Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ahmad Khusairi Azemi | -- | 4655 | 2023-06-27 03:56:35 | | | |
| 2 | Rita Xu | Meta information modification | 4655 | 2023-06-27 04:15:41 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nordin, M.L.; Azemi, A.K.; Nordin, A.H.; Nabgan, W.; Ng, P.Y.; Yusoff, K.; Abu, N.; Lim, K.P.; Zakaria, Z.A.; Ismail, N.; et al. Peptide-Based Vaccine against Breast Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/46085 (accessed on 10 August 2026).
Nordin ML, Azemi AK, Nordin AH, Nabgan W, Ng PY, Yusoff K, et al. Peptide-Based Vaccine against Breast Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/46085. Accessed August 10, 2026.
Nordin, Muhammad Luqman, Ahmad Khusairi Azemi, Abu Hassan Nordin, Walid Nabgan, Pei Yuen Ng, Khatijah Yusoff, Nadiah Abu, Kue Peng Lim, Zainul Amiruddin Zakaria, Noraznawati Ismail, et al. "Peptide-Based Vaccine against Breast Cancer" Encyclopedia, https://encyclopedia.pub/entry/46085 (accessed August 10, 2026).
Nordin, M.L., Azemi, A.K., Nordin, A.H., Nabgan, W., Ng, P.Y., Yusoff, K., Abu, N., Lim, K.P., Zakaria, Z.A., Ismail, N., & Azmi, F. (2023, June 27). Peptide-Based Vaccine against Breast Cancer. In Encyclopedia. https://encyclopedia.pub/entry/46085
Nordin, Muhammad Luqman, et al. "Peptide-Based Vaccine against Breast Cancer." Encyclopedia. Web. 27 June, 2023.
Copy Citation
Breast cancer is considered the second-leading cancer after lung cancer and is the most prevalent cancer among women globally. Cancer immunotherapy via vaccine has gained great attention due to specific and targeted immune cell activity that creates a potent immune response, thus providing long-lasting protection against the disease.
breast cancer
immunotherapy
peptide-based vaccine
1. Introduction
Among women worldwide, breast cancer is considered the most frequently occurring cancer. According to the World Health Organization (WHO), 2.1 million females have been diagnosed with breast cancer every year, which is responsible for approximately 15% of all cancer deaths [1]. Breast cancer is a cancer that develops from the epithelial cells of the mammary gland, duct, or lobules. Breast cancer occurrence also exists in males; however, it is relatively rare (around 1%) [2][3][4]. Although pathophysiological knowledge of breast cancer is minimal, certain established risk factors, such as genetic predisposition (family history), diet, and an unhealthy lifestyle, are unquestionably linked to the development of breast cancer [5][6][7][8]. To highlight, both genetic and environmental factors influence the diversity of breast cancer etiology [9][10].
It is widely accepted that BRCA 1 and BRCA 2 genes are responsible for repairing DNA dysregulation, alteration, and damage. These genes are predicted to be accountable for approximately 40–85% of the risk of hereditary breast cancer when they are mutated. BRCA 1 and BRCA 2 are located on chromosomes 17q and 13q12–13, enabling the inference of more distally mutated loci associated with mutations to affect their functional enhancers and promoters’ actions. Besides BRCA germline families, mutations in p53, PTEN, CHEK2, ATM, PALB2, RAD51C, and RAD51D have also been associated with breast cancer [11][12]. Additionally, mutations in BRCA genes can lead to the acquisition of a multi-drug resistant phenotype, subsequently contributing to a major limitation in clinical treatment for breast cancer [13][14][15].
The condition can worsen if the genes are inherited from one generation to another and become inheritable mutations. It can happen when epigenetically mutated cells accumulate and then further create a microenvironment that improves drug efflux, drug evasion, the anti-apoptotic pathway, and other escape mechanisms from the immune system. Epigenetic mutations, resulting mostly in DNA methylation patterns, histone acetylation, and phosphorylation, are known to have a profound effect on gene expression, resulting in the activation of tumor suppressor genes and leading to the emergence of cancer drug resistance [16]. When malignant cells continue to metastasize, they overly express immune checkpoint inhibition (CPI) signals, resulting in stimulation of inhibitory co-stimulatory molecules (PD1/PD-L1/LAG-3, CTLA4) and anti-apoptotic signaling pathways, causing tumor cells to deactivate immune activation and immune detection; hence, the tumor cells escape and progress to cancerous form [17]. Interactions of cancer in the tumor microenvironment can activate cellular components in the environment, including tumor-associated macrophages (TAM), cancer-associated fibroblasts (CAF), and mesenchymal cells, to protect cancer cells from being susceptible to drugs and promote drug resistance [18]. Some of the cancer biomarkers can also hinder tumor antigen expression, leading to the failure of the intended drug to penetrate cancer due to the unfavorable and diverse mutational landscape possessed by the cancer’s microenvironment, which additionally creates immunometabolism barriers [19].
Myeloid-derived suppressor cells (MDSCs) are one form of immune cell that plays a major role in tumor immunosuppression. These cells consist of immature monocytes and granulocytes released from the bone marrow into the blood during disease conditions, including cancer. Tumor-associated macrophages (TAMs) are another type of cell that functions similarly to MDSCs. The ability of MDSCs and TAMs to suppress the antitumor response is the subject of many recent studies [20][21]. MDSCs could suppress not only natural killer (NK) cells and dendritic cells (DCs) but also T cells. T cells were suppressed through the production of inducible nitric oxide (NO), nitric oxide species (iNOS), reactive oxygen species (ROS), arginine, and cysteine deprivation. Meanwhile, MDSC is able to synergize T regulatory cells (Treg) and TAMs and cause downregulation of IL-12 production by TAMs, which is an important cytokine involved in T cell and NK cell activation through membrane-bound TGF-β [22][23][24]. Figure 1 demonstrates some of the mechanisms of cancer cell evasion via hijacking the immune system.
