Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Clévio Nóbrega | -- | 3615 | 2023-07-21 10:40:09 | | | |
| 2 | Catherine Yang | Meta information modification | 3615 | 2023-07-21 10:50:25 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Paulino, R.; Nóbrega, C. Autophagy in Spinocerebellar Ataxia Type 3. Encyclopedia. Available online: https://encyclopedia.pub/entry/47107 (accessed on 30 June 2026).
Paulino R, Nóbrega C. Autophagy in Spinocerebellar Ataxia Type 3. Encyclopedia. Available at: https://encyclopedia.pub/entry/47107. Accessed June 30, 2026.
Paulino, Rodrigo, Clévio Nóbrega. "Autophagy in Spinocerebellar Ataxia Type 3" Encyclopedia, https://encyclopedia.pub/entry/47107 (accessed June 30, 2026).
Paulino, R., & Nóbrega, C. (2023, July 21). Autophagy in Spinocerebellar Ataxia Type 3. In Encyclopedia. https://encyclopedia.pub/entry/47107
Paulino, Rodrigo and Clévio Nóbrega. "Autophagy in Spinocerebellar Ataxia Type 3." Encyclopedia. Web. 21 July, 2023.
Copy Citation
Machado–Joseph disease (MJD) or spinocerebellar ataxia 3 (SCA3) is a rare, inherited, monogenic, neurodegenerative disease, and the most common SCA worldwide. MJD/SCA3 causative mutation is an abnormal expansion of the triplet CAG at exon 10 within the ATXN3 gene. The gene encodes for ataxin-3, which is a deubiquitinating protein that is also involved in transcriptional regulation. In normal conditions, the ataxin-3 protein polyglutamine stretch has between 13 and 49 glutamines. MJD/SCA3 patients display several signals and symptoms in which the most prominent is ataxia.
Machado–Joseph disease
spinocerebellar ataxia type 3
autophagy
ataxin-3
1. The ATXN3 Gene and Ataxin-3 Protein
The ATXN3 gene, located on chromosome 14q32.1 and under non-pathological circumstances, has a triplet expansion of CAGs on exon 10 [1]. The gene encodes for the ataxin-3 protein, which carries the glutamine expansion in its C-terminal region.
Ataxin-3 protein (Uniprot #P54252) has a molecular weight of approximately 42 kDa and four main domains, including the Josephin domain and three ubiquitin interacting motifs (Figure 1). The Josephin domain is located at the N-terminal of the protein between positions 1 and 180 and is believed to be the catalytic domain of the protein as it is widely conserved among species [2][3]. The three ubiquitin interacting motifs are located at the C-terminal region, are responsible for recognizing and interacting with ubiquitin, and have a higher affinity for ubiquitin chains composed of three or more residues [2]. The polyglutamine tract of ataxin-3 is located from positions 258 to 338 in its normal conformation. Ataxin-3 protein has at least two different splicing forms, both of which contain the polyglutamine domain and have been found in MJD/SCA3 patients [2].
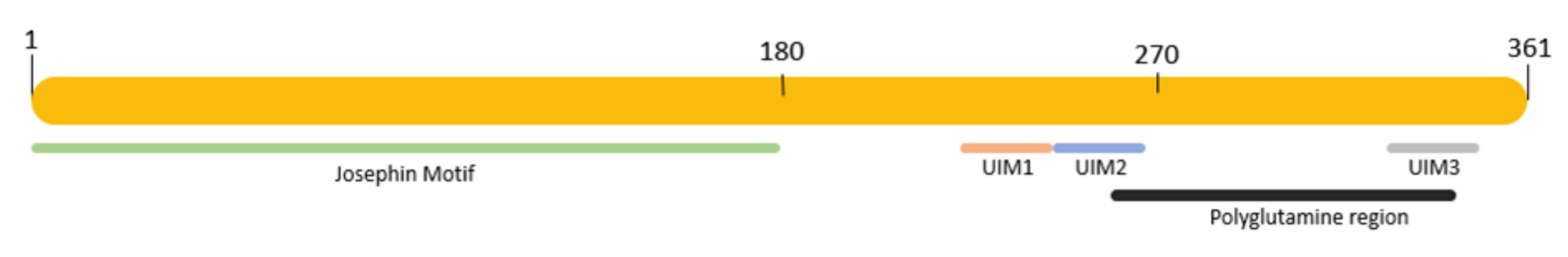
Figure 1. Representation of ataxin-3 protein with its main domains. Green, Josephin motif, positions 1–180; orange, UIM1 motif, positions 224–243; blue, UIM2 motif, positions 244–263; gray, UIM3 motif, positions 331–349; black, polyglutamine region, positions 258–338.
Ataxin-3, under normal conditions, is a cytoplasmatic protein [4]. However, in MJD/SCA3 patients, it is also found in the nucleus, forming aggregates which are a neuropathological hallmark of the disease [5]. This protein uses specific translocators to move in and out of the nucleus, which is a feature that is mediated by nuclear export signals [6]. The reason why the protein is found inside the nucleus in the disease context is still unknown; however, this feature is not exclusive to MJD/SCA3 patients as it is also reported in other PolyQ diseases, such as HD [7].
