Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lucas Fornari Laurindo | -- | 4153 | 2023-07-21 14:35:46 | | | |
| 2 | Peter Tang | + 1 word(s) | 4154 | 2023-07-23 08:54:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Santos, J.P.M.D.; Maio, M.C.D.; Lemes, M.A.; Laurindo, L.F.; Haber, J.F.D.S.; Bechara, M.D.; Prado, P.S.D.; Rauen, E.C.; Costa, F.; Pereira, B.C.D.A.; et al. Non-Alcoholic Steatohepatitis and Organokines. Encyclopedia. Available online: https://encyclopedia.pub/entry/47119 (accessed on 27 July 2026).
Santos JPMD, Maio MCD, Lemes MA, Laurindo LF, Haber JFDS, Bechara MD, et al. Non-Alcoholic Steatohepatitis and Organokines. Encyclopedia. Available at: https://encyclopedia.pub/entry/47119. Accessed July 27, 2026.
Santos, João Paulo Margiotti Dos, Mariana Canevari De Maio, Monike Alves Lemes, Lucas Fornari Laurindo, Jesselina Francisco Dos Santos Haber, Marcelo Dib Bechara, Pedro Sidnei Do Prado, Eduardo Costa Rauen, Fernando Costa, Barbara Cristina De Abreu Pereira, et al. "Non-Alcoholic Steatohepatitis and Organokines" Encyclopedia, https://encyclopedia.pub/entry/47119 (accessed July 27, 2026).
Santos, J.P.M.D., Maio, M.C.D., Lemes, M.A., Laurindo, L.F., Haber, J.F.D.S., Bechara, M.D., Prado, P.S.D., Rauen, E.C., Costa, F., Pereira, B.C.D.A., Flato, U.A.P., Goulart, R.D.A., Chagas, E.F.B., & Barbalho, S.M. (2023, July 21). Non-Alcoholic Steatohepatitis and Organokines. In Encyclopedia. https://encyclopedia.pub/entry/47119
Santos, João Paulo Margiotti Dos, et al. "Non-Alcoholic Steatohepatitis and Organokines." Encyclopedia. Web. 21 July, 2023.
Copy Citation
Non-alcoholic steatohepatitis (NASH) is characterized by steatosis, lobular inflammation, and enlargement of the diameter of hepatocytes (ballooning hepatocytes), with or without fibrosis. It affects 20% of patients with non-alcoholic fatty liver disease (NAFLD). Due to liver dysfunction and the numerous metabolic changes that commonly accompany the condition (obesity, insulin resistance, type 2 diabetes, and metabolic syndrome), the secretion of organokines is modified, which may contribute to the pathogenesis or progression of the disease.
on-alcoholic fatty liver disease
dyslipidemias
oxidative stress
inflammation
organokines
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) can be defined as a macrovesicular steatosis (fat accumulation) in more than 5% of hepatocytes in the absence of a secondary cause such as alcohol and drug use or pre-existing liver disease. Non-alcoholic steatohepatitis (NASH) is a complication of NAFLD. In addition to steatosis, it is characterized by lobular inflammation and enlargement of the diameter of hepatocytes (ballooning hepatocytes), with or without fibrosis, and affects approximately 20% of patients with NAFLD. Such changes are noted on histological examination, and the presence of fibrosis reveals the worsening of the case. Its progression is related to cirrhosis, hepatocellular carcinoma, liver transplantation, and increased mortality from liver causes [1][2].
It should be noted that the metabolic definition of liver disease is primarily defined based on histopathological (pathological) findings or changes; however, recently, many hepatologists have suggested to rename NAFLD as MAFLD (metabolic associated fatty liver disease). The new term would be more faithful to the tight relationship between fatty liver and overfeeding, physical inactivity, and metabolic conditions (such as hypertension, DM2, dyslipidemia, and obesity) [3][4][5].
The understanding of the pathophysiology of NASH evolved substantially from the original hypothesis of two hits. The first is hepatic steatosis due to insulin resistance (IR). The second is oxidative stress, insufficient to describe the multiple molecular pathways and metabolic processes in NASH development. Thus, a multihit model was more recently proposed to explain this phenomenon, as it considers several injuries occurring in parallel in genetically predisposed individuals (polymorphisms in the PNPLA3 and TM6SF2 genes), such as nutritional factors, intestinal microbiota, IR, hormones secreted by adipose tissue, genetic factors, and epigenetics [2][6][7][8]. Figure 1 shows some aspects of NASH.
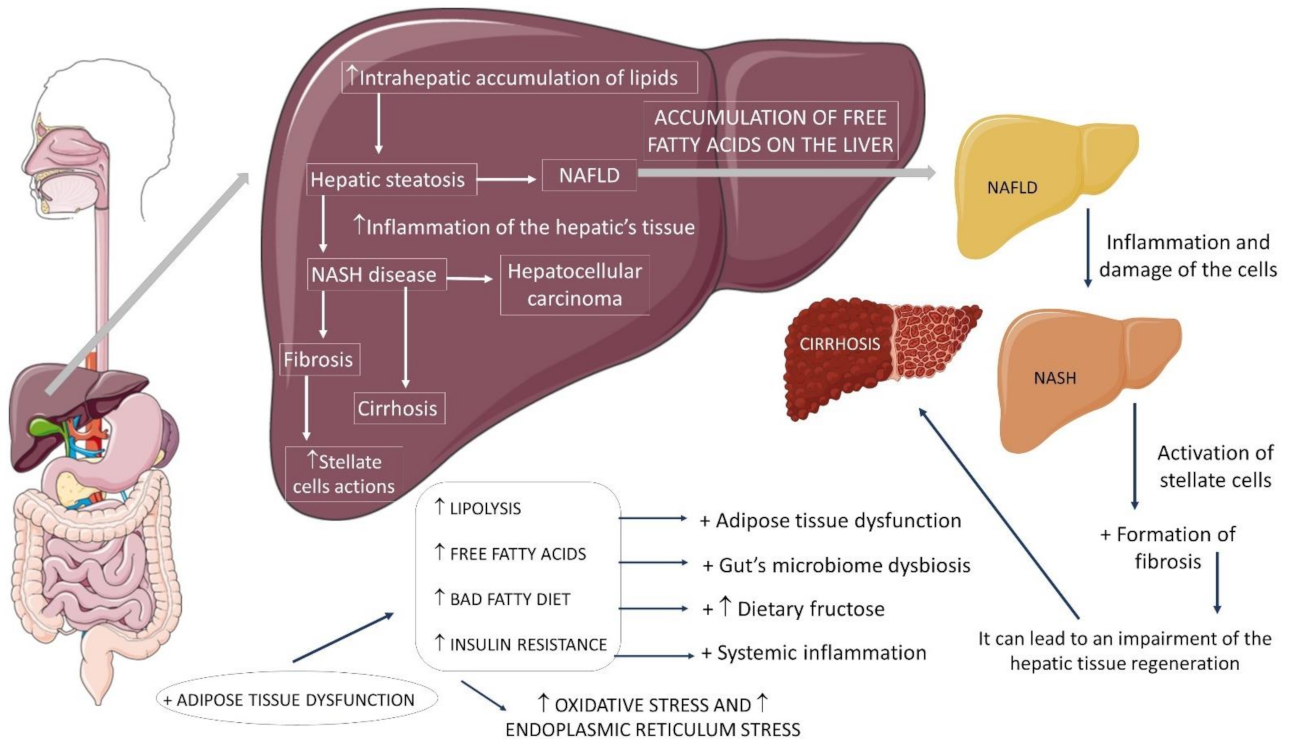
Figure 1. Non-alcoholic fatty liver disease and its progressions. ↑: increase, +: plus, NAFLD: non-alcoholic fatty liver disease, NASH: non-alcoholic steatohepatitis.