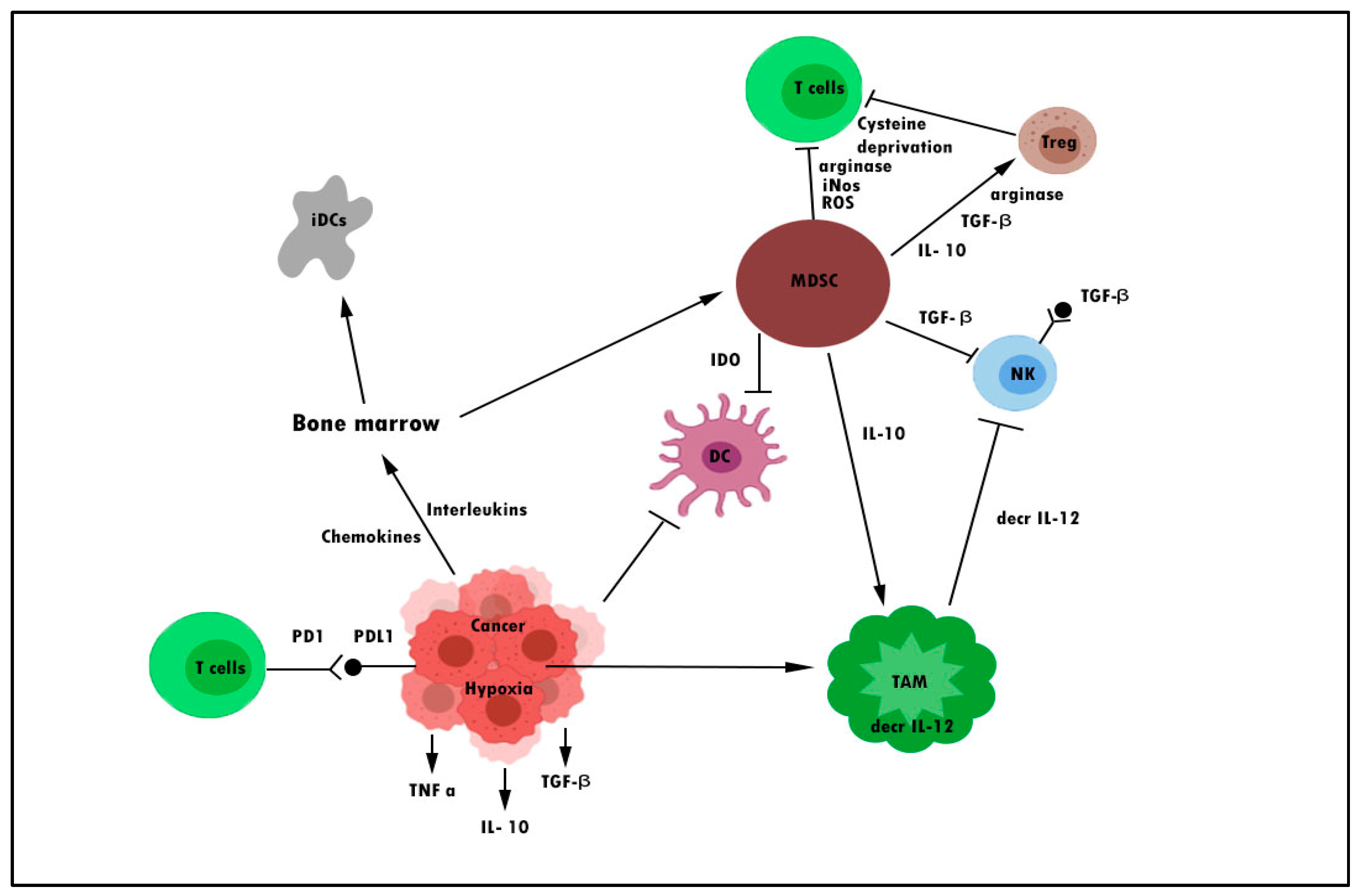
Figure 1. Mechanisms of cancer escaping pathways from the immune system. DC denotes dendritic cell, iDC denotes immature dendritic cell, MDSC denotes myeloid-derived suppressor cell, decr IL-12 denotes a decrease in interleukin-12, TAM denotes tumor-associated macrophage, and Treg denotes regulatory T cell.
The presence of tumor immunogenicity in the breast cancer microenvironment has necessitated the use of immunotherapy as a potential cancer treatment [25][26]. Immunotherapy can target specific cells that are involved in hijacking the immune cells; thus, it seems to be a good idea for the therapy’s success. Immunotherapy in the form of a vaccine functions by utilizing the patient’s immune system to identify and eradicate cancerous cells. Cancer cells produce chemokines, cytokines, and prostaglandins that attract diverse infiltrating immune cells consisting mainly of macrophages, neutrophils, and lymphocytes [27]. These infiltrating immune cells can stimulate tumor necrosis factor (TNF), IFN, matrix metalloproteinases, natural killer (NK) cells, and T cells, leading to the destruction of cancer cells. Most targeted therapies in recent years for cancer immunotherapy involve utilizing and targeting enough tumor-infiltrating T lymphocytes (TILs) and cytotoxic T cells (CTLs), which may correlate with the presence of antigen loads and suppress immune inhibitory signals responsible for local immunosuppression of the tumor microenvironment [28][29]. The discovery of breast cancer tumor-associated antigens (TAAs) or tumor-specific antigens (TSAs), which are expressed in breast cancer cells, has made it possible to develop a vaccine against breast cancer. Therefore, an understanding of the immune cell population may have significant consequences for the prevention of breast cancer, enhanced risk management strategies, and the control of breast carcinogenesis.
2. Cancer Vaccines
The fundamental understanding of tumor immunology and its plausible mechanism of action has opened the route to employing the body’s immunity against cancer [30]. Immunotherapy in the form of a vaccine has great potential for breast cancer treatment over chemotherapy and endocrine therapies due to several issues, including relapse and drug resistance. Recent reports demonstrate that about 80% of treatment failures are due to metastases and drug resistance from several mechanisms of action, such as genetic mutation [13][14]. The management of advanced malignant breast cancer, with median overall survival ranging from 4 to 5 years for luminal-like tumors to 1 year for triple-negative disease, remains minimal and is considered short [31]. Until now, many scientists have tried to discover how to overcome therapeutic resistance because more than one mechanism may be responsible for oncogenesis. Even though there are several immunotherapy forms, including adoptive cell transfer, checkpoint blockage, and antibody-based drugs, vaccines are seemingly more tempting due to their wide safety profile and lifelong protection [32][33][34]. However, until now, no breast cancer vaccine has been authorized by the U.S. Food and Drug Administration (FDA) for either therapeutic or prophylactic purposes. The immune system in humans is incredibly complex. Even though, until now, no breast cancer vaccine has been authorized, many are still in clinical trials. It is only a matter of time. The tumor microenvironment (TME) in tumors remains one of the major hurdles in developing a therapy against breast cancer. Breast cancer is generally infiltrated by immune cells triggered by the cancer cells, which can create an immunosuppressive microenvironment that encourages tumor growth by inhibiting immune cells [35]. Besides TME, the following factors are thought to be responsible for the challenges of breast cancer vaccine development: (i) the stage of breast cancer; (ii) the choice of TAAs to target; and (iii) the vaccine’s low immunogenicity as a result of the antigen selected or as a result of the vaccine delivery platform used [36].
There is, however, increasing attention in clinical research that evaluates vaccines derived from the peptide. The rationale for this interest is based on the aberrant expression of proteins or antigens by breast cancer. With the discovery of breast cancer antigens, the peptide-based vaccine is becoming a potential alternative to conventional therapies, which are known to have serious drawbacks. The vaccines would modulate the immune system of the body to specifically attack cancer cells based on the recognition of tumor associate antigens (TAAs) or tumor-specific antigens (TSAs) on the surface of cancer. Interestingly, recognition of these antigens eventually allows the immune system to recognize these antigens, to have long-lasting immunity, and to solve the relapse issue after completion of the treatment. A robust, fundamental, and precise comprehension mechanism for the action of peptide vaccines is required to establish potent and effective cancer vaccines.
