Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vladimir Chernyshev | -- | 2102 | 2023-07-06 07:12:16 | | | |
| 2 | Catherine Yang | Meta information modification | 2102 | 2023-07-06 07:25:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chernyshev, V.V. SDPD Methods to Pharmaceutical Cocrystals and Salts. Encyclopedia. Available online: https://encyclopedia.pub/entry/46496 (accessed on 24 June 2026).
Chernyshev VV. SDPD Methods to Pharmaceutical Cocrystals and Salts. Encyclopedia. Available at: https://encyclopedia.pub/entry/46496. Accessed June 24, 2026.
Chernyshev, Vladimir V.. "SDPD Methods to Pharmaceutical Cocrystals and Salts" Encyclopedia, https://encyclopedia.pub/entry/46496 (accessed June 24, 2026).
Chernyshev, V.V. (2023, July 06). SDPD Methods to Pharmaceutical Cocrystals and Salts. In Encyclopedia. https://encyclopedia.pub/entry/46496
Chernyshev, Vladimir V.. "SDPD Methods to Pharmaceutical Cocrystals and Salts." Encyclopedia. Web. 06 July, 2023.
Copy Citation
Modern X-ray diffractometers, powerful computers, and the continuously improving methods of structure determination from powder data, so called SDPD methods, provide a reliable basis for conducting such research in laboratories of any level.
X-ray powder diffraction
pharmaceutical cocrystals
structure determination from powder diffraction data
1. Lamivudine Camphorsulfonate (1:1) Salt
The powder diffraction pattern of the title salt, C8H12N3O3S+·C10H15O4S− [1] (Figure 1), used for structure solution was recorded at 153 K in transmission geometry on a STOE Stadi-P diffractometer equipped with a curved Ge monochromator, using CuKα1 radiation. It was indexed with the program DICVOL [2] in a monoclinic unit cell with a volume of 2051 Å3. The Pawley fit [3] confirmed unit cell parameters and chiral space group P21, the asymmetric unit of which contains two formula units—two cations and two anions. The crystal structure was solved with the simulated annealing technique implemented in the DASH program [4] and refined by the Rietveld method, using the program TOPAS [5].
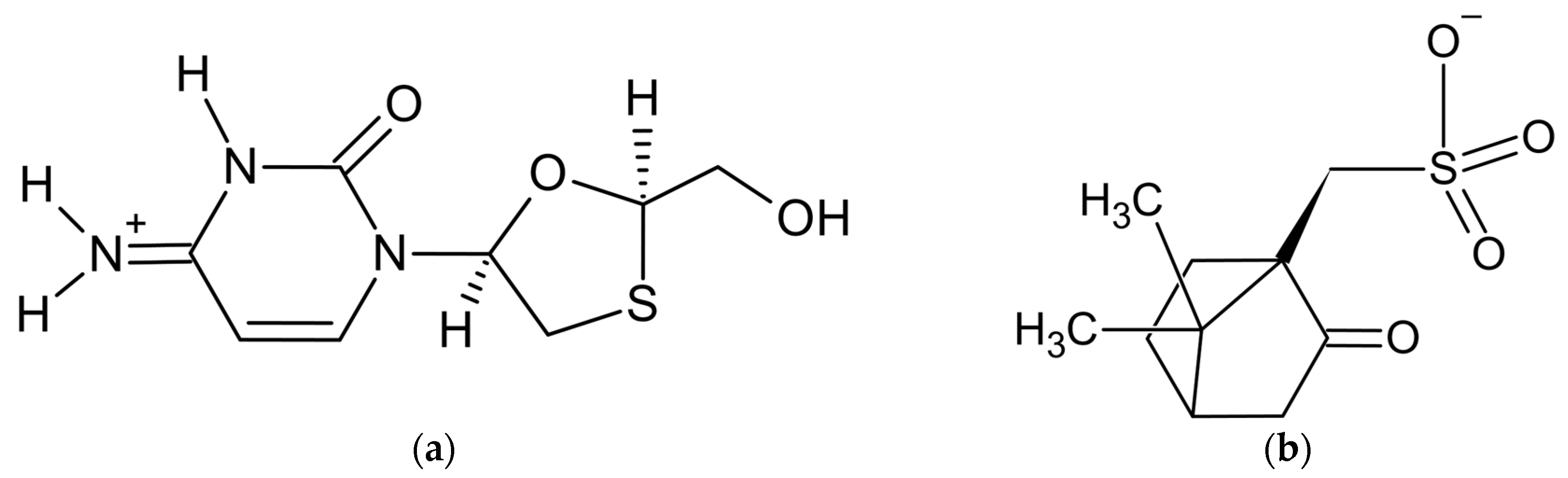
Figure 1. Components of lamivudine camphorsulfonate (1:1) salt: (a) lamivudine cation and (b) camphorsulfonate anion.
