 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Grazia Galleri | -- | 1840 | 2023-07-07 20:29:38 | | | |
| 2 | Jessie Wu | + 28 word(s) | 1868 | 2023-07-10 04:42:48 | | | | |
| 3 | Jessie Wu | Meta information modification | 1868 | 2023-07-10 04:48:26 | | | | |
| 4 | Jessie Wu | -1 word(s) | 1867 | 2023-07-10 04:50:02 | | |
Video Upload Options
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that affects both lower motor neurons in brainstem and spinal cord and upper motor neurons in motor cortex.
1. Background
Amyotrophic lateral sclerosis (ALS) pathogenesis is poorly understood, and currently, there is neither a cure nor a specific diagnostic test for this disease. Approximately 90% of ALS cases are sporadic (sALS), while the remaining 10% of cases are familial (fALS) [1][2]. Several genes involved in ALS pathomechanisms have been identified [3][4][5], and all of these can cause motor neurons death through different mechanisms such as oxidative stress, mitochondrial dysfunctions, protein aggregates formation, glutamate toxicity, and neuroinflammation [5][6][7][8]. Among ALS-causative genes, a point mutation, proline to serine substitution at position 56 (P56S), in a vesicle-associated membrane-protein-associated protein B (VAPB)-encoding gene, responsible for a rare fALS case and classified as ALS-8 [9], has been identified, which is associated with a highly variable clinical course [10]. VAPB belongs to the family of