3. Peptide-Based Vaccine and Key Regulator in Breast Cancer Immunogenicity
A peptide-based cancer vaccine is a short chain of amino acids that contain epitopes that are reactive to T cells. The major objective of peptide-based cancer vaccines is to induce the necessary host immune response to recognize and eliminate targeted cancer cells based on a defined set of TAAs and TSAs. The peptide-based vaccine follows the principle of immunotherapy, which modulates the immune system of the body to specifically attack cancer cells based on the recognition of aberrant expression of tumor antigens or proteins in the cancer cell. Interestingly, recognition of these antigens eventually allows the immune system to recognize these antigens, to have long-lasting immunity, and to solve the relapse issue after completion of the treatment.
Administration of a peptide vaccine functionalized CD8+ cytotoxic T lymphocytes (CTL) cells to attack tumor cells through the release of granozymes, granulysin, perforin, and Fas ligand (FasL) through Fas death receptor binding to cancer cells for apoptosis to occur (Figure 2). Cytokine release helps with lymphocyte migration, B cell development, T cell activation, and expansion. After activation, CD4+ T cells further differentiate to develop dominant anticancer pathways and responses. In order to modulate tumor-specific immune responses and inhibit proliferation in the body, significant interventions have been rendered to identify tumor-expressed antigen cells or those recognized as tumor-associated antigens (TAAs) utilizing T cells [37][38]. Identified as an example of HER2 antigen in breast cancer and transformed into a vaccine component capable of triggering a specific and systemic immune response that may contribute to the suppression, removal, and destruction of cancer in body tissues [39]. When TAAs are found in the body from cancer cells, specific fragments of cancer proteins are expressed on the cell surface and then attached to the MHC 1 complex [40]. It would ultimately be recognized by NK cells and CD8+ cells. The dissimilarity between endogenous and exogenous peptides is crucial for a functional immune response.
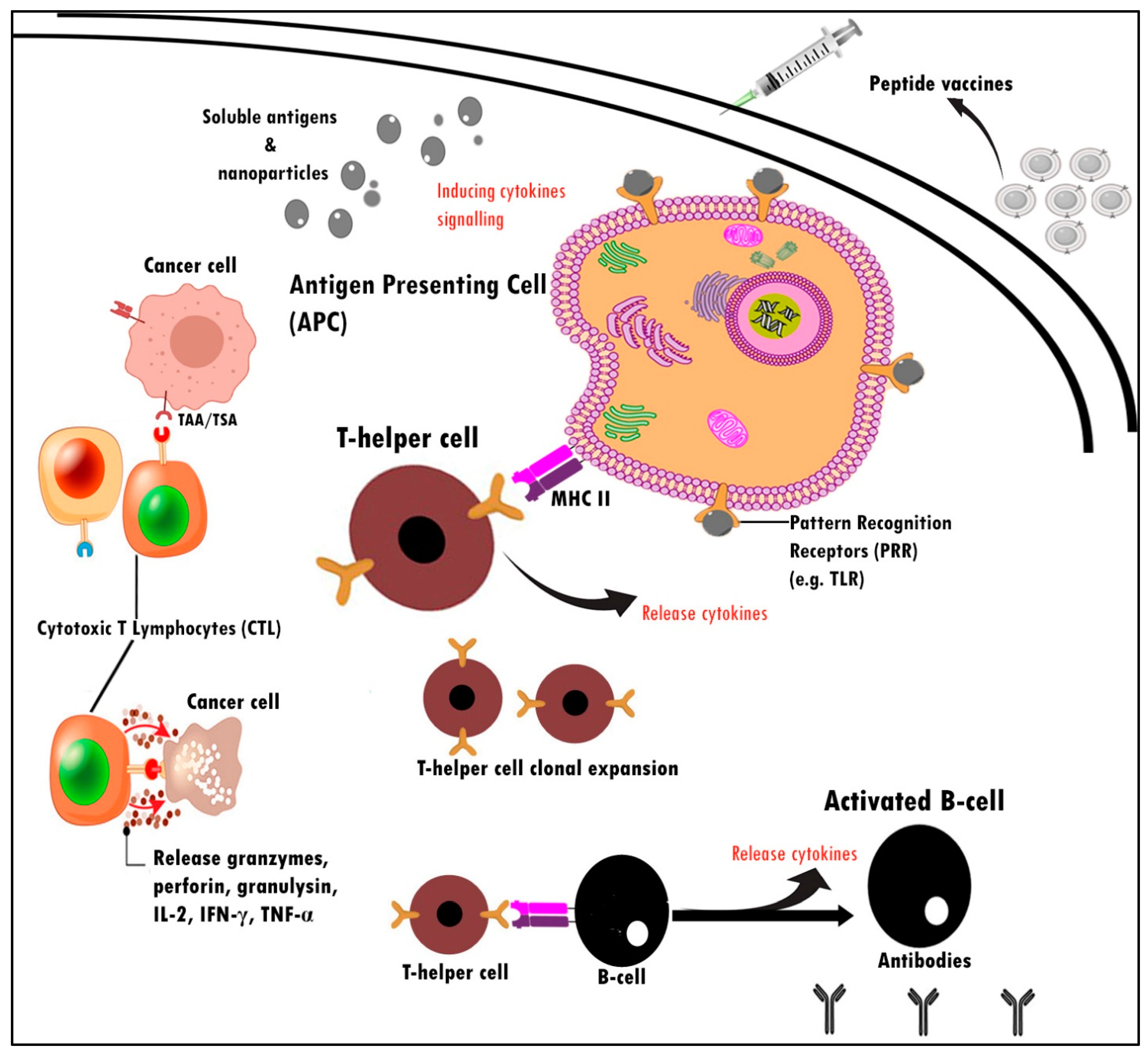
Figure 2. Schematic illustration mechanism of action for peptide vaccines. These immunological events are essential for enhancing the cell-mediated and humoral immune response against peptide vaccines. PRR is expressed by APCs and acts by recognizing and binding with antigens.
T-cell responses are specific and triggered after peptides have been taken up and processed by antigen presentation cells (APCs), which then transit to lymph nodes and expose the antigen on their cell surface as foreign molecules through MHC-I and II [41][42]. Prolonged activation of T cells leads to further differentiation into CD8+ cytotoxic and CD4+ helper T cells through the CD40-CD40L pathway. Studies have shown that CD40-CD40L associations are capable of inducing humoral and cellular thymus-dependent (TD) reactions [43]. When APCs are triggered through CD40-CD40L, it is found that ligation of CD40-CD40L to APCs, particularly dendritic cells, is capable of generating cytotoxic CD8+ T cells [43]. This may grant CD8+ T cells the potential to modulate antigen-specific immune responses similar to the CD4+ T cell response, which is often correlated with CD40L expression [44][45]. The use of TAA-related peptides such as GP2 tends to be an efficient way to stimulate CD8+ and CD4+ production against cancer cells.
Peptide-based vaccines offer several advantages over other types of cancer vaccines. Peptide-based vaccines can be easily customized with minimal epitopes while still being able to induce desired immune responses safely. Peptides are manufactured almost entirely using synthetic chemical approaches. Therefore, peptide antigens can be completely and specifically identified as chemical entities. Hence, all issues associated with the biological contamination of antigens are effectively eliminated.