In the original paper [1], the crystal structure of theophylline benzamide (1:1) cocrystal was successfully determined from synchrotron powder diffraction data. However, the application of SDPD methods to the crystal structure determination of the third compound, aminoglutethimide camphorsulfonate hemihydrate, failed, and its crystal structure was solved later, using a single crystal of suitable size. A detailed description of all the steps of SDPD for two compounds and an analysis of the reasons for failure when working with the third one make this article [1] very useful for scientists who are trying to apply SDPD methods to pharmaceutical substances.
2. Furosemide Urea (1:1) and Carbamazepine Indomethacin (1:1)
The powder diffraction patterns of the title cocrystals, C12H11ClN2O5S·CH4N2O and C15H12N2O·C19H16ClNO4, used for structure solution were recorded at 298 K in transmission geometry on a Bruker D8 Advance diffractometer equipped with a LynxEye detector, using CuKα1 radiation. Both powder patterns were indexed with the program DASH—in a triclinic unit cell with a volume of 836 Å3 for furosemide urea (FUR:URE) and in a monoclinic unit cell with a volume of 2823 Å3 for carbamazepine indomethacin (CBZ:IND). The Pawley fit confirmed unit cell parameters and space groups P-1 and P21/n for FUR:URE and CBZ:IND, respectively, with one formula unit per asymmetric part in each structure. The crystal structures were solved with the simulated annealing technique implemented in the DASH program and subjected to rigid-body Rietveld refinement, using TOPAS. Energy minimization, using the DFT-D theory, was used when finalizing both structures.
In the research of [6], a new polymorph of CBZ:IND (1:1) cocrystal was obtained and described as form II (CCDC ref. code LEZKEY01). Earlier, the same group of authors published the CBZ:IND cocrystal form I (CCDC ref. code LEZKEY, [7]), which crystallizes in a monoclinic unit cell with a volume of 2922 Å3 and space group P21/c. The crystal structure of form I was also determined from laboratory X-ray powder data with the use of SDPD methods. Therefore, two polymorphic modifications of carbamazepine indomethacin (1:1) cocrystal were revealed and structurally characterized by XRPD methods, thus demonstrating the modern abilities of the latter.
In both polymorphic forms, the carbamazepine and indomethacin molecules are paired via acid–amide heterosynthon [8], with slightly different conformations of the indomethacin molecule (Figure 2).

Figure 2. (Prepared with Mercury [9]). The hydrogen-bonded pair of carbamazepine and indomethacin molecules in 1:1 cocrystal of (a) form I and (b) form II. Thin orange lines denote intermolecular N–H…O and O–H…O hydrogen bonds.
The O atom in the chlorobenzoyl (chlb) fragment acts as an H-bond acceptor in further intermolecular interactions. In form I, a N–H…Ochlb hydrogen bond with a N…Ochlb distance of 2.69 Å links these pairs into zigzag chains extended in the [101] direction. In form II, Ochlb acts as an acceptor of two weak hydrogen bonds, namely N–H… Ochlb [N… Ochlb 3.31 Å] and C–H… Ochlb [C… Ochlb 3.36 Å], which link the carbamazepine–indomethacin pairs into corrugated layers parallel to the ac plane. These differences in the crystal packing help us to understand why form II is more thermodynamically stable than form I [6].
3. Three Imidazole-Based (1:1) Salts of Salicylic Acid
To improve the poor solubility of salicylic acid (SA) in water, Emmerling and co-workers obtained its 1:1 salts with imidazole (IMI), 1-methylimidazole (1-MEIM), and 2-methylimidazole (2-MEIM). All three new salts, namely SA:IMI, SA:1-MEIM, and SA:2-MEIM, were mechanochemically synthesized using a vibratory ball mill. Their crystal structures were solved by powder X-ray diffraction.