The prevalence of NASH is difficult to estimate due to the need for liver biopsy, the gold standard for diagnosis, and this practice is infrequent. According to indirect estimates, 3 to 6% of Americans present NASH, being more prevalent in individuals with previously established metabolic diseases, and of these, 20% progress to cirrhosis [2][9][10].
Considering that NAFLD (the precursor of NASH) affects up to 1 billion people worldwide and its prevalence has accompanied the growth of cases of obesity and Metabolic Syndrome (MS), it is projected that the set of both conditions will be responsible for the main causes of liver transplantation [6][10].
Concerning etiology, the primary agents associated with NAFLD and NASH are the presence of insulin resistance (IR), type 2 diabetes (DM2), obesity, and MS. Other endocrine diseases, such as hypothyroidism and polycystic ovary syndrome, can be associated with this condition since they share the fundamental pathophysiological mechanism of NAFLD: IR, obstructive sleep apnea, hypertension, dyslipidemia, changes in the intestinal microbiota, genetic predisposition, menopause, sedentary lifestyle, and excessive consumption of fructose, saturated fatty acids and carbohydrates [2][11][12].
2. Insulin Resistance, the Role of Fructose, Gut Microbiota and Organokines
2.1. Insulin Resistance, Inflammation, and Mitochondrial Dysfunction
IR has a crucial role in the pathophysiology of NAFLD/NASH. Usually, insulin has a regulatory function of hepatic metabolic processes. It acts in other peripheral cells, favoring glucose uptake, storage pathways (glucogenesis), protein, and fatty acid synthesis. On the other hand, it blocks catabolic processes such as lipolysis, proteolysis, glycogenolysis, and gluconeogenesis. Therefore, resistance to its action generates several harms to the metabolism [13][14][15].
With overnutrition (marked by high consumption of carbohydrates, lipids, and fast foods) and a sedentary lifestyle, excessive energy intake is stored in the adipose tissue. By absorbing and storing excess glucose and free fatty acids (FFA), adipose tissue, as a compensatory mechanism, neutralizes the potentially toxic effects of these circulating nutrients, which include adipocyte hypertrophy and hyperplasia. In line with overnutrition, IR increases the flow of fatty acids to the liver by boosting hepatic lipogenesis and lipolysis, and inhibiting FFA esterification, generating an imbalance between synthesis/input versus oxidation/exportation of hepatocellular fat [6][16][17][18][19][20].
Thus, such imbalance is responsible for the excessive intrahepatic accumulation of fatty acids. This accumulation is lipotoxic, as the cells, already incapable of sequestering more reactive lipid molecules, have their mitochondrial beta-oxidation enzymatic system overloaded and suffer mitochondrial damage, endoplasmic reticulum (ER) stress, and autophagy. The results of obesity associated with NASH lead to hypertrophy of adipocytes, cell degeneration and death [2][6][21][22].
The death of hepatocytes is related to the release of damage-associated molecular patterns (DAMPs) to neighboring cells, which, through the inflammasome, are converted into pro-inflammatory cytokines (IL-1β and IL-18). Once secreted, IL-1β attracts neutrophils and activates both neutrophils and hepatic stellate cells (HSCs) and, in turn, IL-18 secretion attracts and activates macrophages. Such responses occur because DAMPs stimulate innate immunity through binding to pattern recognition receptors (PRRs) that activate NFκβ (nuclear factor κβ), initiating the pro-inflammatory action in NASH. Its release results in a cascade of pro-inflammatory chemokines and cytokines, such as TNF-α and IL-6, culminating in ROS formation. The combination of this scenario with liver sinusoidal endothelial cells (LSECs) have a direct action on fibrosis, in addition to providing an environment of chronic regeneration, favoring chromosomal aberrations and, therefore, the development of hepatocellular carcinoma [6][23][24][25].
The increased concentration of ROS can induce cell death in hepatocytes by activating specific pathways and inducing lipid peroxidation at the expense of β-oxidation, resulting in the synthesis of reactive lipids. These reactive molecules can further amplify liver damage, promote the release of more ROS outside the hepatocytes, and contribute to the activation of HSCs and extracellular matrix deposition [22][26].
The peroxidation process generates malondialdehyde, which is also responsible for perisinusoidal and periportal fibrosis, by activating NFκβ and regulating the expression of pro-inflammatory cytokines such as TNF-α and IL-8. These inflammatory biomarkers activate HSCs, contributing to fibrosis. The peroxidation of membrane phospholipids alters their permeability and promotes ballooning hepatocytes. This change may reflect cytoskeletal damage with the inability to complete programmed cell death. This change causes the accumulation of hydrogen peroxide in hepatic peroxisomes, which, together with ferrous iron, produce free hydroxyl (OH) radicals, which are very reactive with membrane phospholipids [22][27][28].