Despite its benefits, peptide-based vaccination also exhibits several drawbacks, including a limited half-life, an insufficient immunogenic response, being easily degradable, and low bioavailability. However, due to the heterogeneity between solid tumors and the external microenvironment, the efficacy of the immune response in solid tumors is not as anticipated [46]. Therefore, by modifying the delivery system of peptides, such as nanocarriers that have a protective layer and are bound to the TAAs, it is then possible to prevent the degradation of these proteases and improve the association between peptide vaccines and cancer. Recent studies and many clinical studies have uncovered the potential use of peptide-based vaccines as immunotherapeutic agents that may weaken or break the immune tolerance of cancer patients. However, several modifications need to be made to the peptide vaccine to reach ideal potency.
The clinical efficacy of peptides can be easily enhanced by covalently conjugating or linking chemically with specific immunostimulatory molecules at specific positions within the peptide sequence. The use of only minimal antigens is capable of triggering humoral and cell-mediated immunities. A peptide-based vaccine depends on mobilizing cytotoxic CD8+ T cells and NK cells to kill the cancerous cells. To stimulate a tumor-specific immune response, TAAs or TSAs must be presented to the APC and make the immune system of the host recognize them. Several TAAs are mainly identified as immune targets for a vaccine against breast cancer. This includes HER2, MUC-1, EphA, Survivin, SART3, CEA, p53, and WT1 [47][48][49]. These antigens have provided convincing evidence as immune targets in preclinical and clinical studies and warrant further research.
Present clinical studies have demonstrated the therapeutic value of peptide-based vaccines to reduce cancer recurrence and enhance overall patient survival [50][51]. NeuVax™ (NCT01479244) is the most mature level of production for a peptide-derived breast cancer vaccine. It was in Phase III clinical trials and was initiated by the US National Cancer Institute in 2011 [52]. Targeting precise TAAs is vital to induce successful T-cell differentiation and alarm signals for tumor destruction mechanisms. In order to obtain a TAA-specific T cell response and upregulate stimulatory signals, APC needs to be provided with enough TAAs and be in a mature state. Otherwise, antigens may not promote oncogenesis and trigger T helper-cell clonal expansion. This is essential because CTLs are a significant cell type responsible for killing cancer cells. Various peptide breast cancer antigens that may trigger an immune response in patients have been identified and used as targets for breast cancer vaccines. However, the reduced immunogenicity of these peptide cancer antigens and cancer immune evasion mechanisms makes the development of breast cancer vaccines challenging. This condition necessitated the need to build an efficient vaccine delivery system with powerful immunostimulatory properties to promote APC activation, thus eliciting a strong T cell response and weakening and breaking the immunotolerance of cancer antigens in the tumor microenvironment.
4. Identified Tumor-Associated Antigens in Peptides Vaccine Development for Breast Cancer
4.1. HER2
HER2, also known as ERBB2, NEU, and CD34, is a human epidermal growth factor receptor 2 and a component of transmembrane glycoprotein that is overexpressed in approximately 20–30% of primary breast carcinomas [47][53] for tyrosine kinase activity. The HER2/neu cell surface receptor is the most frequently targeted TAA; thus, the HER2-derived peptide vaccine has shown excellent potential in developing breast cancer vaccines. Upon dimerization of the antigen receptor, the numerous intracellular signaling pathways are activated by transphosphorylation, which mediates cell proliferation and differentiation. However, when inappropriate activation happens, it contributes to the production of many malignancies [54]. Slamon et al. [55] first discovered the function of HER2 as a marker with a prognosis value for treating breast cancer in 1987. It has been confirmed and proved by several scholars [56][57][58]. Studies conducted by Clynes and colleagues reported monoclonal antibodies targeting HER2 to provide clinical benefits against HER2 overexpressing breast carcinomas [59][60]. To date, the application of HER2 as a therapeutic marker and predictor in invasive breast carcinomas has been commonly utilized and continues to develop.
To sum up, HER2/neu is a well-known therapeutic target that is a hallmark of HER2-positive breast cancer. With HER2-targeted vaccinations, targeting HER2 appears to be a reasonable strategy for the dysregulation of numerous signaling cascades that promote oncogenesis. It has been extensively used with a GM-CSF adjuvant. They offer little or no risk and the chance of producing a memory antibody against the same disease. Ex vivo expansion of cellular immunity, including activation of CD8+ CTL against breast carcinoma, will be enabled by the production of anti-HER2 immunization. Vaccinated patients showed high levels of CD8+ and mediated delayed-type hypersensitivity reactions [60]. Established by Mansourian et al. [61], p5 peptide encapsulated with liposomes co-administrated with CpG-ODN has been shown to decrease tumor size and, at the same time, improve animal survival period in mice of the breast cancer model. This was confirmed by Farzad et al.’s study [53], which displayed another peptide, the P435 HER2-derived peptide, conjugated to liposomes capable of inducing CTL responses, therefore improving prognosis in the TUBO murine breast cancer model. Another study revealed that Nelipepimut-S (E75) was a nine-amino acid peptide extracted from the HER2 protein capable of increasing the patient’s survival rate. Research from recent clinical trials has demonstrated positive effects of HER2-specific vaccinations that, when paired with chemo-drugs, could synergistically inhibit the recurrence of breast cancer, creating robust immunity and sustaining elevated CTL rates. One of the hurdles to the HER2-based vaccine is against TNBC subtypes due to the lack of HER-2 (ERBB2), progesterone, and estrogen receptors. Costa and colleagues suggested that combinations of HER2-based vaccines with pembrolizumab or nivolumab (immune checkpoint inhibitor antibodies) merit promising outcomes [62]. Combinations with other therapies might produce synergistic effects and resensitize other cancer cell death programs.
4.2. MUC-1
Transmembrane mucin-like glycoprotein Mucin 1 (MUC-1) is a type of glycoprotein comprising a single polypeptide chain with multiple oligosaccharide side chains with oxygen linkages to serine, proline, and threonine residues [63] frequently overexpressed in glandular and epithelial mammary, lung, and colon cancers [64]. MUC-1 overexpression can be used as a marker for cancer that suggests that the cancer is progressing [49].
Several studies have demonstrated that targeting MUC-1 was a successful option for the cancer vaccine because it is broadly dispersed in all tumors and cancers, including stem cell cancer [65]. Covalently bound to the TLR agonist, the completely synthetic glycosylated MUC-1 peptide vaccine exhibited good humoral and cellular immune responses [66]. The pioneered MUC-1 peptide vaccine study was performed in 1995 against patients with breast carcinoma. The majority of patients reacted to the medication, and no toxicity was found [67]. The extension of the research was carried out up until the Phase III clinical trial. A pilot Phase III analysis of 31 early Stage II breast cancer patients utilizing oxidized mannan-MUC-1 immunotherapy found that MUC1 immunotherapy is effective [68]. The MUC-1 peptide vaccine candidates have demonstrated an improved survival rate. MUC-1 is proposed as a potential biomarker to be targeted in breast cancer therapy because patients would typically overexpress the MUC-1 biomarker (approximately 90%) for immune system detection. Antibodies against MUC-1 can efficiently cause CTL and TLR. Hence, reasonable disease regulation is accomplished as patients produce strong antibody titers of MUC1 IgG. In clinical studies on women with Stage I and Stage II breast cancer, MUC-1 IgG and IgM antibodies were tested and assessed for their association with disease-specific survival [49][69]. MUC1 is a possible antigen to be utilized as a site-specific target for the deployment of therapeutic agents as a vaccine against MUC1 for breast carcinoma. Recent studies have shown that MUC1 can induce antigen-specific cellular and humoral responses not only to trigger MUC1-specific CD4+ and CD8+ T-cells but also to generate antibodies [65].