vesicle-associated membrane-protein-associated proteins, endoplasmic reticulum (ER) resident proteins [11][12], which play different roles in cells for regulation of calcium homeostasis, vesicle trafficking, bouton formation at a neuromuscular junction, microtubules organization, lipid transport, and unfolded protein response (UPR) [12][13][14][15]. The most frequent mutation in VAPB-encoding genes is P56S [9]. However, other more rare mutations have been identified [10]. The presence of VAPB mutant genes causes an impairment of the protein, resulting in intracellular protein inclusions, already characterized in transgenic mice [9], which entails an abnormal reorganization of an ER structure and alters cellular homeostasis [9][11][13][16][17][18][19][20]. Several studies in human and transgenic mice have documented that VAPB inclusions also occur when other ALS-causative genes are present, such as superoxide dismutase 1 (SOD1) [21], TAR DNA-binding protein (TDP43) [22], and chromosome 9 open reading frame 72 (C9orf72) repeat expansion [23]. Moreover, there are evidences that suggest VAPB involvement in sALS [9][13][20]. In the present paper, researchers studied VAPB as a possible ALS pathologic marker by analyzing peripheral blood mononuclear cells (PBMCs) isolated from sALS-affected patients. In fact, PBMCs share more than 80% of their transcriptomes with other tissues, including the central nervous system, and can be isolated easily from patients