Lipotoxicity is associated with the transition of mitochondrial membrane pores that causes interruption of cellular respiration, generation of oxidative stress, and cytochrome C extravasation from the matrix to the cytosol, activating the apoptosome in apoptosis. Mitochondrial DNA is hit by the excess of free radicals, causing reduced oxidative phosphorylation and depleting ATP stores. The inhibition of electron flows in the phosphorylation chain generates more ROS, further damaging the mitDNA, creating a vicious circle. Therefore, apoptosis is aborted when ATP is depleted, and necrosis or necroptosis occurs—a mechanism involving mitochondrial damage, oxidative stress, and the activation of the c-Jun N-terminal kinase (JNK) [22][23][29][30].
Therefore, various pathological stimuli, including hepatocyte death, molecules secreted by adipose tissue, and intestinal pathogens, can promote inflammation and fibrogenesis by activating resident macrophages (Kupffer cells). Both Kupffer cells and obesity play a critical role in the pathophysiology of NASH. They promote the activation of the phenotype of pro-inflammatory M1 macrophages to the detriment of non-inflammatory M2 macrophages. Activated M1 macrophages produce a variety of cytokines that recruit pro-inflammatory cells, amplifying inflammation. This complex series of events ultimately culminates in the activation of HSCs, followed by excessive synthesis and extracellular matrix deposition. Thus, the loss of function of adipocytes added to adipose inflammation (presence of IL-1, IL-6, and TNF-α) contributes to the development of adipose resistance to insulin [10][21][26][27].
2.2. The Role of Fructose
The role of fructose in NASH is also noteworthy, as it stimulates the accumulation of hepatic fat and is related to the MS risk factors such as high blood pressure, elevated serum triglycerides, and IR. It is noteworthy that the origin of fructose influences this process. The consumption of sweetened fructose drinks and the consumption of fruits show different results, as the latter have a lower fructose content, in addition to containing antioxidants, which combat the effect of this monosaccharide [31][32][33].
Fructose enters the hepatocyte rapidly via glucose transporter 2 (GLUT2) and, at the cellular level, is preferentially converted to fructose-1-phosphate (F1P) by fructokinase. F1P then undergoes the action of aldolase B, producing phosphotriosis, which can be converted into glucose, lactate, and fatty acids. After an acute fructose load, the lipogenic pathway, often underactive, becomes very active as the flow of phosphotriosis increases [27][34]. Figure 2 shows the metabolism of fructose.
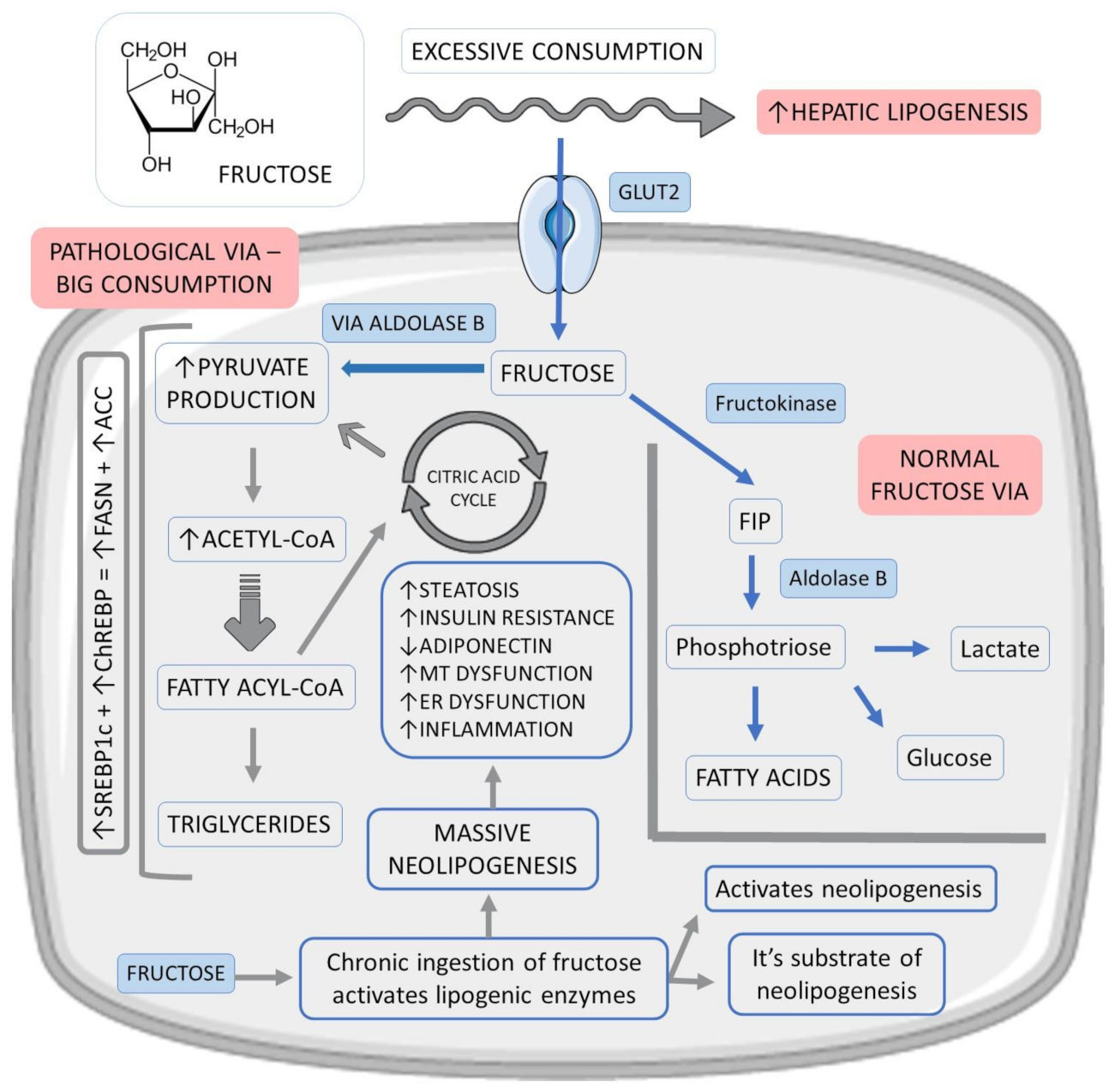
Figure 2. Fructose vias in hepatocytes and their relations with fat liver accumulation. ↑: increase, ↓:decrease, +: plus, ACC: acetyl-CoA carboxylase, ChREBP: carbohydrate-responsive element-binding protein, ER: endoplasmic reticulum, FASN: fatty acid synthase, FIP: fructose-1-phosphate, GLUT2: glucose transporter 2, MT: mitochondrial and SREBP1c: sterol response element-binding protein 1c.