4.3. EphA
EphA is a type of transmembrane glycoprotein with tyrosine kinase (RTK) receptors on the surface that play a significant role as tumor-specific cell-surface receptors for drug-targeting sites. It is the largest group among tyrosine kinase receptor families, and among them, EphA2 is commonly overexpressed in breast cancer. The activation and overexpression of EphA2 frequently lead to its ligand-independent oncogenic and angiogenesis activation, which are triggered by dwindled contact with the ligand, ephrin-A (EphA2). Loss of the ligated EphA2 receptor decreases the intrinsic tumor-suppressive signaling pathways, accompanied by downregulation of the PI3K/Akt and the ERK pathways, thus decreasing the tumor volume and size.
As a therapeutic target, EphA2 receptors remain an essential marker. Overexpression of EphA2 receptors has been correlated with low survival in all patients with breast cancer subtypes due to the EphA2 activity that enhances tumorigenesis and the progression of metastases [70]. A monoclonal antibody (mAb EA5) has been studied to suppress EphA2 receptors in ER-positive breast cancer and to minimize cancer invasiveness [71]. The outcome was promising, and the study proceeded in the presence of tamoxifen. Furthermore, the monoclonal antibody EPhA2 can specifically target antigens and suppress the development of breast cancer cells and tumorigenesis.
YSA and SWL are peptide-based EpHAs that target EpHA2 receptors on the surface of tumor cells. Scarberry et al. [72] reported the success of using a magnetic CoFe2O4 nanoparticle-YSA peptide conjugate to extract ovarian cells from blood and fluid in mice. Even though the EpHA antigens are overexpressed in blood cancer and tumor cells, further exploration with regard to EphA as a peptide-based vaccine is very limited. Perhaps an EpHA-based vaccine does not elicit a potent immune response to eliminate various classifications of breast cancer. This may be triggered by cross-reactions between drugs and other proteins or by incomplete subcellular internalization of antibody-drug conjugates (ADCs). Thus, in order to address this issue, Salem et al. [73] proposed peptide-based targeting drugs that were less harmful but efficient and inexpensive. The aim of breast cancer therapy may be to merge EphA2 expression with carcinogenesis. Strategies focused on EphA2 targeting have been groundbreaking developments in therapeutic discovery. The targeted drug, for example, trastuzumab, is yet to be used; the concern of cardiotoxicity persists. Immunotherapy, similar to a cancer vaccination, tends to be an effective solution to treating metastatic breast carcinoma.
4.4. Survivin
Survivin, a 16.5 kDa intracellular acidic protein of 142 amino acids encoded by the BIRC5 gene, is a multifunctional protein that belongs to the smallest member of the inhibitory apoptosis protein family (IAPs). It regulates cell cycle progression through inhibition of the apoptosis pathway [74][75][76][77]. A high level of survivin expression is significantly associated with breast, urothelial, and colorectal cancer invasiveness and its low prognosis [78]. Survivin is undetectable in healthy tissue, indicating that it is exclusively presented as a biomarker when there is tumor transformation and acts as a transcriptome that is expressed in breast cancer. It seems to play a role in the antiapoptotic function of a protein, preventing the cell program from happening. A study by Ryan et al. [77] showed that a high level of survivin expression patterns is often associated with HER2/neu positive breast cancer and correlates with the prognosis.
This was confirmed by Lyu et al. [48], who reported that dysregulation of survivin was found in HER2/neu breast cancer, and survivin was identified as a desirable therapeutic target for blocking its IAP functions. A gene and immunotherapy named sepantronium bromide (YM155) have been developed to block survival. They provided a positive outcome for in vivo research by lowering the expression of survivin, raising the regression of the tumor, and prolonging the life of the mouse. However, YM155 failed to demonstrate an improvement in treatment response in metastatic non-small-cell lung cancer patients (NSCLC). This failure is probably due to the presence of multiple pathways linking survivin with other regulated proteins, making it more complicated [76][77].
Another research performed by Tanaka et al. [78] found that cytoplasm-responsive nanocarriers conjugated with a functional cell-penetrating peptide could facilitate the delivery of anti-vascular endothelial growth factor siRNA (siVEGF) complexes to tumor tissues after systemic injection and could elicit a potent anti-tumor effect. In a study from Rodel et al. [79], survivin as an antigen vaccine conferred peptide-specific CTL induction of urothelial cancers in patients without significant adverse reactions. On the other hand, the latest research indicated that the presence of the survivin antigen in breast carcinoma revealed a connection between expression and therapeutic outcomes [80].
4.5. SART3
SART3 is a tumor rejection antigen consisting of 3806 bp of nucleotides encoded by a 140-kilodalton (kDa) protein expressed in the cytosol of most of the cell proliferation and has been shown during gene transcription and mRNA synthesis of cancer cells. Similar to survivin, the SART3 oncogene is absent in normal tissues except for the fetal liver and testicles [81]. This antigen exhibits strong binding with HLA-A24-restricted CTL epitopes and may be useful for specific immunotherapy.
A Phase 1 clinical trial was recorded by Miyagi et al. [82] utilizing the SART3 peptide vaccine in colorectal cancer patients. The findings revealed a significant induction of cellular immune responses in 7 out of 11 patients. However, no explicit activation of the humoral immune response (IgG or IgE) has been recorded for peptides. SART3 led to the regulation of pro-inflammatory cytokine expression and the association of the degree of expression with malignancies and the prognosis for patients with breast cancer [83][84]. Given its positive outcome against SART antigen-expressed cancer, no clinical trials of SART-associated breast cancer vaccine goals have been reported.