Ataxin-3 is a deubiquitinating enzyme that acts on proteins that have been targeted for degradation through the ubiquitin–proteasome system, which cleaves the ubiquitin present on those proteins moments before degradation and allows the recycling of ubiquitin [8]. Additionally, the role of ataxin-3 in cytoskeleton regulation due to its interaction with tubulin and the microtubule-associated protein 2 has been described. Along with this, it is also suggested that ataxin-3 plays a role in transcriptional regulation due to its interaction with proteins related to aggresomes, such as dynein and histone deacetylase 6 (HDAC6) [9]. Additionally, the interaction of ataxin-3 with transcription factors and co-regulators has been described. It was shown that it interacts with the forkhead box O (FOXO)-4 transcriptional factor (FOX04), which is a member of the forkhead family of transcriptional factors that are related to gene expression regulation [10]. It also interacts with CBP, which is a co-activator that originates a nuclear response by cascade signaling [11]. It interacts with P300 [12], which is a histone acetyltransferase recruited for the enhancement and regulation of gene expression [13]. It was also shown that it interacts with histone deacetylases (HDAC3), which is a class I HDAC that is recruited, and deacetylase in the gene promoter region, leading to chromatin compaction and the formation of a transcriptionally repressive state preventing the transcriptional machinery from accessing the promoter region [14]. It interacts with the nuclear co-repressor receptor NCor, which contains several nuclear receptor interacting domains that play a role in the recruitment of histone deacetylases to DNA promoter regions that assist with gene down regulation [15]. In addition, ataxin-3 interacts with RAD32, which is a nucleotide excision repair protein that inhibits the degradation of a proteolytic substrate and multi-ubiquitin chain formation [16]. These and other interactions suggest that ataxin-3 is involved in the regulation of a wide range of genes, including those involved in development, metabolism, and disease [5].
Ataxin-3 Pathological Aggregation
PolyQ diseases are characterized by the formation of protein aggregates in neurons, which is a hallmark of the disease [17]. These aggregates form due to the adoption of a non-native conformation that becomes toxic for the cells or induces toxicity by nonspecific interactions [18]. Pathological aggregates can include other proteins and cellular components, such as transcription factors, molecular chaperones, and components of cellular clearance mechanisms [17]. Progressively, these aggregates become insoluble, ultimately contributing to the dysregulation and impairment of several cellular mechanisms, including the dysregulation of calcium homeostasis, ubiquitin–proteasome dysfunction, mitochondrial dysfunction, axonal impairment, aberrant protein interaction, proteolytic cleavage, post-translational modification alterations, autophagy impairment, and others that contribute to neuronal degeneration and death [19].
2. Autophagy
Autophagy is a process by which cells maintain homeostasis by degrading and recycling cellular components in response to the deprivation of nutrients or oxygen or general damage [20]. Currently, three different types of autophagy are described that share a similar role but have differences in their pathways. Despite being different pathways, they all act to promote the proteolytic degradation of cytosolic components at the lysosome. In macro-autophagy, commonly named autophagy, the process of lysosome delivery is assisted by a double membrane-bound vesicle and the autophagosome that fuses with it to form the autolysosome (Figure 2). The autophagosome is made of a portion of the cellular membrane; thus, macro-autophagy is a non-selective degradation system, and this is a major difference when compared with other systems such as the ubiquitin–proteasome system (UPS) which act exclusively on proteins that have been ubiquitinated [21]. However, in micro-autophagy, cellular structures are directly taken up by the lysosome through the invagination of the lysosomal membrane. Finally, chaperone-mediated autophagy (CMA) uses chaperones to select proteins individually through a recognition motif in their amino acid sequences, translocating them to the lysosome for degradation [22].