In addition, dysregulation in the hepatic entry and metabolism of fructose culminates in excess production of acetyl-CoA (glycolytic pathway), above the liver’s oxidative capacity, promoting neolipogenesis through the activation of factors such as sterol response element-binding protein 1c (SREBP1c) and carbohydrate-responsive element-binding protein (ChREBP). As a product of the activation of these factors, there is an increase in the expression of fatty acid synthase (FASN) and acetyl-CoA carboxylase (ACC), which regulate lipid synthesis. Saturation of the glycolytic pathway also promotes an accumulation of glycolysis intermediates, which can be converted into glycerol-3-phosphate, used in triglyceride synthesis (TG) [34][35].
Therefore, fructose stands out as the most potent lipogenic carbohydrate in the development of hepatic steatosis since it is both a substrate and an activator of neolipogenesis. Chronic ingestion of this carbohydrate increases this process by stimulating the expression of lipogenic enzymes. The stimulation of neolipogenesis and the accumulation of lipids in the liver can increase the hepatic IR. Furthermore, fructose is also known to inhibit adiponectin production and release, contributing to increased IR and, therefore, potentiating hepatic steatosis. Such events can promote oxidative stress due to mitochondrial dysfunction and endoplasmic reticulum stress, which are factors that stimulate inflammation and progression from simple steatosis to NASH [34][36][37].
Upon entering the liver, fructose is rapidly phosphorylated by fructokinase C without negative feedback control. This process reduces the production of adenosine triphosphate (ATP) and leads to its consumption because while phosphorylation by fructokinase is fast, the cleavage reaction by aldolase B is relatively slow [34]. Thus, excess fructose, that is, consumption greater than that recommended by the World Health Organization (WHO) which is 5% in relation to the total recommended daily calories, can cause liver phosphate deficiency, with the accumulation of adenosine monophosphate (AMP) and consequent increase in uric acid synthesis, with fructose being the only common carbohydrate that produces uric acid in its metabolism [34][38]. The reduction of intracellular phosphate activates the enzyme AMP deaminase, which converts AMP into inosine monophosphate (IMP), resulting in a nucleotide renewal that stimulates the formation of uric acid, contributing to the formation of ROS [31][36][37].
In this sense, it is significant to observe that even though glucose and fructose are isomers and therefore have the same molecular formula (C6H12O6), the beginning of the metabolism of these molecules differs. The major difference is the involvement of phosphofructose kinase (a regulatory enzyme) in glucose metabolism but not in fructose metabolism. This initial difference leads to decreased intracellular phosphate and ATP levels, and uric acid generation is sufficient to transiently block protein synthesis, induce ROS, and mitochondrial dysfunction, resulting in an MS phenotype. From this stage onwards, the metabolism of these carbohydrates is similar [31][39][40].
The accumulation of uric acid can impair fatty acid oxidation, reduce hepatic ATP levels, and cause the temporary blockage of protein synthesis, inducing oxidative stress and mitochondrial dysfunction. In addition, uric acid also has direct pro-inflammatory effects by activating NFκβ and stimulating the release of inflammatory cytokines. Finally, fructose stimulates the rupture of tight intercellular junctions, contributing to greater intestinal permeability, which allows the entry of endotoxins through the portal vein, worsening the condition of NASH [31][36][41].
In addition, mitochondria contain enzymes known to be sensitive to oxidative stress (aconitase-2 and enoyl CoA hydratase). Together, fructose and uric acid reduce the action of aconitase-2, an enzyme associated with the Krebs cycle, which causes the accumulation of citrate. This excess of citrate is directed to the cytoplasm and activates lipogenesis by stimulating the enzyme ATP citrate lyase. It is also relevant to note that repeated exposure to sugar stimulates glucose transport via GLUT-5 and increases the levels of fructokinase in the liver, resulting in higher absorption of fructose [31][32][42][43].
Excessive and chronic consumption of fructose due to a high glycemic index diet, leads to increased demand for the endoplasmic reticulum (ER) due to the stimulation of lipid metabolism. Therefore, endoplasmic reticulum membrane proteins may be fructolyzed or accumulate lipids contributing to ER stress, generating inflammation, oxidative stress, and apoptosis. ER stress also indirectly stimulates TG accumulation in the liver, inducing hepatic and adipose IR and activating transcription factors related to inflammation and cell death. Fructose promotes the synthesis of saturated fatty acids, which can activate toll-like receptor 4 (TLR4) in the liver. This activation induces oxidative stress resulting from the production of inflammatory cytokines. Furthermore, ER stress and inflammation can lead to the production of DAMPS that signal and contribute to changes throughout metabolism. Excess fructose can change the profile of organokines, with increased production of fetuin A, fibroblast growth factor 21 (FGF-21), leucocyte cell-derived chemotaxin 2 (LECT2) and angiopoietin-like protein (ANGPTL), involved in changes in energy homeostasis, in IR and that can contribute to peripheral organ damage [6][34][44].
2.3. Intestinal Microbiota
The liver receives most of the intestinal blood flow through the portal system. Therefore, it is the first line of defense for intestinal-derived toxins and is exposed to many pathogen-associated molecular patterns (PAMPs); hence, the gut microbiota role in the pathophysiology of NASH is important. Excessive ingestion of foods high in fat or sugar is associated with a significant depletion of bacterial species diversity, with a reduction in the total density of the gut microbiota. In the NASH scenario, the major changes in some groups of bacteria are impressive. This pathology can produce severe depletion of bacterial species diversity, accompanied by a marked reduction in the total density of the microbiota. As a result, beneficial commensal bacteria may have been lost, and pathological microbes are overrepresented. Some examples of beneficial microorganisms are Bifidobacterium, Lactobacillus, Faecalibacterium, Roseburia, Ruminococcus, Bacteroides sp., which have anti-inflammatory effects and favorable action on metabolic patterns. Bacteria that can represent damage to the intestinal microbiota are Clostridium, Enterobacter, and Enterococcus sp. Patients with NASH reduce the amount of bacterioides compared to healthy individuals [45]. Dysbiosis favors the passage of bacteria and bacterial products (endotoxins), such as lipopolysaccharides (which may be related to the development of fibrosis by activation of hepatic stellate cells) to the portal circulation thus contributing to the activation of hepatic stellate cells to the progression of NAFLD to NASH. Diet-induced obesity alters the expression and distribution of tight junctions between intestinal cells, which is directly associated with increased intestinal permeability. Furthermore, patients with NASH compared to healthy or obese patients have an excess of ethanol-producing bacteria, specifically Escherichia, indicating that ethanol-producing microorganisms (Bacteroides, Bifidobacterium, and Clostridium sp.) can act as a risk factor for NASH progression. Therefore, it is notorious that there are pathologies that alter the intestinal microbiota, but its composition is also affected by the diet [22][26][36][45][46][47][48].