4.6. CEA
CEA is a 180-kDa glycoprotein widely recognized as an oncofetal antigen that is found in numerous cancers, including colorectal, breast, gastric, pancreatic, and non-small cell lung cancers. CEA is one of the earliest tumor markers used to identify and anticipate the recurrence of tumors following surgical resection [85]. Its overexpression leads to the progression of the tumor. High secretions of CEA from cancer cells in the blood serum and over-expressed CEA on the surface of tumor cells make it accessible for use as a selective marker for cancer immunotherapy. CEA has been used as the foundation for numerous cancer vaccines, including DNA-based vaccines, dendritic cell-based vaccines, recombinant vector-based vaccines, protein-based vaccines, and anti-idiotype antibody vaccines, with the potential to induce both humoral and cell-mediated immunity that contributes to the killing of cancer cells [86]. Ojima et al. [87] demonstrated that genetically modified dendritic cells that express CEA administered simultaneously with interleukin 12 (IL-12), GM-CSF enhanced the therapeutic effects in CEA transgenic mice through the improvement of CEA-specific T-cell responses. Interestingly, the vaccination therapy eliminated colon cancer up to 2 × 103 mm3 sizes. Furthermore, no detrimental results were found after the experiment. The research performed by Gulley et al. [88] found that 9 out of 16 patients diagnosed with recombinant CEA-MUC-1-TRICOM poxviral-based robust tumor vaccines had an increase in both CD8+ and CD4+ immune responses. To sum up, targeting CEA could be a successful vaccination technique for the clinical application of peptide vaccines to achieve a positive antitumor response.
4.7. p53
p53 is a tumor suppressor protein that plays a vital role in regulating genomic stability by controlling the cell cycle and inducing apoptosis when cell damage is beyond repair. p53 mutations occur in about 18–25% of primary breast cancer, rendering them potential biomarkers for cancer immunotherapy. Missense mutations within the p53 gene could potentially cause the accumulation of mutant proteins within the cell nucleus through posttranscriptional modification. The prognosis value of the patient appeared to be associated with the p53 level. Approximately 80% of TNBC patients have been identified with high p53 gene levels, and so far, no immunotherapeutic medication scientifically used for TNBC has been proven to develop a peptide-derived vaccine [89]. Many 2-sulfonyl pyrimidine compounds, such as PK11007 and PK11000, are successful in killing cancer cells by explicitly attacking mutant P53 thiol groups, thereby reducing oxidative stress levels (e.g., ROS) and eventually retaining a redox state [90][91].
PRIMA-1MET (APR-246) has been clinically studied in a Phase I clinical trial and is currently undergoing more clinical review. It inhibits cancer cell growth by targeting mutant p53 and inactivating it in triple-negative breast cancer (TNBC) [92]. In the Phase I/II clinical trial, ten colorectal cancer patients were vaccinated with p53-derived synthetic long peptides (SLPs). Rapid p53-specific T-cell responses were observed in blood samples obtained six months after the last vaccine [93]. Although the theory suggests that SLP would activate a high level of T cells in vaccinated patients, the clinical findings have unfortunately not been compatible. Perhaps targeting p53 alone is not enough to eliminate breast cancer cells. Thus, multiple peptides that target multiple antigens while stimulating multi-antigenic immune responses tend to be the right approach to improving the immunogenicity and clinical efficacy of p53-directed immunotherapies [94][95].
References
- World Health Organization. Breast Cancer: Prevention and Control. Available online: https://www.who.int/cancer/prevention/diagnosis-screening/breast-cancer/en/ (accessed on 10 December 2020).
- Reddington, R.; Galer, M.; Hagedorn, A.; Liu, P.; Barrack, S.; Husain, E.; Sharma, R.; Speirs, V.; Masannat, Y. Incidence of Male Breast Cancer in Scotland over a Twenty-Five-Year Period (1992–2017). Eur. J. Surg. Oncol. 2020, 46, 1546–1550.
- Giordano, S.H. Breast Cancer in Men. N. Engl. J. Med. 2018, 378, 2311–2320.
- Gargiulo, P.; Pensabene, M.; Milano, M.; Arpino, G.; Giuliano, M.; Forestieri, V.; Condello, C.; Lauria, R.; De Placido, S. Long-Term Survival and BRCA Status in Male Breast Cancer: A Retrospective Single-Center Analysis. BMC Cancer 2016, 16, 375.
- Gaddam, S.; Heller, S.L.; Babb, J.S.; Gao, Y. Male Breast Cancer Risk Assessment and Screening Recommendations in High-Risk Men Who Undergo Genetic Counseling and Multigene Panel Testing. Clin. Breast Cancer 2021, 21, e74–e79.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29.
- Shah, R.; Rosso, K.; David Nathanson, S. Pathogenesis, Prevention, Diagnosis and Treatment of Breast Cancer. World J. Clin. Oncol. 2014, 5, 283–298.
- Ly, D.; Forman, D.; Ferlay, J.; Brinton, L.A.; Cook, M.B. An International Comparison of Male and Female Breast Cancer Incidence Rates. Int. J. Cancer 2013, 132, 1918–1926.
- Chakraborty, S.; Rahman, T. The Difficulties in Cancer Treatment. Ecancermedicalscience 2012, 6, ed16.
- Ekici, S.; Jawzal, H. Breast Cancer Diagnosis Using Thermography and Convolutional Neural Networks. Med. Hypotheses 2020, 137, 109542.
- Sánchez-Bermúdez, A.I.; Sarabia-Meseguer, M.D.; García-Aliaga, A.; Marín-Vera, M.; Macías-Cerrolaza, J.A.; Henaréjos, P.S.; Guardiola-Castillo, V.; la Peña, F.A.-D.; Alonso-Romero, J.L.; Noguera-Velasco, J.A.; et al. Mutational Analysis of RAD51C and RAD51D Genes in Hereditary Breast and Ovarian Cancer Families from Murcia (Southeastern Spain). Eur. J. Med. Genet. 2018, 61, 355–361.
- Karami, F.; Mehdipour, P. A Comprehensive Focus on Global Spectrum of BRCA1 and BRCA2 Mutations in Breast Cancer. Biomed. Res. Int. 2013, 2013, 928562.
- Ayala de la Peña, F.; Andrés, R.; Garcia-Sáenz, J.A.; Manso, L.; Margelí, M.; Dalmau, E.; Pernas, S.; Prat, A.; Servitja, S.; Ciruelos, E. SEOM Clinical Guidelines in Early Stage Breast Cancer (2018). Clin. Transl. Oncol. 2019, 21, 18–30.
- Suter, R.; Marcum, J.A. The Molecular Genetics of Breast Cancer and Targeted Therapy. Biologics 2007, 1, 241–258.
- Townsend, D.M.; Tew, K.D. The Role of Glutathione-S-Transferase in Anti-Cancer Drug Resistance. Oncogene 2003, 22, 7369–7375.
- Aziz, M.H.; Ahmad, A. Epigenetic Basis of Cancer Drug Resistance. Cancer Drug Resist. 2020, 3, 113–116.
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel Immune Checkpoint Targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155.
- Ji, X.; Lu, Y.; Tian, H.; Meng, X.; Wei, M.; Cho, W.C. Chemoresistance Mechanisms of Breast Cancer and Their Countermeasures. Biomed. Pharmacother. 2019, 114, 108800.
- Crespo, I.; Götz, L.; Liechti, R.; Coukos, G.; Doucey, M.A.; Xenarios, I. Identifying Biological Mechanisms for Favorable Cancer Prognosis Using Non-Hypothesis-Driven Iterative Survival Analysis. NPJ Syst. Biol. Appl. 2016, 2, 16037.
- Anfray, C.; Ummarino, A.; Andón, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2020, 9, 46.