by venipuncture [24]. Previous studies demonstrated that PBMCs are a valid cell model to study ALS [24][25][26][27]. Based on this and on the new vision of ALS as a pathology affecting not only motor neurons exclusively but also other cell systems [28][29][30], researchers investigated the possible role of VAPB as a pathologic marker in PBMCs from patients with sALS.
2. Cellular Model Analysis
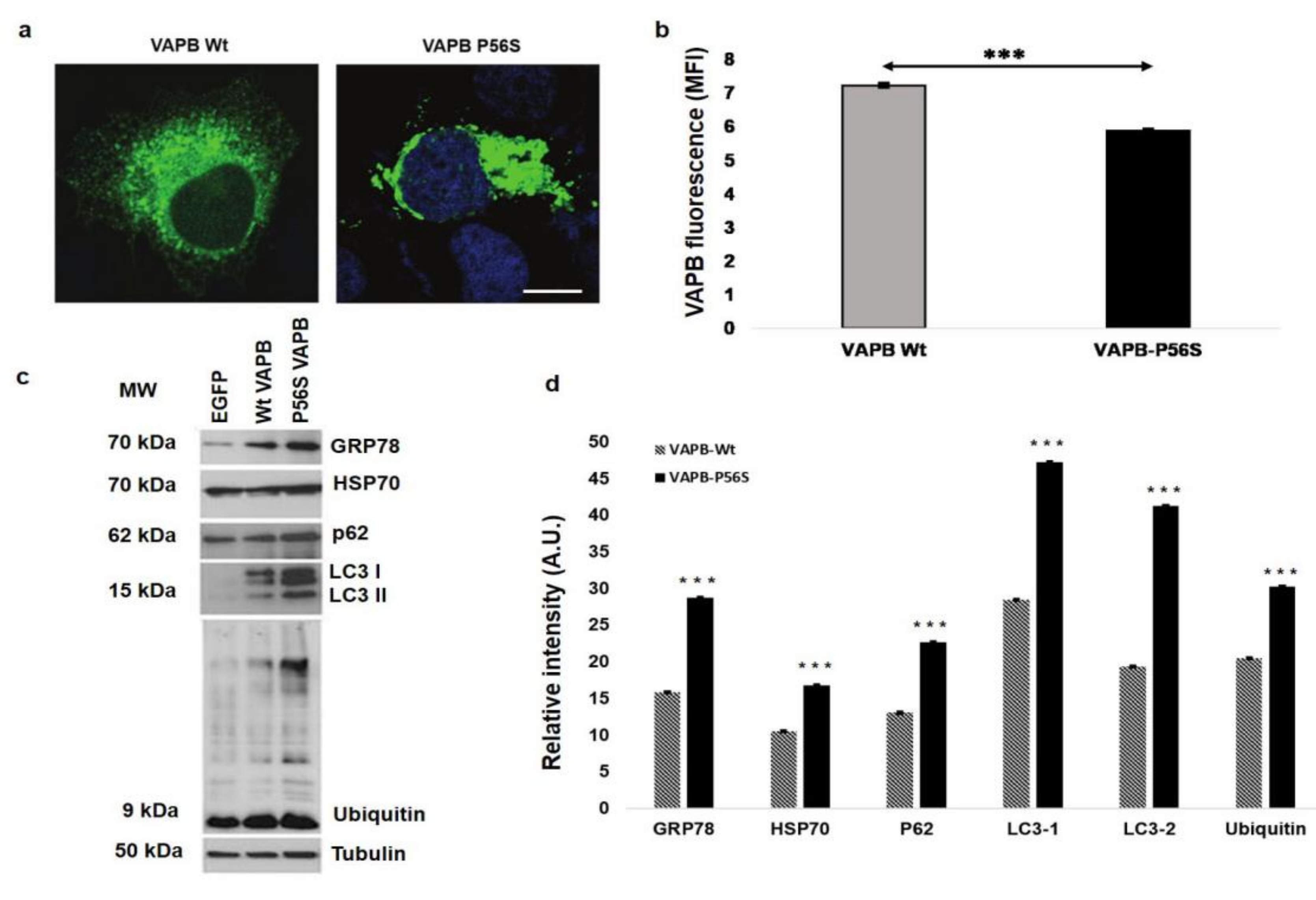
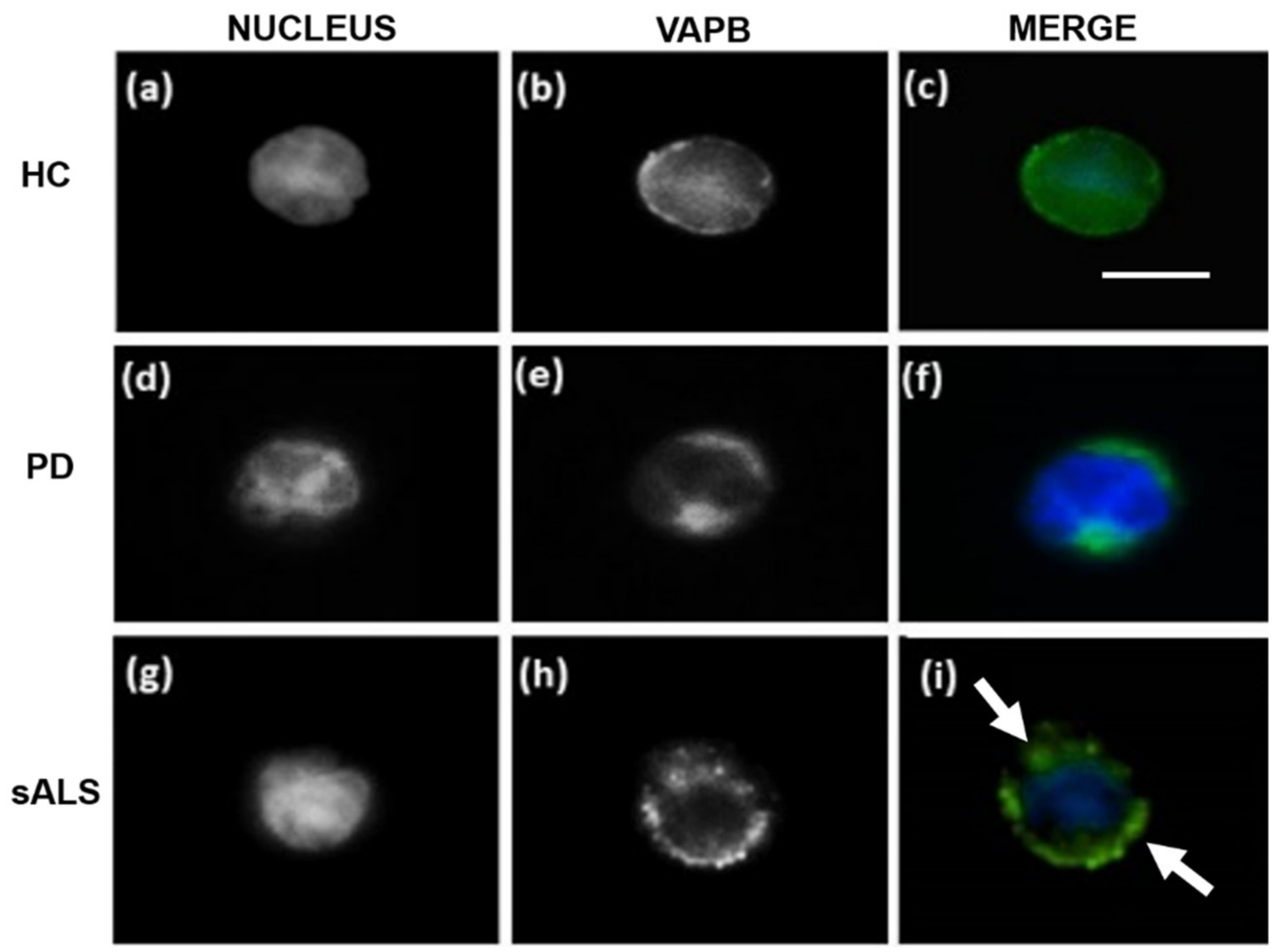
3. Vesicle-Associated Membrane-Protein-Associated Protein B ER-Aggregates in Sporadic Amyotrophic Lateral Sclerosis Patients Peripheral Blood Mononuclear Cells
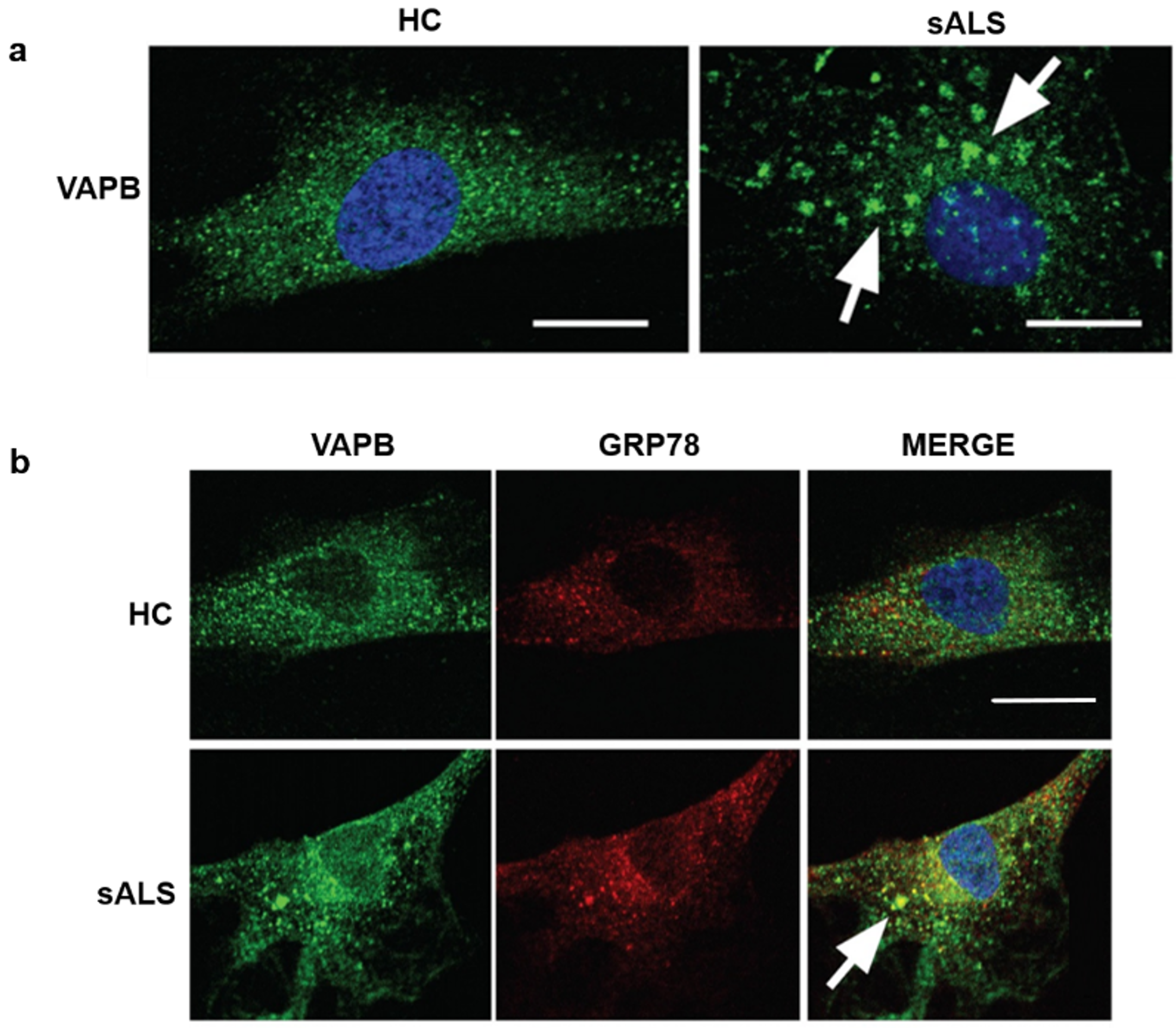