The powder diffraction patterns that were used for structure solutions were recorded at room temperature in Debye–Scherrer geometry on a Bruker D8 Discover diffractometer equipped with a position-sensitive LynxEye detector and a Johansson monochromator, using CuKα1 radiation. Powder patterns were indexed with DICVOL [2] in orthorhombic unit cells with the volumes of 2020, 2192, and 2239 Å3 for SA:IMI, SA:1-MEIM, and SA:2-MEIM, respectively. The Pawley fits confirmed unit cell parameters and space groups Pbca, Pbca, and Pbcn, respectively, thus assuming one formula unit per asymmetric part in each structure. The crystal structures were solved with the simulated annealing technique implemented in the DASH program and refined by the Rietveld method, using the program TOPAS and restraints applied for all bond lengths, angles, and planar rings.
The subsequent physicochemical studies revealed that all three new cocrystal salts present remarkably higher solubility and a faster dissolution rate than free salicylic acid [10]. It is noteworthy that the orthorhombic crystal structure of SA:IMI (CCDC ref. code NIJDOB02) presents a new polymorph of this compound in addition to the earlier established tetragonal polymorph with the CCDC ref. code NIJDOB [11]. Different crystal packings in two polymorphs are shown in Figure 3.

Figure 3. Crystal packings of two polymorphs of SA:IMI (1:1) cocrystal viewed along the c-axis: (A) tetragonal polymorph (NIJDOB) and (B) orthorhombic polymorph (NIJDOB02).
The same SDPD methodology was used later by Martins and Emmerling in the crystal structure determination of three new 1:1 cocrystals of carbamazepine with 2,4-, 2,5-, and 2,6-dihydroxybenzoic acids as coformers [12].
4. Metaxalone Nicotinamide (1:1)
The powder diffraction pattern used for the structure solution of the title cocrystal, C12H15NO3·C6H6N2O [13], was recorded at 295 K in transmission geometry on a Guinier-Huber camera G670 equipped with a curved Ge monochromator, using CuKα1 radiation. It was indexed with the program ITO [14] in a triclinic unit cell, with a volume of 872 Å3. The Pawley fit confirmed the unit cell parameters. The crystal structure was solved in space group P-1 with the simulated annealing technique [15] and refined by the Rietveld method, using the program MRIA [16].
Metaxalone (MET) is a poor aqueous soluble drug, while its cocrystals with nicotinamide (NAM), salicylamide, and 4-hydroxybenzoic acid (hydrate) exhibit better solubility of the native drug, as followed from the dissolution experiments in pH 6.8 phosphate buffer [13]. Dissolution experiments indicated the superior solubility of the MET-NAM cocrystal, and this was correlated with its lower melting point, higher solubility of NAM, and weaker intermolecular interaction between MET and NAM. The improved solubility of MET–NAM is anticipated to enhance the drug’s bioavailability and decrease side effects [13].
In the MET-NAM crystal structure, the drug and coformer form hydrogen-bonded imide and amide homodimers of the R22(8) ring, respectively, which are further linked into chains in [110] via N–H…O hydrogen bonds (Figure 4).

Figure 4. The hydrogen-bonded chain extended in [110] of imide and syn-amide homodimers of MET and NAM molecules, respectively, in MET-NAM 1:1 cocrystal. Thin orange lines denote intermolecular N–H…O hydrogen bonds.
The aforementioned experimental facilities and SDPD methodology were used in the crystal structure determination of 1:1 cocrystals of dexamethasone with catechol and resorcinol [17], and cephalexin with serine [18].
5. Acridine Diclofenac (1:1) Salt
The powder diffraction pattern of the title salt, C13H10N+·C14H10Cl2NO2− [19], used for the structure solution was recorded at 295 K in transmission geometry on a Bruker D8 Advance diffractometer equipped with a LynxEye XE-T detector, using CuKα radiation. It was indexed with the programs EXPO2014 [20] and Topas Academic [5] in a monoclinic unit cell with a volume of 2247 Å3. The crystal structure was solved in space group P21/n, with the simulated annealing technique, and refined by the Rietveld method, using Topas Academic.
The authors [19] paid special attention to the correct positioning of the hydrogen atom between the N atom of acridine and O atom of diclofenac separated at 2.77(8) Å to make a choice between the salt (H atom attached to N) and cocrystal (H atom attached to O). The final choice in favor of salt was made after the energy framework calculations performed with CrystalExplorer 17.5 [21], which led to the following total lattice energies: −513.4 kJ mol−1 for the case of salt (N–H bond) and −215.2 kJ mol−1 for cocrystal (O–H bond). The content of asymmetric unit of the final structural model of the title salt is shown in Figure 5.