- Davis, R.J.; Van Waes, C.; Allen, C.T. Overcoming Barriers to Effective Immunotherapy: MDSCs, TAMs, and Tregs as Mediators of the Immunosuppressive Microenvironment in Head and Neck Cancer. Oral Oncol. 2016, 58, 59–70.
- Lin, Y.; Xu, J.; Lan, H. Tumor-Associated Macrophages in Tumor Metastasis: Biological Roles and Clinical Therapeutic Applications. J. Hematol. Oncol. 2019, 12, 76.
- Schmitt, N.; Bustamante, J.; Bourdery, L.; Bentebibel, S.E.; Boisson-Dupuis, S.; Hamlin, F.; Tran, M.V.; Blankenship, D.; Pascual, V.; Savino, D.A.; et al. IL-12 Receptor Β1 Deficiency Alters in Vivo T Follicular Helper Cell Response in Humans. Blood 2013, 121, 3375–3385.
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The Immunosuppressive Tumour Network: Myeloid-Derived Suppressor Cells, Regulatory T Cells and Natural Killer T Cells. Immunology 2013, 138, 105–115.
- Li, S.; Yao, M.; Niu, C.; Liu, D.; Tang, Z.; Gu, C.; Zhao, H.; Ke, J.; Wu, S.; Wang, X.; et al. Inhibition of MCF-7 Breast Cancer Cell Proliferation by a Synthetic Peptide Derived from the C-Terminal Sequence of Orai Channel. Biochem. Biophys. Res. Commun. 2019, 516, 1066–1072.
- Gatti-Mays, M.E.; Balko, J.M.; Gameiro, S.R.; Bear, H.D.; Prabhakaran, S.; Fukui, J.; Disis, M.L.; Nanda, R.; Gulley, J.L.; Kalinsky, K.; et al. If We Build It They Will Come: Targeting the Immune Response to Breast Cancer. NPJ Breast Cancer 2019, 5, 37.
- Gun, S.Y.; Lee, S.W.L.; Sieow, J.L.; Wong, S.C. Targeting Immune Cells for Cancer Therapy. Redox Biol. 2019, 25, 101174.
- Zhang, Y.; Zhang, Z. The History and Advances in Cancer Immunotherapy: Understanding the Characteristics of Tumor-Infiltrating Immune Cells and Their Therapeutic Implications. Cell Mol. Immunol. 2020, 17, 807–821.
- Criscitiello, C.; Viale, G.; Curigliano, G. Peptide Vaccines in Early Breast Cancer. Breast 2019, 44, 128–134.
- Yamaguchi, Y.; Yamaue, H.; Okusaka, T.; Okuno, K.; Suzuki, H.; Fujioka, T.; Otsu, A.; Ohashi, Y.; Shimazawa, R.; Nishio, K.; et al. Guidance for Peptide Vaccines for the Treatment of Cancer. Cancer Sci. 2014, 105, 924–931.
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA J. Am. Med. Assoc. 2019, 321, 288–300.
- Shi, W.; Qiu, Q.; Tong, Z.; Guo, W.; Zou, F.; Feng, Z.; Wang, Y.; Huang, W.; Qian, H. Synthetic Tumor-Specific Antigenic Peptides with a Strong Affinity to HLA-A2 Elicit Anti-Breast Cancer Immune Response through Activating CD8+ T Cells. Eur. J. Med. Chem. 2020, 189, 112051.
- Nguyen, T.L.; Choi, Y.; Kim, J. Mesoporous Silica as a Versatile Platform for Cancer Immunotherapy. Adv. Mater. 2019, 31, e1803953.
- De Temmerman, M.L.; Rejman, J.; Demeester, J.; Irvine, D.J.; Gander, B.; De Smedt, S.C. Particulate Vaccines: On the Quest for Optimal Delivery and Immune Response. Drug Discov. Today 2011, 16, 569–582.
- Nejad, A.E.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Javanmard, S.H.; Taherian, M.; Ahmadlou, M.; et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Intl. 2021, 21, 62.
- Solinas, C.; Aiello, M.; Migliori, E.; Willard-Gallo, K.; Emens, L.A. Breast cancer vaccines: Heeding the lessons of the past to guide a path forward. Cancer Treat. Rev. 2020, 84, 101947.
- de Paula Peres, L.; da Luz, F.A.C.; dos Anjos Pultz, B.; Brígido, P.C.; de Araújo, R.A.; Goulart, L.R.; Silva, M.J.B. Peptide Vaccines in Breast Cancer: The Immunological Basis for Clinical Response. Biotechnol. Adv. 2015, 33, 1868–1877.
- Criscitiello, C. Tumor-Associated Antigens in Breast Cancer. Breast Care 2012, 7, 262–266.
- Thundimadathil, J. Cancer Treatment Using Peptides: Current Therapies and Future Prospects. J. Amino Acids 2012, 2012, 1–13.
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of Antigen Processing. Annu. Rev. Immunol. 2013, 31, 443–473.
- Zhang, L.; Huang, Y.; Lindstrom, A.R.; Lin, T.Y.; Lam, K.S.; Li, Y. Peptide-Based Materials for Cancer Immunotherapy. Theranostics 2019, 9, 7807–7825.
- Santoni, D. Viral Peptides-MHC Interaction: Binding Probability and Distance from Human Peptides. J. Immunol. Methods 2018, 459, 35–43.
- Jones, N.D.; Van Maurik, A.; Hara, M.; Spriewald, B.M.; Witzke, O.; Morris, P.J.; Wood, K.J. CD40-CD40 Ligand-Independent Activation of CD8+ T Cells Can Trigger Allograft Rejection. J. Immunol. 2000, 165, 1111–1118.
- Tay, N.Q.; Lee, D.C.P.; Chua, Y.L.; Prabhu, N.; Gascoigne, N.R.J.; Kemeny, D.M. CD40L Expression Allows CD8+ T Cells to Promote Their Own Expansion and Differentiation through Dendritic Cells. Front. Immunol. 2017, 8, 1484.
- Wong, K.L.; Tang, L.F.M.; Lew, F.C.; Wong, H.S.K.; Chua, Y.L.; MacAry, P.A.; Kemeny, D.M. CD44high Memory CD8 T Cells Synergize with CpG DNA to Activate Dendritic Cell IL-12p70 Production. J. Immunol. 2009, 183, 41–50.
- Ikeda, H.; Shiku, H. Immunotherapy of Solid Tumor: Perspectives on Vaccine and Cell Therapy. Nihon Rinsho 2012, 70, 2043–2050.
- Costa, R.L.B.; Czerniecki, B.J. Clinical Development of Immunotherapies for HER2+ Breast Cancer: A Review of HER2-Directed Monoclonal Antibodies and Beyond. NPJ Breast Cancer 2020, 6, 10.
- Lyu, H.; Huang, J.; He, Z.; Liu, B. Epigenetic Mechanism of Survivin Dysregulation in Human Cancer. Sci. China Life Sci. 2018, 61, 808–814.