In this mechanism, a substance that deserves attention is fructose. It stimulates changes in the composition of the small intestine microbiota, reducing the expression of cell junction proteins and interfering with cell adhesion. Such processes allow the entry of endotoxins through the portal vein, triggering upregulation of lipogenic genes, as well as pro-inflammatory genes. This higher permeability allows bacterial translocation from the intestine to the bloodstream, providing pathogenic signals to various organs, including the liver, an event that can stimulate the innate immune response and further increase the pro-inflammatory response [22][36][49][50].
Scientific evidence indicates that changes in lifestyle—with adherence to diet and exercise—and reduction in body weight improve NAFLD. Increased dietary fiber intake, probiotics and prebiotics, and caloric restriction are examples of recommendations. Dietary fiber is the main non-digestible element in most diets, influencing the modulation of digestion as a substrate for microbial fermentation. Furthermore, the degradation of fibers that act as prebiotics provides short-chain fatty acids, which act as protectors of the structure and function of the intestinal barrier. A high-fiber diet increases the amount of Bifidobacterium and decreases Firmicutes/Bacteroidetes in humans, in addition to reducing the frequency of feeding by increasing satiety, which contributes to caloric restriction as a factor in improving NASH. Some studies show that a higher intake of insoluble fiber (≥7.5 g/day) can improve three different liver fibrosis scores (hepatic fibrosis index, fatty liver index, and liver fat index NAFLD) [22][45][51].
2.4. The Role of Diacylglycerols
Diacylglycerols (DAGs) also play an essential role in the pathophysiology of NASH, causing hepatic IR and the progression of liver steatosis. DAGs are the penultimate intermediates for triglyceride production and have been investigated to mediate IR among hepatocytes by activating protein kinase C epsilon type (PKCƐ). PKCƐ phosphorylates a threonine residue on insulin receptors of hepatocytes, impairing insulin signaling in these cells and leading to IR-derived lipid deposition stimuli. Moreover, in individuals with NAFLD and NASH, DAGs can be expressed in the liver under higher concentrations, augmenting triglycerides production extensively. Increased hepatic IR and liver lipid production are assessed as DAGs-mediated contributors of liver fatty accumulation and steatohepatitis [52][53]. In a trial, Loomba et al. [54] evaluated if the antisense inhibition of diacylglycerol O-acyltransferase 2 (DGAT2) could effectively reduce liver fatting in individuals with diabetes and NAFLD and showed that the inhibition reduced liver fat content without causing hyperlipidemia.
3. Organokines
The endocrine function of the liver, adipose, and hepatic tissues is of great value in NASH development. These tissues can produce biomarker peptides named organokines (hepatokines, adipokines, and myokines, respectively) that crosstalk through autocrine, endocrine, and paracrine pathways. The combined action of organokines is related to health or to the genesis of several diseases. With the increase in adipose tissue, there is an increase in the secretion of pro-inflammatory organokines, contributing to metabolic disorders such as IR, DM2, and MS [46][55].
The proximate relationship between skeletal muscle and bone tissue goes beyond anatomy: these tissues are also physiologically connected by the endocrine system through organokines, which completes this biochemical crosstalk. As discussed above, when there is a chronic excess of energy intake, there is an increase in adiposity and lipid deposition both in the bone marrow and in nearby myocytes, causing the release of FFA, which are lipotoxic to muscle and bone cells present there. This process triggers a vicious cycle, with low-grade systemic inflammation (LGI) and impairing metabolism. The load applied to skeletal muscle in resistance exercise is transferred to bone, which in addition to initiating muscle protein production, also signals a high energy requirement to facilitate bone formation. Some systemic mediators, including leptin, can initiate muscle hypertrophy and bone formation, proving that such tissues receive endocrine signals, and bone and muscle are now a bidirectional pathway for biochemical signals. Such signals are commanded by myokines, osteokines and adipokines, exerting autocrine, paracrine, and endocrine effects, thus regulating muscle and bone metabolism [56][57].
Since the liver is a producer of essential hormones or hormone precursors, such as insulin-like growth factor I (IGF-1), angiotensinogen, thrombopoietin, and hepcidin, the lipid imbalance resulting from the damage and death of hepatocytes is also responsible for the dysregulation of production of organokines. The expression of the insulin-sensitizing anti-inflammatory hormone adiponectin is decreased in NASH. Although leptin is essential to regulate appetite and increase energy expenditure, its levels are increased in obesity. Leptin promotes fibrogenesis in stellate cells, stimulating the production of fibrogenic genes and inflammation in T cells [14][58].
Several myokines, such as irisin, IL-6, IGF-1, brain-derived neurotrophic factor (BDNF), and myostatin, exert effects on bone metabolism, both anabolic and catabolic. In contrast, osteokines such as osteocalcin and sclerostin induce muscle anabolism and catabolism, respectively. Adipokines such as resistin, leptin, adiponectin, and TNF-α can also interfere with bone and muscle metabolism. Furthermore, lipolytic myokines such as irisin and IL-6 released after exercise stimulate thermogenesis and darkening adipocytes [57][59].
4. Paths to Unravel
Several exciting questions can arise since these organokines affect each other and communicate through various endocrine, paracrine and autocrine pathways. It is known that several environmental factors (such as diet, exercise, stress, sleep quality, and microbiota) and genetic factors are related to the expression of organokines, but how much and what lifestyle changes can modulate the expression of these mediators?
How do patients with NASH, with some degree of therapeutic intervention (diet, physical activity, pharmacological treatment) differ qualitatively and quantitatively in producing these organokines (hepatokines) compared to sedentary and untreated patients?
4.1. Genetics
How can individual genetic variations interfere in the production (expression, synthesis) of these organokines and, therefore, the susceptibility to developing metabolic diseases of the liver and other organs?