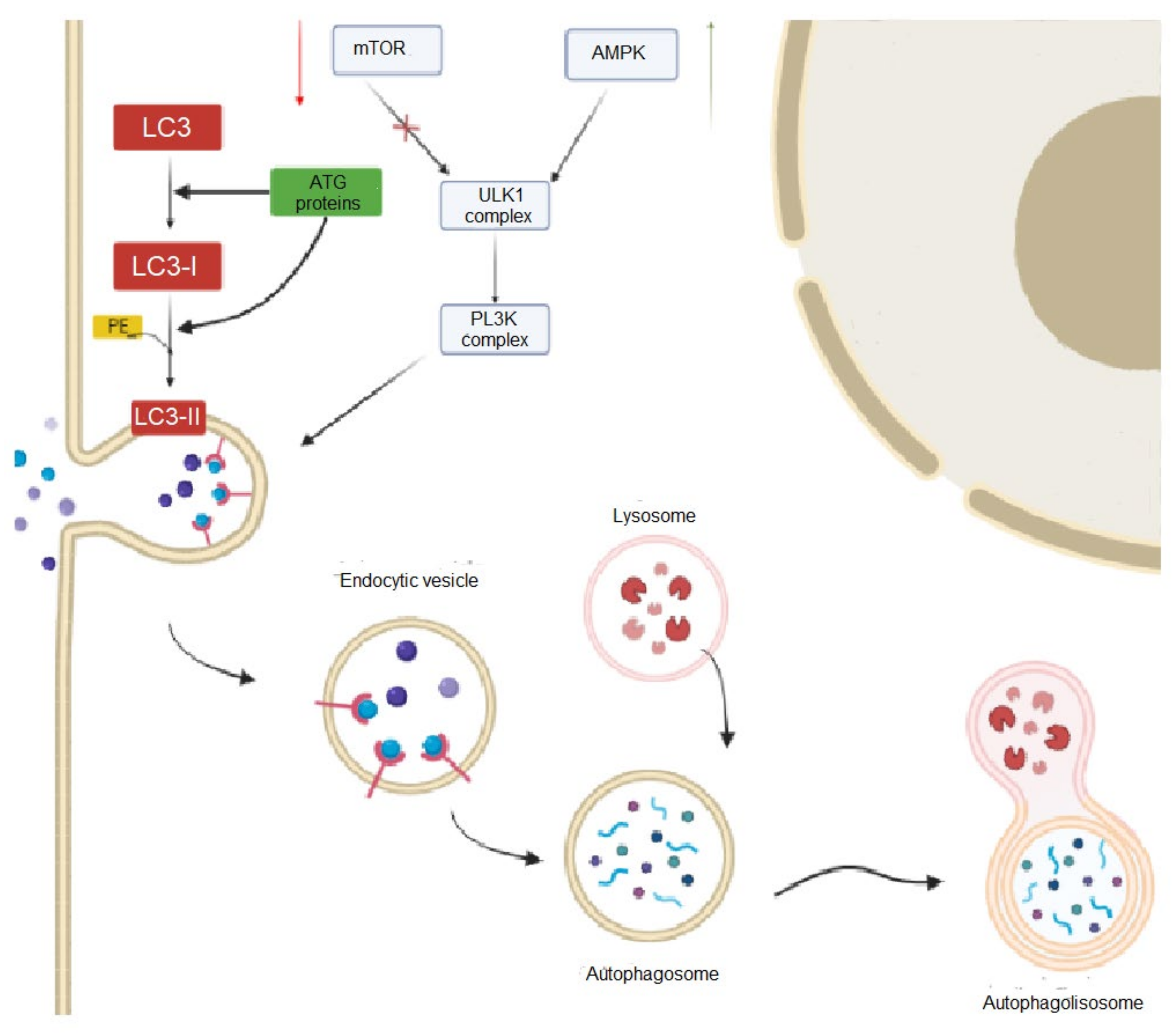
Figure 2. Overview of the main steps of macro-autophagy in mammal cells. In this pathway, the process of lysosome delivery is assisted by a double membrane-bound vesicle and the autophagosome that fuses with it to form the autolysosome.
Autophagy’s initial steps are crucial for the correct development of this degradation pathway. In these initial steps, the Unc-51-like kinase 1 (ULK1) complex plays an important role. The complex is formed by ULK1, ATG13, FIP200, and ATG101 proteins which are regulated by the mTOR complex 1 (mTORC1) signaling mechanism. mTORC1 inhibits autophagy according to the availability of nutrients in the cells, namely under nutrient-rich conditions. On the contrary, in depleted nutrient conditions, mTORC1 is inhibited and ULK1 is activated, initiating the autophagy process [20]. After, the autophagy elongation step involves the recruitment of ATG proteins to the phagophore. The formation of the phagophore comprises the formation of ubiquitin-like protein conjugated systems, including ATG12, ATG5, ATL161L1, the Ublike LC3 family, and the phosphatidylethanolamine (PE) which are present on the phagophore membrane. The elongation phase ends with the closure of the phagophore membrane, resulting in the formation of the autophagosome [20]. During the maturation phase, the autophagosome fuses with the lysosome, which is a process regulated by rab7 and LAMP-2, creating the autolysosome. Defects in the autophagy process have been linked to a range of diseases, including cancer, neurodegenerative diseases, and metabolic disorders.
2.1. Autophagy in MJD/SCA3
Autophagy is the main pathway responsible for removing oligomers or fibrils in the brain of neurodegenerative diseases patients as the UPS system is not able to degrade such large protein aggregates. However, the impairment of autophagy is a common feature in neurodegenerative diseases which compromises this degradation. Mutations in genes related to the autophagy pathway are linked to the development of familial neurodegenerative diseases. Moreover, in idiopathic neurodegenerative diseases, the presence of protein aggregates hinders autophagic degradation even without mutations [23]. In polyglutamine diseases, there is much evidence of an impairment of autophagy, which plays an important role in disease pathogenesis [24][25].
