020, 64, 687–703.
- Meng, H.; Cao, Y.; Qin, J.; Song, X.; Zhang, Q.; Shi, Y.; Cao, L. DNA methylation, its mediators and genome integrity. Int. J. Biol. Sci. 2015, 11, 604–617.
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38.
- Edwards, J.R.; Yarychkivska, O.; Boulard, M.; Bestor, T.H. DNA methylation and DNA methyltransferases. Epigenetics Chromatin 2017, 10, 23.
- Ehrlich, M. DNA hypermethylation in disease: Mechanisms and clinical relevance. Epigenetics 2019, 14, 1141–1163.
- Martisova, A.; Holcakova, J.; Izadi, N.; Sebuyoya, R.; Hrstka, R.; Bartosik, M. DNA Methylation in Solid Tumors: Functions and Methods of Detection. Int. J. Mol. Sci. 2021, 22, 4247.
- Wilson, A.S.; Power, B.E.; Molloy, P.L. DNA hypomethylation and human diseases. Biochim. Biophys. Acta 2007, 1775, 138–162.
- Xu, X. DNA methylation and cognitive aging. Oncotarget 2015, 6, 13922–13932.
- Agrawal, K.; Das, V.; Vyas, P.; Hajduch, M. Nucleosidic DNA demethylating epigenetic drugs—A comprehensive review from discovery to clinic. Pharmacol. Ther. 2018, 188, 45–79.
- Mehdipour, P.; Murphy, T.; De Carvalho, D.D. The role of DNA-demethylating agents in cancer therapy. Pharmacol. Ther. 2020, 205, 107416.
- Diesch, J.; Zwick, A.; Garz, A.K.; Palau, A.; Buschbeck, M.; Gotze, K.S. A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin. Epigenetics 2016, 8, 71.
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13.
- Pan, D.; Rampal, R.; Mascarenhas, J. Clinical developments in epigenetic-directed therapies in acute myeloid leukemia. Blood Adv. 2020, 4, 970–982.
- Palacios-Berraquero, M.L.; Alfonso-Pierola, A. Current Therapy of the Patients with MDS: Walking towards Personalized Therapy. J. Clin. Med. 2021, 10, 2107.
- Platzbecker, U. Treatment of MDS. Blood 2019, 133, 1096–1107.
- Howell, P.M.; Liu, Z.; Khong, H.T. Demethylating Agents in the Treatment of Cancer. Pharmaceuticals 2010, 3, 2022–2044.
- Sato, T.; Issa, J.J.; Kropf, P. DNA Hypomethylating Drugs in Cancer Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026948.
- Schmiedel, B.J.; Arelin, V.; Gruenebach, F.; Krusch, M.; Schmidt, S.M.; Salih, H.R. Azacytidine impairs NK cell reactivity while decitabine augments NK cell responsiveness toward stimulation. Int. J. Cancer 2011, 128, 2911–2922.
- Hasserjian, R.P. Myelodysplastic Syndrome Updated. Pathobiology 2019, 86, 7–13.
- De Kouchkovsky, I.; Abdul-Hay, M. ‘Acute myeloid leukemia: A comprehensive review and 2016 update’. Blood Cancer J. 2016, 6, e441.
- Hu, C.; Liu, X.; Zeng, Y.; Liu, J.; Wu, F. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: Mechanism and clinical application. Clin. Epigenetics 2021, 13, 166.
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341.
- Bogenberger, J.M.; Kornblau, S.M.; Pierceall, W.E.; Lena, R.; Chow, D.; Shi, C.X.; Mantei, J.; Ahmann, G.; Gonzales, I.M.; Choudhary, A.; et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia 2014, 28, 1657–1665.
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740 e724.
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179.
- Pathania, R.; Ramachandran, S.; Mariappan, G.; Thakur, P.; Shi, H.; Choi, J.H.; Manicassamy, S.; Kolhe, R.; Prasad, P.D.; Sharma, S.; et al. Combined Inhibition of DNMT and HDAC Blocks the Tumorigenicity of Cancer Stem-like Cells and Attenuates Mammary Tumor Growth. Cancer Res. 2016, 76, 3224–3235.
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528.
- Pulliam, N.; Fang, F.; Ozes, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 2018, 24, 3163–3175.
- Dhillon, S. Correction to: Decitabine/Cedazuridine: First Approval. Drugs 2021, 81, 179.
- Patel, A.A.; Cahill, K.; Saygin, C.; Odenike, O. Cedazuridine/decitabine: From preclinical to clinical development in myeloid malignancies. Blood Adv. 2021, 5, 2264–2271.
- Thota, S.; Oganesian, A.; Azab, M.; Griffiths, E.A. Role of cedazuridine/decitabine in the management of myelodysplastic syndrome and chronic myelomonocytic leukemia. Future Oncol. 2021, 17, 2077–2087.
- Stahl, M.; Goldberg, A.D. Immune Checkpoint Inhibitors in Acute Myeloid Leukemia: Novel Combinations and Therapeutic Targets. Curr. Oncol. Rep. 2019, 21, 37.
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986.
More
Information
Subjects:
Genetics & Heredity
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
599
Revision:
1 time
(View History)
Update Date:
23 Jul 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No