4.2. Diet and Microbiota
Some dietary compounds are metabolized by the gut microbiota and result in metabolites capable of modulating the host metabolism. On the other hand, diet can modify the microbiota composition and consequently exert beneficial or harmful effects on the host. These interferences can be localized in the gut, but others have systemic effects, leading, for example, to IR and can be related to the MetS risk factors [60]. Furthermore, diet regulates some organokines, such as FGF-21 [61].
Some authors have also shown the link between the microbiome, NAFLD and NASH pathogenesis and the interaction between the gut and liver, named the gut–liver axis, play a critical role in NASH development and evolution [62][63]. Microbiome-derived components can affect the liver via biomarkers that lead to interconnected effects such as gene expression, pro-inflammatory signaling, and modifications in metabolism and toxicity, resulting in liver inflammation and fibrosis [62].
The researchers can think of several associations: diet interferes with the microbiota, which interferes with the production of metabolites related to MetS. Diet high in sugars and fats alone is associated with MetS (a condition directly associated with NAFLD/NASH). In a liver inflammation scenario, the secretion of organokines (especially hepatokines) may be altered, thus altering the crosstalk between other organokines and possibly amplifying the process of a vicious cycle. Therefore, is there a possibility that the intestinal microbiota modulates the synthesis of organokines/hepatokines? How does the microbiota interfere with the expression of hepatokines in patients with liver disease? If the gut liver axis is involved with the pathogenesis and progression of NASH, most likely the composition or changes in the gut microbiota influence the establishment of NASH and indirectly the secretion of hepatokines. As hepatokines participate in crosstalk, then indirectly, the metabolism of other organs/tissues such as the pancreas, adipose tissue, and muscle may also be affected.
4.3. Exercise
Some authors have shown that exercise and maintained muscle mass can modify the circulating amounts of several organokines. Exercise can prevent liver steatosis and fibrosis in patients with NAFLD, independent of weight loss. The benefits of exercise seem to be associated with modifying inter-organs crosstalk, resulting in organokine balance and decreased inflammation and oxidative stress [64].
4.4. Sleep, Melatonin and Organokines
It is known that there is a relationship between physical activity and melatonin release, which in turn influences both the composition of skeletal muscle (type of fibers) and the metabolism of this tissue. Since skeletal muscle secretes myokines and crosstalks with other organs, it is believed that there is a possible causal relationship between melatonin production, organokine secretion and the degree of inflammation. This set would be related to the development of NAFLD/NASH.
Sleep restriction and poor sleep quality are profoundly implicated in the pathogenesis of metabolic dysfunctions, including NAFLD [65]. In the presence of NASH or NAFLD, it is plausible to admit that it will have an alteration in the release of hepatokines. Consequently, it may alter the release of other organokines and crosstalk.
Some authors have explored the relationship between melatonin and skeletal muscle metabolism [66]. As is known, muscle secretes myokines with different metabolic effects. The question is: is there a link between melatonin and myokine secretion? If it exists, can endogenous production or therapeutic use of melatonin interfere with organokine production? Could this have any therapeutic benefit? How can sleep quality interfere qualitatively and quantitatively on the output of organokines/hepatokines?
4.5. Other Metabolic Conditions
It is plausible that other conditions such as liver iron overload (hyperferritinemia/dysmetabolic iron overload syndrome) can alter the synthesis of these hepatokines and consequently crosstalk and thus play a role in the pathogenesis or progression of NASH (circuit and feedback)?
4.6. Intervention, Management, and Therapy
As discussed above, for the development of NASH, the two hits hypothesis is accepted. Most organokines mentioned in this entry participate in the first or second hit and therefore influence the onset of liver disease. Besides that, as organokines may be involved in the origin of NAFLD and its progression to NASH, it is possible to use the dosage of these biomarkers as a marker for liver disease and its progression.
References
- Ajmera, V.; Loomba, R. Imaging biomarkers of NAFLD, NASH, and fibrosis. Mol. Metab. 2021, 50, 101167.
- Gariani, K.; Jornayvaz, F.R. Pathophysiology of NASH in endocrine diseases. Endocr. Connect. 2021, 10, R52–R65.
- Rinaldi, L.; Pafundi, P.C.; Galiero, R.; Caturano, A.; Morone, M.V.; Silvestri, C.; Giordano, M.; Salvatore, T.; Sasso, F.C. Mechanisms of Non-Alcoholic Fatty Liver Disease in the Metabolic Syndrome. A Narrative Review. Antioxidants 2021, 10, 270.
- Ayada, I.; van Kleef, L.A.; Alferink, L.J.M.; Li, P.; de Knegt, R.J.; Pan, Q. Systematically comparing epidemiological and clinical features of MAFLD and NAFLD by meta-analysis: Focusing on the non-overlap groups. Liver. Int. 2021.
- Tilg, H.; Effenberger, M. From NAFLD to MAFLD: When pathophysiology succeeds. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 387–388.
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428.
- Jiang, X.; Zheng, J.; Zhang, S.; Wang, B.; Wu, C.; Guo, X. Advances in the Involvement of Gut Microbiota in Pathophysiology of NAFLD. Front. Med. 2020, 7, 361.
- Lonardo, A.; Mantovani, A.; Lugari, S.; Targher, G. Epidemiology and pathophysiology of the association between NAFLD and metabolically healthy or metabolically unhealthy obesity. Ann. Hepatol. 2020, 19, 359–366.
- Baltieri, L.; Chaim, E.; Chaim, F.; Utrini, M.; Gestic, M.; Cazzo, E. Correlation between nonalcoholic fatty liver disease features and levels of adipokines and inflammatory cytokines among morbidly obese individuals. Arq. Gastroenterol. 2018, 55, 247–251.
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183.
- DiStefano, J.K. NAFLD and NASH in Postmenopausal Women: Implications for Diagnosis and Treatment. Endocrinology 2020, 161, bqaa134.
- Zarghamravanbakhsh, P.; Frenkel, M.; Poretsky, L. Metabolic causes and consequences of nonalcoholic fatty liver disease (NAFLD). Metab. Open 2021, 12, 100149.
- Kolb, H.; Kempf, K.; Röhling, M.; Martin, S. Insulin: Too much of a good thing is bad. BMC Med. 2020, 18, 224.
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393.
- Armandi, A.; Rosso, C.; Caviglia, G.P.; Bugianesi, E. Insulin Resistance across the Spectrum of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 155.