4. Vesicle-Associated Membrane-Protein-Associated Protein B Accumulations in Sporadic Amyotrophic Lateral Sclerosis Patients Fibroblasts
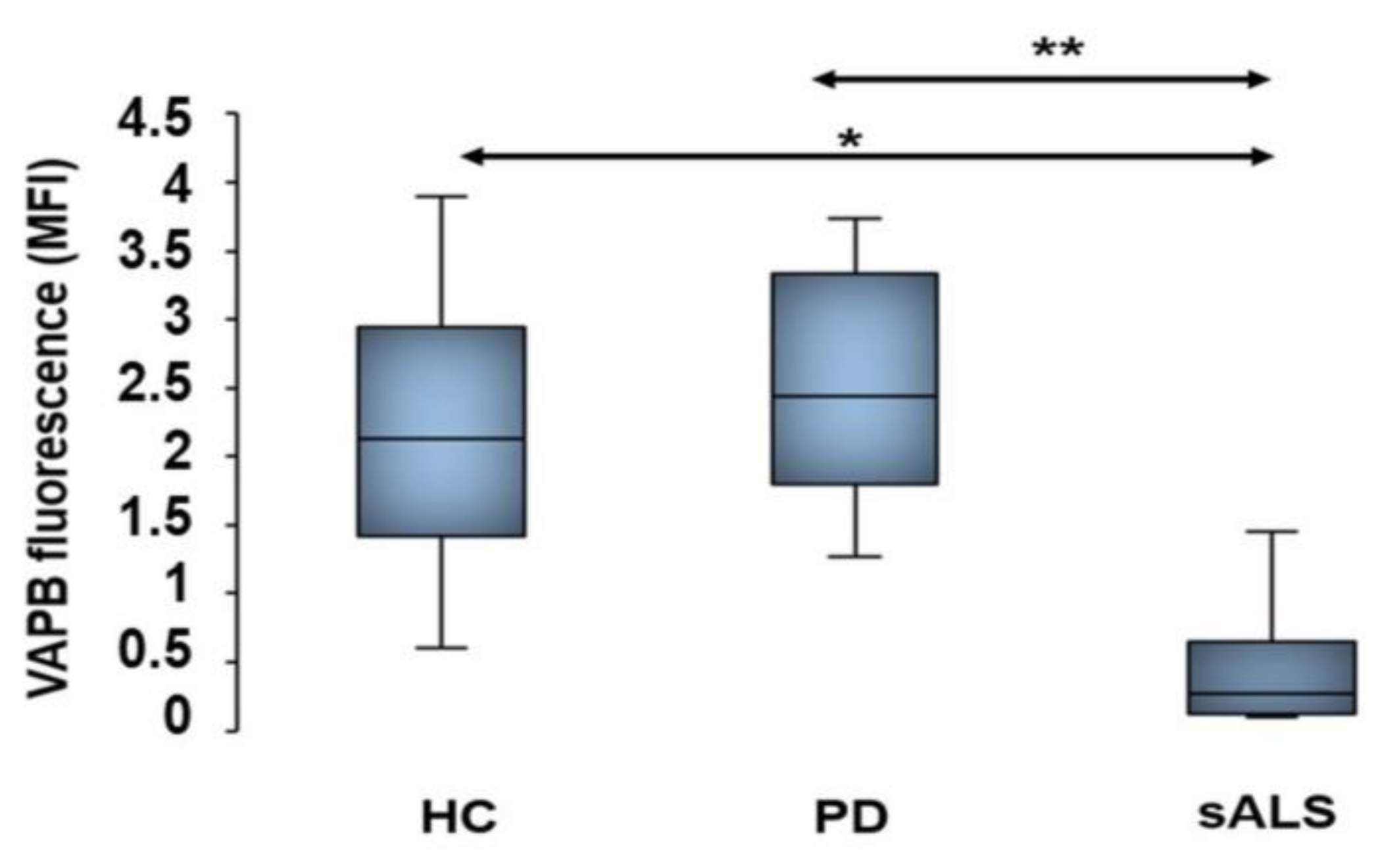
5. Decrease of Vesicle-Associated Membrane-Protein-Associated Protein B Fluorescent Signals in Sporadic Amyotrophic Lateral Sclerosis Patients
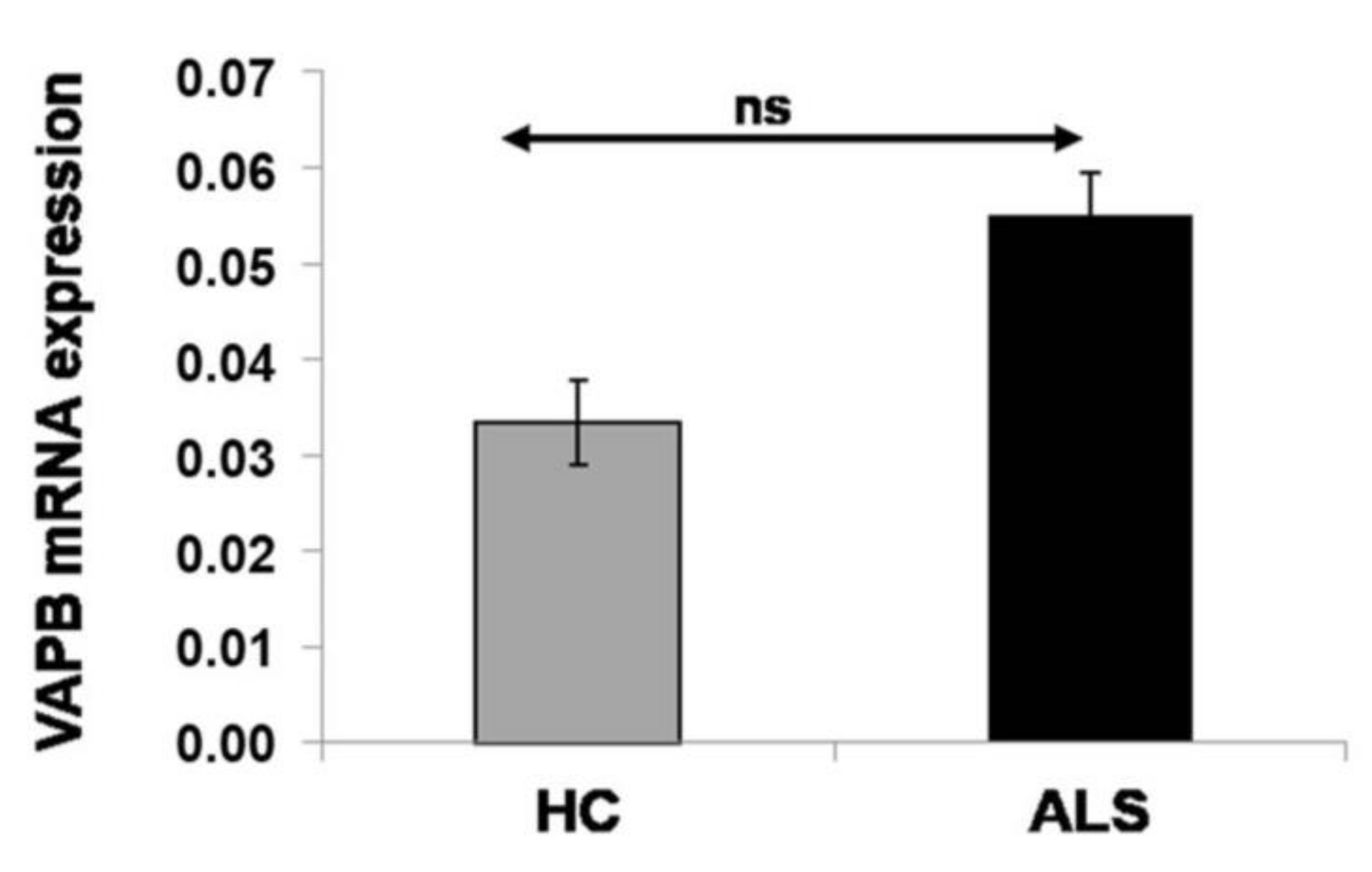
6. Nonalteration of Vesicle-Associated Membrane-Protein-Associated Protein B mRNA Expression in Sporadic Amyotrophic Lateral Sclerosis Patients
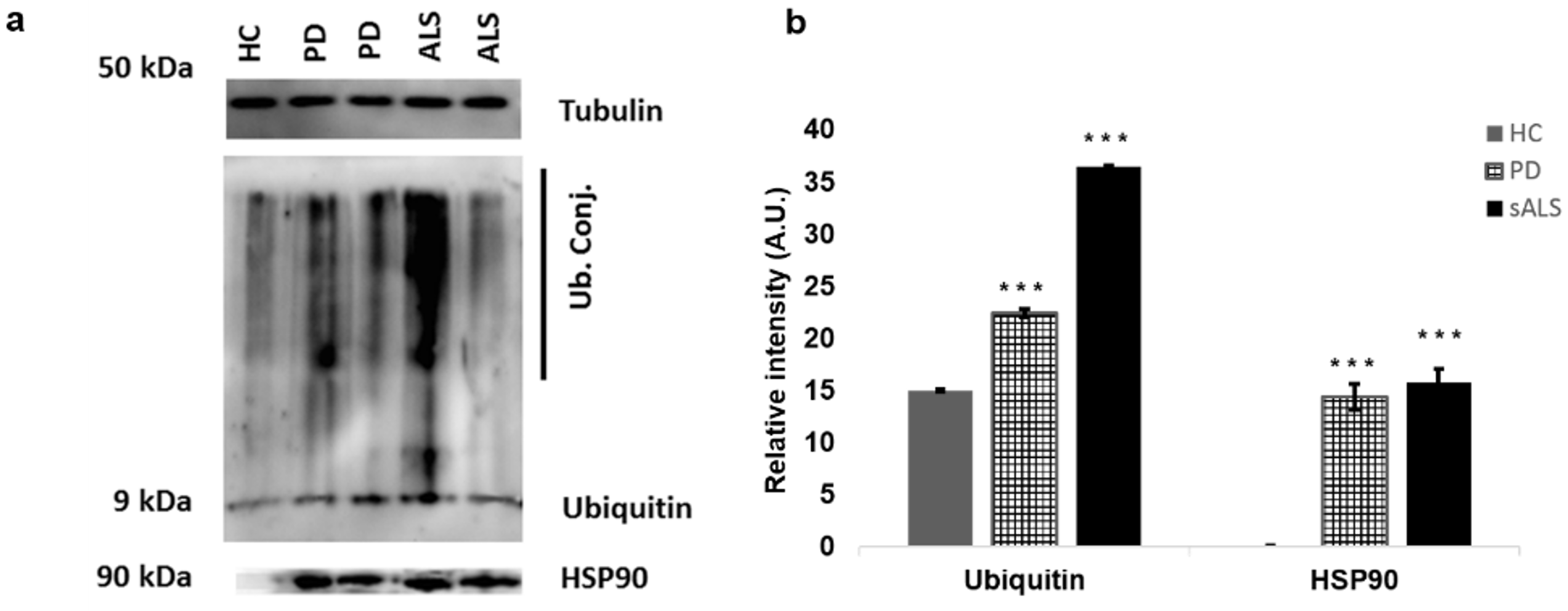
7. Western Blot Analysis of Peripheral Blood Mononuclear Cells
8. Genetic Analysis
References
- Saberi, S.; Stauffer, J.E.; Schulte, D.J.; Ravits, J. Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol. Clin. 2015, 33, 855–876.