As described, autophagy is a highly regulated pathway in which several proteins are involved. For example, beclin-1 is important for the localization of autophagic proteins to a pre-autophagosomal structure [26]. Ataxin-3 protein was shown to protect beclin-1 from proteasomal degradation by interacting with it through the ataxin-3 polyQ stretch, thus enabling autophagy [27]. This study also showed that cells depleted of ataxin-3 showed partial inhibition of autophagy. Moreover, results suggest that a lack of or malfunction of ataxin-3 causes decreased autophagy as no interaction exists with beclin-1 [27].
Previously, it was shown that several autophagic proteins abnormally accumulate in the brain of MJD/SCA3 patients [28], including autophagic proteins LC3-II and p62/SQSTM1 [29]. Later, Onofre and colleagues used fibroblasts derived from MJD/SCA3 patients and reported that beclin-1 mRNA and protein levels were reduced in patients compared with healthy controls [30]. LC3 is a protein that is present on the lumen and cytosolic surface of mature autophagosomes. It normally exists as LC3-I form until it is conjugated with phosphatidylethanolamine (PE), forming LC3-II. This latter form is recruited to the membrane of the autophagosome to assist the process of fusion between the autophagosome and lysosome [31]. However, there is no consensus about the expression levels of LC3-II in MJD/SCA3 as some findings found increased levels, while other studies showed reduced LC3-II expression levels [30]. p62/SQSTM1 is involved in several signal transduction pathways shuttling cargo for autophagic degradation. It was shown to have an abnormal accumulation in MJD/SCA3 patients’ fibroblasts [30]. It is an autophagy substrate which acts as an adapter protein that binds to ubiquitinated protein aggregates and targets them for autophagic degradation. Impairments in autophagy could be related to increased levels of p62/SQSTM1, which resulted in an increase of polyubiquitinated aggregates in Drosophila and mammal models [32].
2.2. Targeting Autophagy through Pharmacological Approaches
Several pre-clinical studies and clinical trials were conducted in the last decades to try to find pharmacological therapies for MJD/SCA3 by targeting the activation of autophagy. However, generally, the results obtained show a mild improvement in motor deficits and neuropathological abnormalities. In 2002, the efficacy of rapamycin was investigated as an inducer of cell clearance mechanisms, especially autophagy, targeting the degradation of aggregate-prone polyglutamine proteins. Using a cellular model, the efficacy of rapamycin treatment was evaluated at 24 and 48 h after transfection with plasmids that were encoded for an expanded polyQ protein. The 24 h treatment with rapamycin led to a decrease in cell death and in the proportion of cells containing aggregates. However, in the 48 h treatment, no significant improvement was observed upon rapamycin treatment. These results suggest that in advanced stages, the consolidated structure of aggregates adopt a stable form that confers resistance to autophagy induced by rapamycin [33][34]. In the same study, researchers used an adrenal phaeochromocytoma cell line (PC12) expressing EGFP-Q74 to evaluate the efficacy of rapamycin in reducing protein levels and aggregates. The rate of protein clearance improved when it was treated with rapamycin and showed a reduction in GFP-positive fluorescence. Through western blot, the study showed a reduction both in the soluble and aggregated protein with rapamycin treatment [33]. It was hypothesized that rapamycin could be acting as a slow inhibitor of protein synthesis. However, when using cycloheximide, which is a protein synthesis inhibitor, it was possible to obtain an increase in protein levels and a decrease in autophagy activity [32].
Based on previous findings, a study used temsirolimus, which is a rapamycin ester that is soluble in water, inhibits mTORC, and increases and upregulates protein degradation by autophagy. The study used primary cortical cells and MJD/SCA3 transgenic mice at 5 weeks of age [35]. As temsirolimus acts as a kinase mTOR inhibitor, researchers investigated if the mTOR pathway was in fact inhibited in the brains of treated mice. Under normal conditions, LC3-II levels could be used to evaluate whether autophagy was being induced as LC3-II is a key component in the formation of autophagosomes. However, LC3-II levels in vivo were impossible to access, probably because neurons clear the autophagosomes. Therefore, researchers focused on the expression of two different mTOR pathway substrates, including p7056 kinase and eukaryotic initiation factor 4E-binding-protein-1, which were assessed by western blot. The results showed a decreased expression in the temsirolimus-treated group compared with the control animals. Additionally, an improvement in treated mice performance was observed with a higher latency to fall in the rotarod compared with the controls. The neuropathological analyses showed a reduced number of aggregates in the motor cortex and a reduction in the cytoplasmatic levels of expanded ataxin-3 in the treated animals. However, the levels of nuclear inclusions remained the same in the treated and control groups, probably because autophagy is a cytoplasmic process [36].