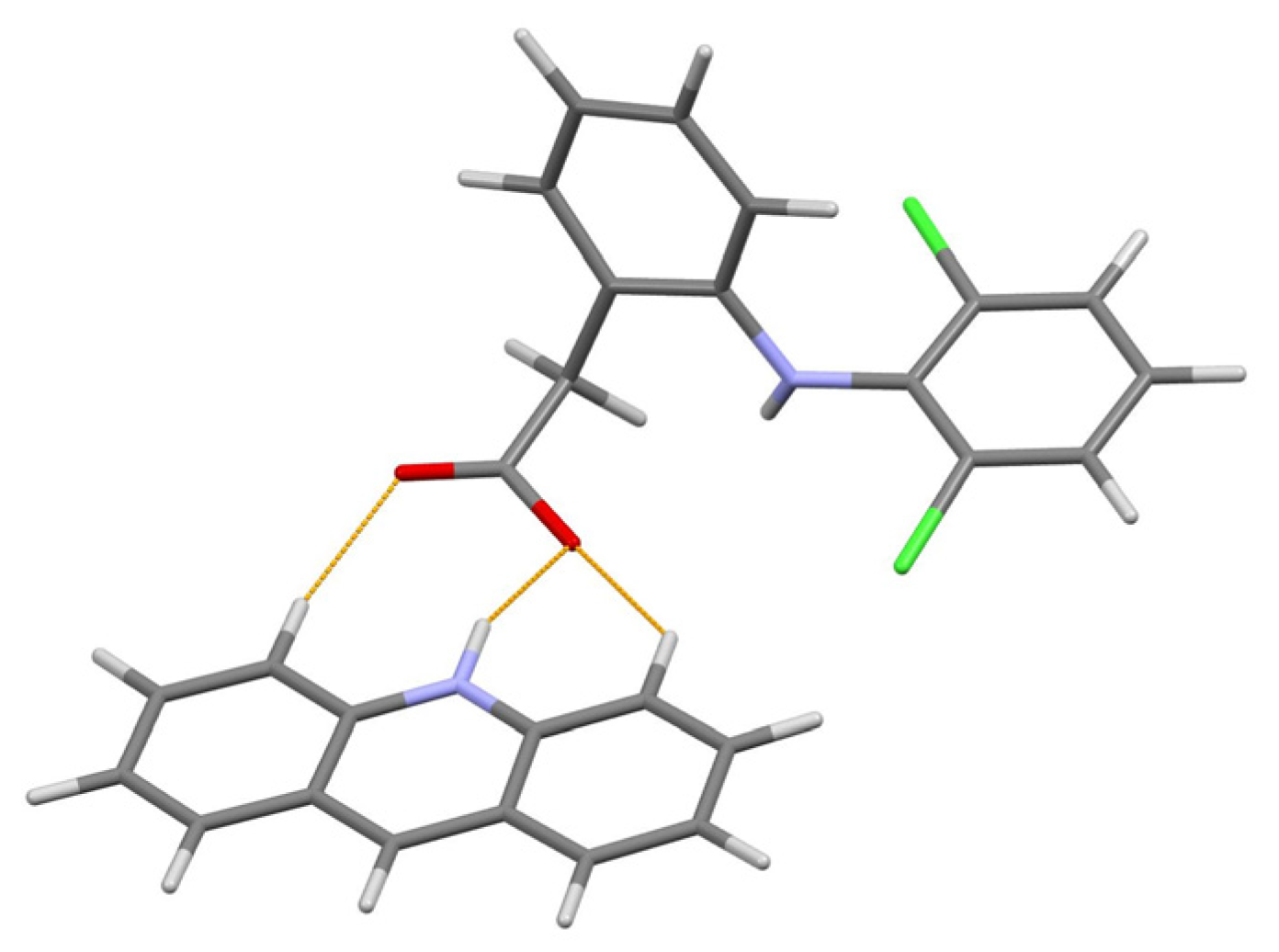
Figure 5. The content of asymmetric unit of the acridine diclofenac (1:1) salt. Thin orange lines denote intermolecular N–H…O and weak non-classical C–H…O hydrogen bonds.