- Attia, S.L.; Softic, S.; Mouzaki, M. Evolving Role for Pharmacotherapy in NAFLD/NASH. Clin. Transl. Sci. 2021, 14, 11–19.
- Rives, C.; Fougerat, A.; Ellero-Simatos, S.; Loiseau, N.; Guillou, H.; Gamet-Payrastre, L.; Wahli, W. Oxidative Stress in NAFLD: Role of Nutrients and Food Contaminants. Biomolecules 2020, 10, 1702.
- Kimura, T.; Singh, S.; Tanaka, N.; Umemura, T. Role of G Protein-Coupled Receptors in Hepatic Stellate Cells and Approaches to Anti-Fibrotic Treatment of Non-Alcoholic Fatty Liver Disease. Front. in Endocrinol. 2021, 12, 773432.
- Fujii, H.; Kawada, N.; Japan Study Group of NAFLD. The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863.
- Secor, J.D.; Fligor, S.C.; Tsikis, S.T.; Yu, L.J.; Puder, M. Free Fatty Acid Receptors as Mediators and Therapeutic Targets in Liver Disease. Front. Physiol. 2021, 12, 656441.
- Albhaisi, S.; Noureddin, M. Current and Potential Therapies Targeting Inflammation in NASH. Front. Endocrinol. 2021, 12, 767314.
- Xiao, X.; Hu, Q.; Deng, X.; Shi, K.; Zhang, W.; Jiang, Y.; Ma, X.; Zeng, J.; Wang, X. Old wine in new bottles: Kaempferol is a promising agent for treating the trilogy of liver diseases. Pharmacol. Res. 2021, 175, 106005.
- Farrell, G.C.; Haczeyni, F.; Chitturi, S. Pathogenesis of NASH: How Metabolic Complications of Overnutrition Favour Lipotoxicity and Pro-Inflammatory Fatty Liver Disease. Adv. Exp. Med. Biol. 2018, 1061, 19–44.
- Longo, M.; Paolini, E.; Meroni, M.; Dongiovanni, P. Remodeling of Mitochondrial Plasticity: The Key Switch from NAFLD/NASH to HCC. Int. J. Mol. Sci. 2021, 22, 4173.
- Bates, J.; Vijayakumar, A.; Ghoshal, S.; Marchand, B.; Yi, S.; Kornyeyev, D.; Zagorska, A.; Hollenback, D.; Walker, K.; Liu, K.; et al. Acetyl-CoA carboxylase inhibition disrupts metabolic reprogramming during hepatic stellate cell activation. J. Hepatol. 2020, 73, 896–905.
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression From NAFLD to NASH. Transplantation 2019, 103, e1–e13.
- Softic, S.; Meyer, J.G.; Wang, G.X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab. 2019, 30, 735–753.e734.
- Gonzalez, A.; Huerta-Salgado, C.; Orozco-Aguilar, J.; Aguirre, F.; Tacchi, F.; Simon, F.; Cabello-Verrugio, C. Role of Oxidative Stress in Hepatic and Extrahepatic Dysfunctions during Nonalcoholic Fatty Liver Disease (NAFLD). Oxid. Med. Cell. Longev. 2020, 2020, 1617805.
- Dong, J.; Viswanathan, S.; Adami, E.; Singh, B.K.; Chothani, S.P.; Ng, B.; Lim, W.W.; Zhou, J.; Tripathi, M.; Ko, N.S.J.; et al. Hepatocyte-specific IL11 cis-signaling drives lipotoxicity and underlies the transition from NAFLD to NASH. Nat. Commun. 2021, 12, 66.
- Rada, P.; González-Rodríguez, Á.; García-Monzón, C.; Valverde, Á.M. Understanding lipotoxicity in NAFLD pathogenesis: Is CD36 a key driver? Cell Death Dis. 2020, 11, 802.
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075.
- Roeb, E.; Weiskirchen, R. Fructose and Non-Alcoholic Steatohepatitis. Front. Pharmacol. 2021, 12.
- Schwingshackl, L.; Neuenschwander, M.; Hoffmann, G.; Buyken, A.E.; Schlesinger, S. Dietary sugars and cardiometabolic risk factors: A network meta-analysis on isocaloric substitution interventions. Am. J. Clin. Nutr. 2020, 111, 187–196.
- Jegatheesan, P.; De Bandt, J.-P. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients 2017, 9, 230.
- DiStefano, J.K. Fructose-mediated effects on gene expression and epigenetic mechanisms associated with NAFLD pathogenesis. Cell. Mol. Life Sci. 2020, 77, 2079–2090.
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2020, 2, 1034–1045.
- Muriel, P.; López-Sánchez, P.; Ramos-Tovar, E. Fructose and the Liver. Int. J. Mol. Sci. 2021, 22, 6969.
- Powell, E.S.; Smith-Taillie, L.P.; Popkin, B.M. Added Sugars Intake Across the Distribution of US Children and Adult Consumers: 1977–2012. J. Acad. Nutr. Diet. 2016, 116, 1543–1550.e1541.
- Sievenpiper, J.L.; de Souza, R.J.; Cozma, A.I.; Chiavaroli, L.; Ha, V.; Mirrahimi, A. Fructose vs. glucose and metabolism: Do the metabolic differences matter? Curr. Opin. Lipidol. 2014, 25, 8–19.
- Chan, A.M.L.; Ng, A.M.H.; Mohd Yunus, M.H.; Idrus, R.B.H.; Law, J.X.; Yazid, M.D.; Chin, K.-Y.; Shamsuddin, S.A.; Lokanathan, Y. Recent Developments in Rodent Models of High-Fructose Diet-Induced Metabolic Syndrome: A Systematic Review. Nutrients 2021, 13, 2497.
- Eshraghian, A.; Nikeghbalian, S.; Geramizadeh, B.; Kazemi, K.; Shamsaeefar, A.; Malek-Hosseini, S.A. Characterization of biopsy proven non-alcoholic fatty liver disease in healthy non-obese and lean population of living liver donors: The impact of uric acid. Clin. Res. Hepatol. Gastroenterol. 2020, 44, 572–578.
- Brennan, P.; Clare, K.; George, J.; Dillon, J.F. Determining the role for uric acid in non-alcoholic steatohepatitis development and the utility of urate metabolites in diagnosis: An opinion review. World J. Gastroenterol. 2020, 26, 1683–1690.