The activation of the monophosphate-activated protein kinase (AMPK) pathway is one of several pathways able to induce autophagy. A study used cordycepin, which is an AMPK inducer, to active autophagy in cellular and mouse models of MJD/SCA3. Researchers reported that N2a cells treated with cordycepin presented increased levels of phosphorylated-AMPK (P-AMPK) and a decrease in general protein synthesis. As the AMPK pathway can induce autophagy by itself, the authors evaluated whether cordycepin activated the AMPK pathway and autophagy. In neuroblastoma cells (N2a) expressing mutant ataxin-3, cordycepin treatment (20 µM) reduced the levels of the protein, while it did not interfere with the levels of non-expanded ataxin-3 (both wild-type ataxin-3 human and mouse endogenous ataxin-3). In addition, the levels of LC3B-II and p62/SQSTM1 reduced in the treated cells, indicating an increase in autophagy clearance. Treatment with cordycepin was also evaluated in induced pluripotent stem cells (iPSC) derived from MJD/SCA3 patients. The results were in line with those reported for the N2a cells, showing fewer aggregates and higher cell viability upon cordycepin treatment. Moreover, in an MJD/SCA3 lentiviral mouse model [37], cordycepin administration (20 mg/Kg) was able to reduce the levels of insoluble and soluble expanded ataxin-3. In addition to that, a decrease in the total levels of ubiquitinated aggregates and their size was observed due to an increase in the autophagy flux upon cordycepin treatment. These results were accompanied by preservation in the neuronal marker NeuN, showing signs of neuroprotection [38]. Finally, in a transgenic mouse model [39], cordycepin treatment for six weeks (starting from 15 mg/Kg to 25 mg/Kg) showed a higher number of Purkinje cells and a reduction in the number of aggregates compared with the control animals that were treated with saline solution. The results also showed an increased latency to fall on the rotarod and improvements in the footprint patterns of the cordycepin-treated animals [38].
In 2020, another study evaluated the trehalose efficacy as an autophagy inducer in the treatment of MJD/SCA3. In this study, 10 mM of trehalose was applied to N2a cells expressing mutant ataxin-3 for different periods of time. The levels of LC3B as an autophagy marker were measured, along with the levels of the human mutant ataxin-3. Western blot analyses showed an increase in the expression of LC3B-I and LC3B-II in the conditions treated with trehalose, indicating the activation of autophagy. The results also showed a decrease in the levels of mutant ataxin-3 at 24 h, 48 h, and 72 h post-treatment with trehalose. In vivo, the study reported the rescue of motor and coordination of an MJD/SCA3 transgenic mouse model [39] treated with a 2% trehalose solution for 28 weeks. The trehalose-treated animals showed an improvement in their rotarod performance and a decrease in their ataxia phenotype compared with the control animals. These results are in line with neuropathological findings as trehalose-treated animals showed the preservation of Purkinje cells and a decrease in the number and size of mutant ataxin-3 aggregates. However, is worth mentioning that the behavior tests and neuropathological analyses were performed just in female mice which are characterized by a weaker phenotype [40].
Carbamazepine (CBZ), which is a medicine used for the treatment of epilepsy, nerve pain, and bipolar disorder, was investigated as a therapeutic option for MJD/SCA3 through autophagy activation. Vasconcelos-Ferreira and colleagues showed that carbamazepine acts through an mTOR-independent pathway, leading to AMPK activation. In this study, three different strategies were tested considering the length of the treatment and the administration frequency. CBZ treatment was administered to MJD/SCA3 transgenic mice for a short period of time (1 week), a long period of time (4 weeks), and intermittently (three times per week) for 10 weeks [41]. The authors found that the animals that were treated with carbamazepine for a continuous long treatment (50 mg/Kg) did not present significant improvements in neuropathology or behavior deficits. Moreover, the results showed that both p62 and AMPK phosphorylation levels were not changed, suggesting that autophagy was not induced. However, the short and intermittent administrations were more efficient. In vitro, cells treated with CBZ (100 µM) showed a significant reduction in the levels of high molecular weight species of mutant ataxin-3. In vivo, the short treatment led to a reduction in the high molecular weight species of mutant ataxin-3 both in the striatum and in the cerebellum. In the intermittent treatment, animals that were treated with 50 mg/Kg of CBZ showed significant improvements in their rotarod performance from 4 weeks to 8 weeks of treatment. This was accompanied by a significant improvement in footprint overlapping at 4 weeks of treatment, suggesting a rescue of balance and coordination. Along with this, a decrease in the number of aggregates, a higher number of Purkinje cells, and the preservation of molecular layer thickness were observed in the CBZ-intermittently treated mice compared with the control animals [41].
The small molecule n-Butylidenephthalide (n-BP) isolated from the plant Angelica sinensis was investigated as a therapeutic approach for MJD/SCA3 using a transgenic mouse model [42]. In this study, the animals that started presenting motor deficiency at 13–14 weeks of age were treated with n-BP at 23 weeks of age until the animals reached 28 weeks of age. The treated and control animals were subjected to behavior tests to assess their balance and coordination through accelerated rotarod and footprint patterns tests. The animals treated with n-BP presented a significant improvement in their distance traveled, latency to fall, and speed when compared with the non-treated group, yet they never reached the performance of wild-type mice. The footprint patterns results reinforced these results as n-BP treated mice presented a closer relationship with a normal phenotype, showing a shorter footprint overlap and a longer stride length compared with the control animals [43]. The highest improvement observed in all the tests was in the fourth and fifth weeks of treatment, indicating that longer exposure to treatment is beneficial. The neuropathological analysis showed that the treated animals had a larger brain volume compared with the non-treated group and preserved the number of Purkinje cells. Moreover, the neurite network in the cerebellum was preserved, showing non-fragmented signs of neurite in the animals treated with n-BP, which is similar to what is observed in wild-type animals [43].