Among the advantages of this research, the quantitative phase analysis should also be noted, as it allowed one to estimate the excess of reactants in the sample (3.5% of acridine and a 4.5% of diclofenac) obtained with the liquid-assisted grinding, and thus define the yield of final product 92%.
The aforementioned experimental facilities and SDPD methodology were used in the crystal structure determination of orthorhombic form of naproxen acridine 1:1 cocrystal [22].
6. Acefylline Nicotinamide (1:2) Cocrystal
The powder diffraction pattern used for the structure solution of the title cocrystal, C9H10N4O4·2(C6H6N2O) [23], was recorded at 295 K in transmission geometry on a Guinier-Huber camera G670, using CuKα1 radiation. It was indexed with the program TREOR [24] in a triclinic unit cell with a volume of 1127 Å3. The Pawley fit confirmed the unit cell parameters. The crystal structure was solved in space group P-1 and refined using the program MRIA.
The asymmetric unit of the title 1:2 cocrystal contains one acefylline (ACF) and two independent isonicotinamide (INA) molecules. One independent INA molecule, labeled with the letter A, being translated along the short parameter c = 5.0288(7) Å, generates hydrogen-bonded chain, while another independent INA molecule, labeled with the letter B, forms centrosymmetric amide homodimer (Figure 4). Interestingly, the acefylline molecule acts as a bridge, which links the INA chains and dimers via intermolecular N–H…N and N–H…O hydrogen bonds (Figure 6).
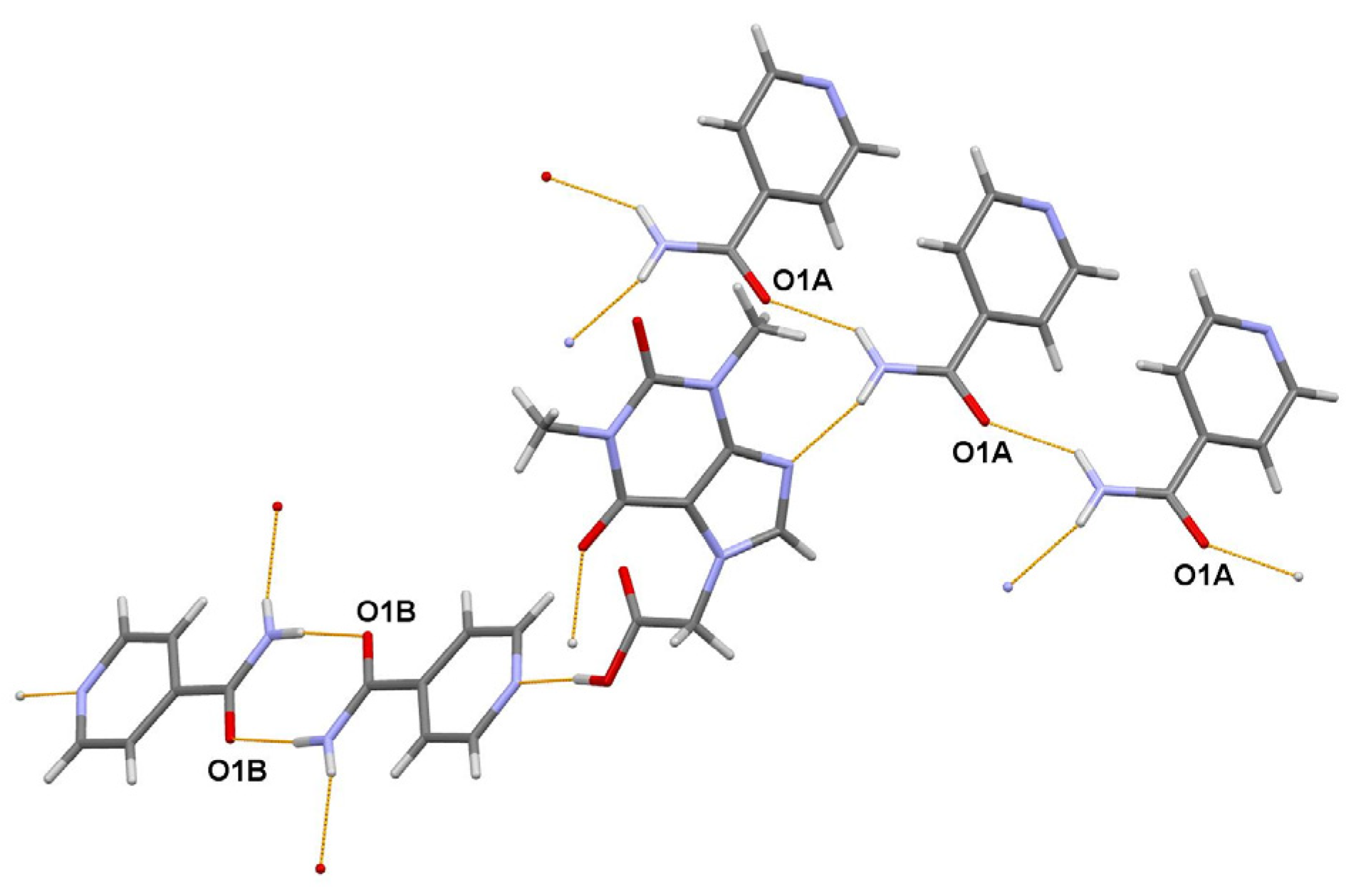
Figure 6. A portion of the crystal packing of ACF:INA (1:2) cocrystal showing the intermolecular N–H…N and N–H…O hydrogen bonds as thin orange lines.