- Kanekura, K.; Nishimoto, I.; Aiso, S.; Matsuoka, M. Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8). J. Biol. Chem. 2006, 281, 30223–30233.
- Ince, P.G.; Highley, J.R.; Kirby, J.; Wharton, S.B.; Takahashi, H.; Strong, M.J.; Shaw, P.J. Molecular pathology and genetic advances in amyotrophic lateral sclerosis: An emerging molecular pathway and the significance of glial pathology. Acta Neuropathol. 2011, 122, 657–671.
- Yamashita, S.; Ando, Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl. Neurodegener. 2015, 4, 13.
- Bozzoni, V.; Pansarasa, O.; Diamanti, L.; Nosari, G.; Cereda, C.; Ceroni, M. Amyotrophic lateral sclerosis and environmental factors. Funct. Neurol. 2016, 31, 7–19.
- Hu, Y.; Cao, C.; Qin, X.Y.; Yu, Y.; Yuan, J.; Zhao, Y.; Cheng, Y. Increased peripheral blood inflammatory cytokine levels in amyotrophic lateral sclerosis: A meta-analysis study. Sci. Rep. 2017, 7, 12–15.
- Carrì, M.T.; D’Ambrosi, N.; Cozzolino, M. Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochem. Biophys. Res. Commun. 2017, 483, 1187–1193.
- Groen, E.J.N.; Pasterkamp, R.J.; van den Berg, L.H.; Koppers, M.; Blokhuis, A.M. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794.
- Fasana, E.; Fossati, M.; Ruggiano, A.; Brambillasca, S.; Hoogenraad, C.C.; Navone, F.; Francolini, M.; Borgese, N. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 2010, 24, 1419–1430.
- Mitne-Neto, M.; Machado-Costa, M.; Marchetto, M.C.N.; Bengtson, M.H.; Joazeiro, C.A.; Tsuda, H.; Bellen, H.J.; Silva, H.C.A.; Oliveira, A.S.B.; Lazar, M.; et al. Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum. Mol. Genet. 2011, 20, 3642–3652.
- Teuling, E.; Ahmed, S.; Haasdijk, E.; Demmers, J.; Steinmetz, M.O.; Akhmanova, A.; Jaarsma, D.; Hoogenraad, C.C. Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. J. Neurosci. 2007, 27, 9801–9815.
- Lev, S.; Halevy, D.B.; Peretti, D.; Dahan, N. The VAP protein family: From cellular functions to motor neuron disease. Trends Cell Biol. 2008, 18, 282–290.
- Kim, S.; Leal, S.S.; Ben Halevy, D.; Gomes, C.M.; Lev, S. Structural requirements for VAP-B oligomerization and their implication in amyotrophic lateral sclerosis-associated VAP-B(P56S) neurotoxicity. J. Biol. Chem. 2010, 285, 13839–13849.
- Gkogkas, C.; Middleton, S.; Kremer, A.M.; Wardrope, C.; Hannah, M.; Gillingwater, T.H.; Skehel, P. VAPB interacts with and modulates the activity of ATF6. Hum. Mol. Genet. 2008, 17, 1517–1526.
- Mórotz, G.M.; De Vos, K.J.; Vagnoni, A.; Ackerley, S.; Shaw, C.E.; Miller, C.C.J. Amyotrophic lateral sclerosis-associated mutant VAPBP56s perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum. Mol. Genet. 2012, 21, 1979–1988.
- Suzuki, H.; Kanekura, K.; Levine, T.P.; Kohno, K.; Olkkonen, V.M.; Aiso, S.; Matsuoka, M. ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J. Neurochem. 2009, 108, 973–985.
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.A.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.M.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831.
- Marques, V.D.; Barreira, A.A.; Davis, M.B.; Abou-Sleiman, P.M.; Silva, W.A.; Zago, M.A.; Sobreira, C.; Fazan, V.; Marques, W. Expanding the phenotypes of the Pro56Ser VAPB mutation: Proximal SMA with dysautonomia. Muscle Nerve 2006, 34, 731–739.
- Chen, H.-J.; Anagnostou, G.; Chai, A.; Withers, J.; Morris, A.; Adhikaree, J.; Pennetta, G.; de Belleroche, J.S. Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 40266–40281.