2.3. Targeting Autophagy through Gene-Based Approaches
As described above, several studies were performed using pharmacological strategies to activate activation autophagy. Similarly, recent gene-based approaches were also used in a preclinical setting to activate autophagy as a therapeutic option for MJD/SCA3. Overall, gene delivery strategies to activate autophagy improve neuropathological abnormalities and behavior deficits in MJD/SCA3 mouse models. In 2011, a study analyzed levels of the autophagy protein beclin-1 in MJD/SCA3 patients’ fibroblasts. The results showed a decrease in beclin-1 levels compared with fibroblasts from healthy controls. Based on that, the authors prompted to investigate whether the up-regulation of beclin-1 could lead to an improvement in the MJD/SCA3 disease phenotype [28]. In primary striatal cells, beclin-1 expression led to a drastic reduction of mutant ataxin-3 aggregates, oligomers, and soluble protein. Additionally, in an MJD/SCA3 lentiviral mouse model [37], beclin-1 expression mediated by lentiviral vectors showed a reduction in the number of aggregates at 4 weeks and 8 weeks post-injection. Similar findings were observed in the neuronal dysfunction upon beclin-1 up-regulation. Altogether, these results suggest that the overexpression of beclin-1 mitigates MJD/SCA3 neuropathology [28].
In MJD/SCA3 patients, several molecular mechanisms are affected in neurons and underlie disease pathogenesis. In 2019, a study was published focusing on cholesterol 24-hydroxylase (CYP46A1). This is an enzyme that is responsible for converting cholesterol into 24S-hydroxycholesterol, allowing the efflux of brain cholesterol, and activating brain cholesterol turnover. When CYP46A1 levels are reduced, it can lead to a malfunction causing the accumulation of cholesterol in the brain. The reduction of this enzyme seems to occur in several neurodegenerative diseases, including MJD/SCA3 [44]. In this study, Nóbrega and colleagues evaluated whether restoring CYP46A1 expression could lead to improvement in the MJD/SCA3 disease phenotype. In an MJD/SCA3 lentiviral mouse model [37], the overexpression of CYP46A1 promoted the clearance of mutant ataxin-3 aggregates and neuroprotection [44]. Similarly, in a transgenic MJD/SCA3 mouse model [39], the upregulation of CYP46A1 in the cerebellum led to an alleviation of the motor phenotype and neuropathological abnormalities. CYP46A1-injected mice had a better performance in the rotarod test and in the footprint patterns measures compared with the GFP-injected control mice. Neuropathologically, there was a reduction in the number of mutant ataxin-3 aggregates and an increase in the number of Purkinje cells upon CYP46A1 expression mediated by lentiviral vectors [44].
Recently, targeted ULK 1/2 expression as an autophagy inducer was investigated as a possible therapeutic strategy for MJD/SCA3. Using an MJD/SCA3 transgenic mouse model [39], ULK 1/2 was injected using lentiviral vectors in the cerebellum. The results showed a rescue of the motor performance of treated mice at 9 weeks post-injection for the swimming and beamwalk tests. Along with this, a major rescue of motor coordination in the rotarod test and an improvement in the ataxic phenotype were observed at 6 weeks post-injection compared with the control group that was injected with GFP. Neuropathological findings showed an increase in the cerebellar volume and in the number of Purkinje cells. Moreover, aggregates were also reduced in the ULK 1/2 injected animals [45].
References
- Matos, C.A.; de Almeida, L.P.; Nóbrega, C. Machado-Joseph disease/spinocerebellar ataxia type 3: Lessons from disease pathogenesis and clues into therapy. J. Neurochem. 2019, 148, 8–28.
- Paulson, H. Machado-Joseph disease/spinocerebellar ataxia type 3. Handb. Clin. Neurol. 2012, 103, 437–449.
- Chow, M.K.; Mackay, J.P.; Whisstock, J.C.; Scanlon, M.J.; Bottomley, S.P. Structural and functional analysis of the Josephin domain of the polyglutamine protein ataxin-3. Biochem. Biophys. Res. Commun. 2004, 322, 387–394.
- Evers, M.M.; Toonen, L.J.; van Roon-Mom, W.M. Ataxin-3 protein and RNA toxicity in spinocerebellar ataxia type 3: Current insights and emerging therapeutic strategies. Mol. Neurobiol. 2014, 49, 1513–1531.