In the research [23] devoted to acefylline binary and ternary cocrystals, salt–cocrystals, and their polymorphs co-crystallized with different coformers, the crystal structure determination of the title ternary cocrystal from the laboratory X-ray powder pattern is a particular result, although it demonstrates the routine capabilities of SDPD methods.
7. Salt–Cocrystal Continuum of the Agomelatine: Phosphoric Acid (1:1) System
The existence of two polymorphic forms of the title compound, C15H17NO2·H3PO4 [25], referred to as RT and HT, was observed from the DSC analysis. The powder diffraction patterns of the RT and HT polymorphs were measured at 298 K in transmission geometry on a Bruker D8 Advance diffractometer, using CuKα radiation, and indexed with the program X-Cell [26] in monoclinic unit cells, with the volumes of 1644 and 1682 Å3, respectively. The Pawley fit confirmed unit cell parameters and space group P21/c, the asymmetric unit of which contains one formula unit. The crystal structures were solved with the simulated annealing technique implemented in the DASH program [4], refined by the Rietveld method, using the program TOPAS [2], and optimized using plane wave-periodic DFT-D2 calculations implemented in the MedeA software [27].
The authors of [25] concluded that the obtained crystal structures of RT and HT forms of the title compound in combination with DFT-D2 analysis allowed them to discriminate between the salt and cocrystal unambiguously. Moreover, detailed characterization using a set of complementary techniques, including DSC/TGA, solid state NMR, FTIR, and chemical crystallographic database analysis, confirmed that the nature of polymorphic form transformation is a solid-state phase transition, exhibiting an enantiotropic and reversible relationship. The portions of the DFT-D2 optimized crystal packings of RT and HT forms are shown in Figure 7.
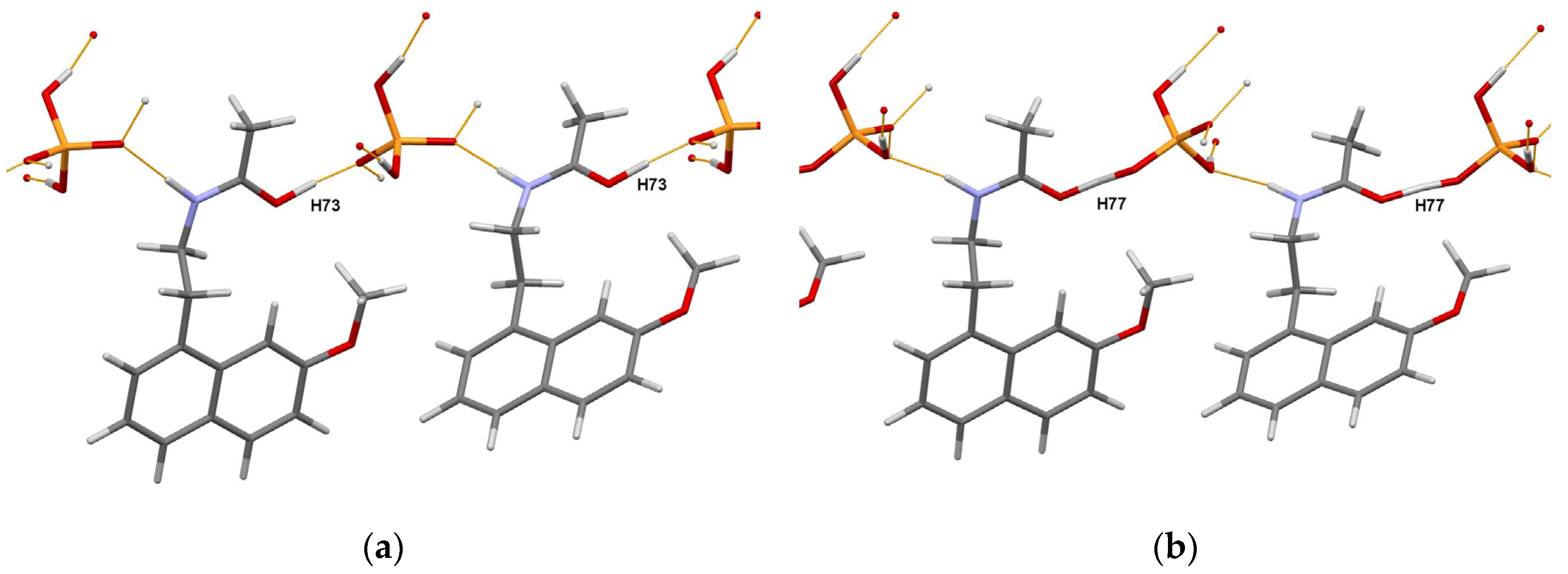
Figure 7. Portions of the crystal packings in the DFT-D2 optimized RT (a) and HT (b) forms of the agomelatine:phosphoric acid (1:1) system showing the definite position of H73 atom in RT form, and intermediate position of H77 atom (migrating proton) in HT form. Thin orange lines denote intermolecular N–H…O and O–H…O hydrogen bonds.