- Anagnostou, G.; Paul, P.; Akbar, M.T.; Steiner, T.J.; Angelinetta, C.; de Belleroche, J. Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol. Aging 2010, 31, 969–985.
- Qiu, L.; Qiao, T.; Beers, M.; Tan, W.; Wang, H.; Yang, B.; Xu, Z. Widespread aggregation of mutant VAPB associated with ALS does not cause motor neuron degeneration or modulate mutant SOD1 aggregation and toxicity in mice. Mol. Neurodegener. 2013, 8, 1.
- Tudor, E.L.; Galtrey, C.M.; Perkinton, M.S.; Lau, K.-F.; De Vos, K.J.; Mitchell, J.C.; Ackerley, S.; Hortobágyi, T.; Vámos, E.; Leigh, P.N.; et al. Amyotrophic lateral sclerosis mutant vesicle-associated membrane protein-associated protein-B transgenic mice develop TAR-DNA-binding protein-43 pathology. Neuroscience 2010, 167, 774–785.
- van Blitterswijk, M.; van Es, M.A.; Koppers, M.; van Rheenen, W.; Medic, J.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; Veldink, J.H.; van den Berg, L.H. VAPB and C9orf72 mutations in 1 familial amyotrophic lateral sclerosis patient. Neurobiol. Aging 2012, 33, 2950-e1.
- Liew, C.C.; Ma, J.; Tang, H.C.; Zheng, R.; Dempsey, A.A. The peripheral blood transcriptome dynamically reflects system wide biology: A potential diagnostic tool. J. Lab. Clin. Med. 2006, 147, 126–132.
- Cereda, C.; Leoni, E.; Milani, P.; Pansarasa, O.; Mazzini, G.; Guareschi, S.; Alvisi, E.; Ghiroldi, A.; Diamanti, L.; Bernuzzi, S.; et al. Altered Intracellular Localization of SOD1 in Leukocytes from Patients with Sporadic Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e75916.
- Deidda, I.; Galizzi, G.; Passantino, R.; Cascio, C.; Russo, D.; Colletti, T.; La Bella, V.; Guarneri, P. Expression of vesicle-associated membrane-protein-associated protein B cleavage products in peripheral blood leukocytes and cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Eur. J. Neurol. 2014, 21, 478–485.
- Nardo, G.; Pozzi, S.; Pignataro, M.; Lauranzano, E.; Spano, G.; Garbelli, S.; Mantovani, S.; Marinou, K.; Papetti, L.; Monteforte, M.; et al. Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS ONE 2011, 6, e25545.
- Silani, V.; Ludolph, A.; Fornai, F. The emerging picture of ALS: A multisystem, not only a “motor neuron disease”. Arch. Ital. Biol. 2017, 155, 153–158.
- Vijayakumar, U.G.; Milla, V.; Stafford, M.Y.C.; Bjourson, A.J.; Duddy, W.; Duguez, S.M.R. A systematic review of suggested molecular strata, biomarkers and their tissue sources in ALS. Front. Neurol. 2019, 10, 400.
- Musarò, A. Understanding ALS: New therapeutic approaches. FEBS J. 2013, 280, 4315–4322.
- Papiani, G.; Ruggiano, A.; Fossati, M.; Raimondi, A.; Bertoni, G.; Francolini, M.; Benfante, R.; Navone, F.; Borgese, N. Restructured endoplasmic reticulum generated by mutant amyotrophic lateral sclerosis-linked VAPB is cleared by the proteasome. J. Cell Sci. 2012, 125, 3601–3611.
- Kiriyama, Y.; Nochi, H. The Function of Autophagy in Neurodegenerative Diseases. Int. J. Mol. Sci. 2015, 16, 26797–26812.
- Ciechanover, A.; Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003, 40, 427–446.
- Jaronen, M.; Goidsteins, G.; Koistinaho, J. ER stress and unfolded protein response in amyotrophic lateral sclerosis—A controversial role of protein disulphide isomerase. Front. Cell. Neurosci. 2014, 8, 402.
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316.
- Chakrabarti, A.; Chen, A.W.; Varner, J.D. A review of the mammalian unfolded protein response. Biotechnol. Bioeng. 2011, 108, 2777–2793.
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and autophagy. Methods Mol. Biol. 2008, 445, 77–88.
- Nedelsky, N.B.; Todd, P.K.; Taylor, J.P. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta Mol. Basis Dis. 2008, 1782, 691–699.