- Evert, B.O.; Araujo, J.; Vieira-Saecker, A.M.; de Vos, R.A.; Harendza, S.; Klockgether, T.; Wüllner, U. Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J. Neurosci. 2006, 26, 11474–11486.
- Macedo-Ribeiro, S.; Cortes, L.; Maciel, P.; Carvalho, A.L. Nucleocytoplasmic shuttling activity of ataxin-3. PLoS ONE 2009, 4, e5834.
- Lieberman, A.P.; Shakkottai, V.G.; Albin, R.L. Polyglutamine Repeats in Neurodegenerative Diseases. Annu. Rev. Pathol. 2019, 14, 1–27.
- Mao, Y.; Senic-Matuglia, F.; Di Fiore, P.P.; Polo, S.; Hodsdon, M.E.; De Camilli, P. Deubiquitinating function of ataxin-3: Insights from the solution structure of the Josephin domain. Proc. Natl. Acad. Sci. USA 2005, 102, 12700–12705.
- Matos, C.A.; de Macedo-Ribeiro, S.; Carvalho, A.L. Polyglutamine diseases: The special case of ataxin-3 and Machado-Joseph disease. Prog. Neurobiol. 2011, 95, 26–48.
- Mohan, R.D.; Abmayr, S.M.; Workman, J.L. The expanding role for chromatin and transcription in polyglutamine disease. Curr. Opin. Genet. Dev. 2014, 26, 96–104.
- McCampbell, A.; Taylor, J.P.; Taye, A.A.; Robitschek, J.; Li, M.; Walcott, J.; Merry, D.; Chai, Y.; Paulson, H.; Sobue, G.; et al. CREB-binding protein sequestration by expanded polyglutamine. Hum. Mol. Genet. 2000, 9, 2197–2202.
- Li, F.; Macfarlan, T.; Pittman, R.N.; Chakravarti, D. Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities. J. Biol. Chem. 2002, 277, 45004–45012.
- Schulze, S.R.; Wallrath, L.L. Gene regulation by chromatin structure: Paradigms established in Drosophila melanogaster. Annu. Rev. Entomol. 2007, 52, 171–192.
- Watson, P.J.; Fairall, L.; Schwabe, J.W. Nuclear hormone receptor co-repressors: Structure and function. Mol. Cell Endocrinol. 2012, 348, 440–449.
- Ortolan, T.G.; Tongaonkar, P.; Lambertson, D.; Chen, L.; Schauber, C.; Madura, K. The DNA repair protein rad23 is a negative regulator of multi-ubiquitin chain assembly. Nat. Cell Biol. 2000, 2, 601–608.
- Breuer, P.; Haacke, A.; Evert, B.O.; Wüllner, U. Nuclear aggregation of polyglutamine-expanded ataxin-3: Fragments escape the cytoplasmic quality control. J. Biol. Chem. 2010, 285, 6532–6537.
- Nóbrega, C.; Simões, A.T.; Duarte-Neves, J.; Duarte, S.; Vasconcelos-Ferreira, A.; Cunha-Santos, J.; Pereira, D.; Santana, M.; Cavadas, C.; de Almeida, L.P. Molecular Mechanisms and Cellular Pathways Implicated in Machado-Joseph Disease Pathogenesis. Adv. Exp. Med. Biol. 2018, 1049, 349–367.
- Nobrega, C.; Carmo-Silva, S.; Albuquerque, D.; Vasconcelos-Ferreira, A.; Vijayakumar, U.G.; Mendonça, L.; Hirai, H.; Pereira de Almeida, L. Re-establishing ataxin-2 downregulates translation of mutant ataxin-3 and alleviates Machado-Joseph disease. Brain 2015, 138 Pt 12, 3537–3554.
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42.
- Mizushima, N. Autophagy: Process and function. Genes. Dev. 2007, 21, 2861–2873.
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12.
- Chandran, A.; Rochet, J.C. Shining a light on autophagy in neurodegenerative diseases. J. Biol. Chem. 2022, 298, 101437.
- Cortes, C.J.; La Spada, A.R. Autophagy in polyglutamine disease: Imposing order on disorder or contributing to the chaos? Mol. Cell Neurosci. 2015, 66 Pt A, 53–61.
- Marcelo, A.; Afonso, I.T.; Afonso-Reis, R.; Brito, D.V.; Costa, R.G.; Rosa, A.; Alves-Cruzeiro, J.; Ferreira, B.; Henriques, C.; Nobre, R.J.; et al. Autophagy in Spinocerebellar ataxia type 2, a dysregulated pathway, and a target for therapy. Cell. Death Dis. 2021, 12, 1117.
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell. Death Differ. 2011, 18, 571–580.
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111.
- Nascimento-Ferreira, I.; Santos-Ferreira, T.; Sousa-Ferreira, L.; Auregan, G.; Onofre, I.; Alves, S.; Dufour, N.; Colomer Gould, V.F.; Koeppen, A.; Déglon, N.; et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado-Joseph disease. Brain 2011, 134 Pt 5, 1400–1415.