This work [25] once again demonstrated the obstacles that researchers face in the choice between the salt and cocrystal. It also emphasized the special role of DFT-D optimization, which complements experimental methods such as X-ray photoelectron spectroscopy [28] or solid-state NMR [29][30], thus helping to make an informed choice.
References
- Schlesinger, C.; Bolte, M.; Schmidt, M.U. Challenging structure determination from powder diffraction data: Two pharmaceutical salts and one cocrystal with Z′ = 2. Z. Krist.-Cryst. Mater. 2019, 234, 257–268.
- Boultif, A.; Louer, D. Indexing of powder diffraction patterns for low-symmetry lattices by the successive dichotomy method. J. Appl. Crystallogr. 1991, 24, 987–993.
- Pawley, G.S. Unit-cell refinement from powder diffraction scans. J. Appl. Crystallogr. 1981, 14, 357–361.
- David, W.I.F.; Shankland, K.; van de Streek, J.; Pidcock, E.; Motherwell, W.D.S.; Cole, J.C. DASH: A program for crystal structure determination from powder diffraction data. J. Appl. Crystallogr. 2006, 39, 910–915.
- Coelho, A.A. TOPAS and TOPAS-Academic: An optimization program integrating computer algebra and crystallographic objects written in C++. J. Appl. Crystallogr. 2018, 51, 210–218.
- Rahal, O.A.; Majumder, M.; Spillman, M.J.; van de Streek, J.; Shankland, K. Co-crystal structures of furosemide: Urea and carbamazepine:indomethacin determined from powder X-ray diffraction data. Crystals 2020, 10, 42.
- Majumder, M.; Buckton, G.; Rawlinson-Malone, C.; Williams, A.C.; Spillman, M.J.; Shankland, N.; Shankland, K. A carbamazepine: Indomethacin (1:1) cocrystal produced by milling. CrystEngComm 2011, 13, 6327–6328.
- Bolla, G.; Sarma, B.; Nangia, A. Crystal engineering of pharmaceutical cocrystals in the discovery and development of improved drugs. Chem. Rev. 2022, 122, 11514–11603.
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470.
- Martins, I.C.B.; Al-Sabbagh, D.; Meyer, K.; Maiwald, M.; Scholz, G.; Emmerling, F. Insight into the structure and properties of novel imidazole-based salts of salicylic acid. Molecules 2019, 24, 4144.
- Nabulsi, N.A.R.; Gandour, R.D.; Fronczek, F.R. CSD Communication (Private Communication). 2013.