- Cui, Y.; Liu, J.; Shi, H.; Hu, W.; Song, L.; Zhao, Q. Serum uric acid is positively associated with the prevalence of nonalcoholic fatty liver in non-obese type 2 diabetes patients in a Chinese population. J. Diabetes Complicat. 2021, 35, 107874.
- Federico, A.; Rosato, V.; Masarone, M.; Torre, P.; Dallio, M.; Romeo, M.; Persico, M. The Role of Fructose in Non-Alcoholic Steatohepatitis: Old Relationship and New Insights. Nutrients 2021, 13, 1314.
- Pérez-Montes de Oca, A.; Julián, M.T.; Ramos, A.; Puig-Domingo, M.; Alonso, N. Microbiota, Fiber, and NAFLD: Is There Any Connection? Nutrients 2020, 12, 3100.
- Carter, J.K.; Bhattacharya, D.; Borgerding, J.N.; Fiel, M.I.; Faith, J.J.; Friedman, S.L. Modeling dysbiosis of human NASH in mice: Loss of gut microbiome diversity and overgrowth of Erysipelotrichales. PLoS ONE 2021, 16, e0244763.
- Martinez, K.B.; Leone, V.; Chang, E.B. Western diets, gut dysbiosis, and metabolic diseases: Are they linked? Gut Microb. 2017, 8, 130–142.
- Tsai, M.-C.; Liu, Y.-Y.; Lin, C.-C.; Wang, C.-C.; Wu, Y.-J.; Yong, C.-C.; Chen, K.-D.; Chuah, S.-K.; Yao, C.-C.; Huang, P.-Y.; et al. Gut Microbiota Dysbiosis in Patients with Biopsy-Proven Nonalcoholic Fatty Liver Disease: A Cross-Sectional Study in Taiwan. Nutrients 2020, 12, 820.
- Ferro, D.; Baratta, F.; Pastori, D.; Cocomello, N.; Colantoni, A.; Angelico, F.; Del Ben, M. New Insights into the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Gut-Derived Lipopolysaccharides and Oxidative Stress. Nutrients 2020, 12, 2762.
- Vasques-Monteiro, I.M.L.; Silva-Veiga, F.M.; Miranda, C.S.; de Andrade Gonçalves, É.C.B.; Daleprane, J.B.; Souza-Mello, V. A rise in Proteobacteria is an indicator of gut-liver axis-mediated nonalcoholic fatty liver disease in high-fructose-fed adult mice. Nutr. Res. 2021, 91, 26–35.
- Semmler, G.; Datz, C.; Reiberger, T.; Trauner, M. Diet and exercise in NAFLD/NASH: Beyond the obvious. Liver. Int. 2021, 41, 2249–2268.
- Lyu, K.; Zhang, Y.; Zhang, D.; Kahn, M.; Ter Horst, K.W.; Rodrigues, M.R.S.; Gaspar, R.C.; Hirabara, S.M.; Luukkonen, P.K.; Lee, S.; et al. A Membrane-Bound Diacylglycerol Species Induces PKCϵ-Mediated Hepatic Insulin Resistance. Cell Metab. 2020, 32, 654–664.e655.
- Marušić, M.; Paić, M.; Knobloch, M.; Liberati Pršo, A.-M. NAFLD, Insulin Resistance, and Diabetes Mellitus Type 2. Can. J. Gastroenterol. Hepatol. 2021, 2021, 6613827.
- Loomba, R.; Morgan, E.; Watts, L.; Xia, S.; Hannan, L.A.; Geary, R.S.; Baker, B.F.; Bhanot, S. Novel antisense inhibition of diacylglycerol O-acyltransferase 2 for treatment of non-alcoholic fatty liver disease: A multicentre, double-blind, randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol. Hepatol. 2020, 5, 829–838.
- Kim, H.; Lee, D.S.; An, T.H.; Park, H.J.; Kim, W.K.; Bae, K.H.; Oh, K.J. Metabolic Spectrum of Liver Failure in Type 2 Diabetes and Obesity: From NAFLD to NASH to HCC. Int. J. Mol. Sci. 2021, 22, 4495.
- Chung, H.S.; Choi, K.M. Chapter Six—Organokines in disease. In Advances in Clinical Chemistry; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 94, pp. 261–321.
- Kirk, B.; Feehan, J.; Lombardi, G.; Duque, G. Muscle, Bone, and Fat Crosstalk: The Biological Role of Myokines, Osteokines, and Adipokines. Curr. Osteoporos Rep. 2020, 18, 388–400.
- Kucukoglu, O.; Sowa, J.P.; Mazzolini, G.D.; Syn, W.K.; Canbay, A. Hepatokines and adipokines in NASH-related hepatocellular carcinoma. J. Hepatol. 2021, 74, 442–457.
- Choi, K.M. The Impact of Organokines on Insulin Resistance, Inflammation, and Atherosclerosis. Endocrinol. Metab. 2016, 31, 1–6.
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56.
- Janzi, S.; González-Padilla, E.; Najafi, K.; Ramne, S.; Ahlqvist, E.; Borné, Y.; Sonestedt, E. Single Nucleotide Polymorphisms in Close Proximity to the Fibroblast Growth Factor 21 (FGF21) Gene Found to be Associated with Sugar Intake in a Swedish Population. Nutrients 2021, 13, 3954.
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302.
- Safari, Z.; Gérard, P. The links between the gut microbiome and non-alcoholic fatty liver disease (NAFLD). Cell. Mol. Life Sci. CMLS 2019, 76, 1541–1558.
- Oh, S.; Tsujimoto, T.; Kim, B.; Uchida, F.; Suzuki, H.; Iizumi, S.; Isobe, T.; Sakae, T.; Tanaka, K.; Shoda, J. Weight-loss-independent benefits of exercise on liver steatosis and stiffness in Japanese men with NAFLD. JHEP Rep. Innov. Hepatol. 2021, 3, 100253.
- Marjot, T.; Ray, D.W.; Williams, F.R.; Tomlinson, J.W.; Armstrong, M.J. Sleep and liver disease: A bidirectional relationship. Lancet. Gastroenterol. Hepatol. 2021, 6, 850–863.
- Stacchiotti, A.; Favero, G.; Rodella, L.F. Impact of Melatonin on Skeletal Muscle and Exercise. Cells 2020, 9, 288.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
821
Revisions:
2 times
(View History)
Update Date:
23 Jul 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No