- Onofre, I.; Mendonça, N.; Lopes, S.; Nobre, R.; de Melo, J.B.; Carreira, I.M.; Januário, C.; Gonçalves, A.F.; de Almeida, L.P. Fibroblasts of Machado Joseph Disease patients reveal autophagy impairment. Sci. Rep. 2016, 6, 28220.
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.I.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163.
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88.
- Aparicio, R.; Rana, A.; Walker, D.W. Upregulation of the Autophagy Adaptor p62/SQSTM1 Prolongs Health and Lifespan in Middle-Aged Drosophila. Cell. Rep. 2019, 28, 1029–1040.e5.
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222.
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117.
- Bichelmeier, U.; Schmidt, T.; Hübener, J.; Boy, J.; Rüttiger, L.; Häbig, K.; Poths, S.; Bonin, M.; Knipper, M.; Schmidt, W.J.; et al. Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: In vivo evidence. J. Neurosci. 2007, 27, 7418–7428.
- Menzies, F.M.; Huebener, J.; Renna, M.; Bonin, M.; Riess, O.; Rubinsztein, D.C. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 2010, 133 Pt 1, 93–104.
- Alves, S.; Régulier, E.; Nascimento-Ferreira, I.; Hassig, R.; Dufour, N.; Koeppen, A.; Carvalho, A.L.; Simoes, S.; de Lima, M.C.P.; Brouillet, E.; et al. Striatal and nigral pathology in a lentiviral rat model of Machado-Joseph disease. Hum. Mol. Genet. 2008, 17, 2071–2083.
- Marcelo, A.; Brito, F.; Carmo-Silva, S.; Matos, C.A.; Alves-Cruzeiro, J.; Vasconcelos-Ferreira, A.; Koppenol, R.; Mendonça, L.; de Almeida, L.P.; Nóbrega, C. Cordycepin activates autophagy through AMPK phosphorylation to reduce abnormalities in Machado-Joseph disease models. Hum. Mol. Genet. 2019, 28, 51–63.
- Torashima, T.; Koyama, C.; Iizuka, A.; Mitsumura, K.; Takayama, K.; Yanagi, S.; Oue, M.; Yamaguchi, H.; Hirai, H. Lentivector-mediated rescue from cerebellar ataxia in a mouse model of spinocerebellar ataxia. EMBO Rep. 2008, 9, 393–399.
- Santana, M.M.; Paixão, S.; Cunha-Santos, J.; Silva, T.P.; Trevino-Garcia, A.; Gaspar, L.S.; Nóbrega, C.; Nobre, R.J.; Cavadas, C.; Greif, H.; et al. Trehalose alleviates the phenotype of Machado-Joseph disease mouse models. J. Transl. Med. 2020, 18, 161.
- Vasconcelos-Ferreira, A.; Carmo-Silva, S.; Codêsso, J.M.; Silva, P.; Martinez, A.R.M.; França, M.C., Jr.; Nóbrega, C.; Pereira de Almeida, L. The autophagy-enhancing drug carbamazepine improves neuropathology and motor impairment in mouse models of Machado-Joseph disease. Neuropathol. Appl. Neurobiol. 2022, 48, e12763.
- Cemal, C.K.; Carroll, C.J.; Lawrence, L.; Lowrie, M.B.; Ruddle, P.; Al-Mahdawi, S.; King, R.H.; Pook, M.A.; Huxley, C.; Chamberlain, S. YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum. Mol. Genet. 2002, 11, 1075–1094.
- Lee, J.H.; Lin, S.Y.; Liu, J.W.; Lin, S.Z.; Harn, H.J.; Chiou, T.W. n-Butylidenephthalide Modulates Autophagy to Ameliorate Neuropathological Progress of Spinocerebellar Ataxia Type 3 through mTOR Pathway. Int. J. Mol. Sci. 2021, 22, 6339.
- Nascimento-Ferreira, I.; Nobrega, C.; Vasconcelos-Ferreira, A.; Onofre, I.; Albuquerque, D.; Aveleira, C.; Hirai, H.; Deglon, N.; Pereira de Almeida, L. Beclin 1 mitigates motor and neuropathological deficits in genetic mouse models of Machado-Joseph disease. Brain 2013, 136 Pt 7, 2173–2188.
- Vasconcelos-Ferreira, A.; Martins, I.M.; Lobo, D.; Pereira, D.; Lopes, M.M.; Faro, R.; Lopes, S.M.; Verbeek, D.; Schmidt, T.; Nóbrega, C.; et al. ULK overexpression mitigates motor deficits and neuropathology in mouse models of Machado-Joseph disease. Mol. Ther. 2022, 30, 370–387.
- Zhang, L.; Chen, D.; Song, D.; Liu, X.; Zhang, Y.; Xu, X.; Wang, X. Clinical and translational values of spatial transcriptomics. Signal. Transduct. Target. Ther. 2022, 7, 111.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
646
Revisions:
2 times
(View History)
Update Date:
21 Jul 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No