- Martins, I.C.B.; Emmerling, F. Carbamazepine dihydroxybenzoic acid cocrystals: Exploring packing interactions and reaction kinetics. Cryst. Growth Des. 2021, 21, 6961–6970.
- Gohel, S.K.; Palanisamy, V.; Sanphui, P.; Prakash, M.; Singh, G.P.; Chernyshev, V. Isostructural cocrystals of metaxalone with improved dissolution characteristics. RSC Adv. 2021, 11, 30689–30700.
- Visser, J.W. A fully automatic program for finding the unit cell from powder data. J. Appl. Crystallogr. 1969, 2, 89–95.
- Zhukov, S.G.; Babaev, E.V.; Chernyshev, V.V.; Rybakov, V.B.; Sonneveld, E.J.; Schenk, H. Crystal structure determination of 2-oxo-3-benzoyloxazolopyridine from X-ray powder data. Z. Krist.-Cryst. Mater. 2000, 215, 306–308.
- Zlokazov, V.B.; Chernyshev, V.V. MRIA—A program for a full profile analysis of powder multiphase neutron-diffraction time-of-flight (direct and Fourier) spectra. J. Appl. Crystallogr. 1992, 25, 447–451.
- Varsa, S.R.B.; Sanphui, P.; Chernyshev, V. Polymorphs and isostructural cocrystals of dexamethasone: Towards the improvement of aqueous solubility. CrystEngComm 2022, 24, 6045–6058.
- Fayaz, T.K.S.; Palanisamy, V.; Sanphui, P.; Chernyshev, V. Multicomponent solid forms of antibiotic cephalexin towards improved chemical stability. CrystEngComm 2023, 25, 1252–1262.
- Mirocki, A.; Conterosito, E.; Palin, L.; Sikorski, A.; Milanesio, M.; Lopresti, M. Crystal structure of a new 1:1 acridine-diclofenac salt, obtained with high yield by a mechanochemical approach. Crystals 2022, 12, 1573.
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Crystallogr. 2013, 46, 1231–1235.
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587.
- Mirocki, A.; Lopresti, M.; Palin, L.; Conterosito, E.; Sikorski, A.; Milanesio, M. Exploring the molecular landscape of multicomponent crystals formed by naproxen drug and acridines. CrystEngComm 2022, 24, 6839–6853.
- Allu, S.; Garai, A.; Chernyshev, V.V.; Nangia, A.K. Synthesis of ternary cocrystals, salts, and hydrates of acefylline with enhanced dissolution and high permeability. Cryst. Growth Des. 2022, 22, 4165–4181.
- Werner, P.-E.; Eriksson, L.; Westdahl, M. TREOR, a semi-exhaustive trial-and-error powder indexing program for all symmetries. J. Appl. Crystallogr. 1985, 18, 367–370.
- Voguri, R.S.; Ranga, S.; Dey, A.; Ghosal, S. Solid-state phase transition of agomelatine-phosphoric acid molecular complexes along the salt-cocrystal continuum: Ab initio powder X-ray diffraction structure determination and DFT-D2 analysis. Cryst. Growth Des. 2020, 20, 7647–7657.
- Neumann, M.A. X-Cell: A novel indexing algorithm for routine tasks and difficult cases. J. Appl. Crystallogr. 2003, 36, 356–365.
- MedeA, Materials Design, Inc.: San Diego, CA, USA, 2015.
- Stevens, J.S.; Byard, S.J.; Schroeder, S.L.M. Salt or co-crystal? Determination of protonation state by X-ray photoelectron spectroscopy (XPS). J. Pharm. Sci. 2010, 99, 4453–4457.
- Harris, K.D.M. NMR Crystallography as a vital tool in assisting crystal structure determination from powder XRD data. Crystals 2022, 12, 1277.
- Smalley, C.J.H.; Logsdail, A.J.; Hughes, C.E.; Iuga, D.; Young, M.T.; Harris, K.D.M. Solid-state structural properties of alloxazine determined from powder XRD data in conjunction with DFT-D calculations and solid-state NMR spectroscopy: Unraveling the tautomeric identity and pathways for tautomeric interconversion. Cryst. Growth Des. 2022, 22, 524–534.
More
Information
Subjects:
Crystallography
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
762
Revisions:
2 times
(View History)
Update Date:
06 Jul 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No