- Genitsch, V.; Zlobec, I.; Thalmann, G.N.; Fleischmann, A. MUC1 Is Upregulated in Advanced Prostate Cancer and Is an Independent Prognostic Factor. Prostate Cancer Prostatic. Dis. 2016, 19, 242–247.
- Malonis, R.J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020, 120, 3210–3229.
- Kim, I.; Sanchez, K.; McArthur, H.L.; Page, D. Immunotherapy in Triple-Negative Breast Cancer: Present and Future. Curr. Breast Cancer Rep. 2019, 11, 259–271.
- Mittendorf, E.A.; Lu, B.; Melisko, M.; Hiller, J.P.; Bondarenko, I.; Brunt, A.M.; Sergii, G.; Petrakova, K.; Peoples, G.E. Efficacy and Safety Analysis of Nelipepimut-S Vaccine to Prevent Breast Cancer Recurrence: A Randomized, Multicenter, Phase III Clinical Trial. Clin. Cancer Res. 2019, 25, 4248–4254.
- Farzad, N.; Barati, N.; Momtazi-Borojeni, A.A.; Yazdani, M.; Arab, A.; Razazan, A.; Shariat, S.; Mansourian, M.; Abbasi, A.; Saberi, Z.; et al. P435 HER2/Neu-Derived Peptide Conjugated to Liposomes Containing DOPE as an Effective Prophylactic Vaccine Formulation for Breast Cancer. Artif. Cells Nanomed. Biotechnol. 2019, 47, 664–672.
- Furrer, D.; Sanschagrin, F.; Jacob, S.; Diorio, C. Advantages and Disadvantages of Technologies for HER2 Testing in Breast Cancer Specimens: Table 1. Am. J. Clin. Pathol. 2015, 144, 686–703.
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human Breast Cancer: Correlation of Relapse and Survival with Amplification of the HER-2/Neu Oncogene. Science 1987, 235, 177–182.
- Ayoub, N.M.; Al-Shami, K.M.; Yaghan, R.J. Immunotherapy for HER2-Positive Breast Cancer: Recent Advances and Combination Therapeutic Approaches. Breast Cancer Targets Ther. 2019, 11, 53–69.
- Krasniqi, E.; Barchiesi, G.; Pizzuti, L.; Mazzotta, M.; Venuti, A.; Maugeri-Saccà, M.; Sanguineti, G.; Massimiani, G.; Sergi, D.; Carpano, S.; et al. Immunotherapy in HER2-Positive Breast Cancer: State of the Art and Future Perspectives. J. Hematol. Oncol. 2019, 12, 1–26.
- Katzorke, N.; Rack, B.K.; Haeberle, L.; Neugebauer, J.K.; Melcher, C.A.; Hagenbeck, C.; Forstbauer, H.; Ulmer, H.U.; Soeling, U.; Kreienberg, R.; et al. Prognostic Value of HER2 on Breast Cancer Survival. J. Clin. Oncol. 2013, 31, 640.
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc Receptors Modulate in Vivo Cytoxicity against Tumor Targets. Nat. Med. 2000, 6, 443–446.
- Wang, J.; Xu, B. Targeted Therapeutic Options and Future Perspectives for Her2-Positive Breast Cancer. Signal Transduct. Target. Ther. 2019, 4, 3305.
- Mansourian, M.; Badiee, A.; Jalali, S.A.; Shariat, S.; Yazdani, M.; Amin, M.; Jaafari, M.R. Effective Induction of Anti-Tumor Immunity Using P5 HER-2/Neu Derived Peptide Encapsulated in Fusogenic DOTAP Cationic Liposomes Co-Administrated with CpG-ODN. Immunol. Lett. 2014, 162, 87–93.
- Costa, R.L.B.; Soliman, H.; Czerniecki, B.J. The Clinical Development of Vaccines for HER2+ Breast Cancer: Current Landscape and Future Perspectives. Cancer Treat. Rev. 2017, 61, 107–115.
- Richman, C.M.; DeNardo, S.J. Systemic Radiotherapy in Metastatic Breast Cancer Using 90Y-Linked Monoclonal MUC-1 Antibodies. Crit. Rev. Oncol. Hematol. 2001, 38, 25–35.
- Nath, S.; Mukherjee, P. MUC1: A Multifaceted Oncoprotein with a Key Role in Cancer Progression. Trends Mol. Med. 2014, 20, 332–342.
- Kovjazin, R.; Horn, G.; Smorodinsky, N.I.; Shapira, M.Y.; Carmon, L. Cell Surface-Associated Anti-MUC1-Derived Signal Peptide Antibodies: Implications for Cancer Diagnostics and Therapy. PLoS ONE 2014, 9, e85400.
- Lakshminarayanan, V.; Thompson, P.; Wolfert, M.A.; Buskas, T.; Bradley, J.M.; Pathangey, L.B.; Madsen, C.S.; Cohen, P.A.; Gendler, S.J.; Boons, G.J. Immune Recognition of Tumor-Associated Mucin MUC1 Is Achieved by a Fully Synthetic Aberrantly Glycosylated MUC1 Tripartite Vaccine. Proc. Natl. Acad. Sci. USA 2012, 109, 261–266.
- Xing, P.X.; Michael, M.; Apostolopoulos, V.; Prenzoska, J.; Marshall, C.; Bishop, J.; McKenzie, I.F.C. Phase I Study of Synthetic MUC1 Peptides in Breast Cancer. Int. J. Oncol. 1995, 6, 1283–1289.
- Apostolopoulos, V.; Pietersz, G.A.; Tsibanis, A.; Tsikkinis, A.; Drakaki, H.; Loveland, B.E.; Piddlesden, S.J.; Plebanski, M.; Pouniotis, D.S.; Alexis, M.N.; et al. Pilot Phase III Immunotherapy Study in Early-Stage Breast Cancer Patients Using Oxidized Mannan-MUC1 . Breast Cancer Res. 2006, 8, R27.
- Jeong, S.; Park, M.J.; Song, W.; Kim, H.S. Current Immunoassay Methods and Their Applications to Clinically Used Biomarkers of Breast Cancer. Clin. Biochem. 2020, 78, 43–57.
- Brantley-Sieders, D.M.; Zhuang, G.; Hicks, D.; Wei, B.F.; Hwang, Y.; Cates, J.M.M.; Coffman, K.; Jackson, D.; Bruckheimer, E.; Muraoka-Cook, R.S.; et al. The Receptor Tyrosine Kinase EphA2 Promotes Mammary Adenocarcinoma Tumorigenesis and Metastatic Progression in Mice by Amplifying ErbB2 Signaling. J. Clin. Investig. 2008, 118, 64–78.
- Gökmen-Polar, Y.; Toroni, R.A.; Hocevar, B.A.; Badve, S.; Zhao, Q.; Shen, C.; Bruckheimer, E.; Kinch, M.S.; Miller, K.D. Dual Targeting of EphA2 and ER Restores Tamoxifen Sensitivity in ER/EphA2-Positive Breast Cancer. Breast Cancer Res. Treat. 2011, 127, 375–384.
- Scarberry, K.E.; Dickerson, E.B.; McDonald, J.F.; Zhang, Z.J. Magnetic Nanoparticle-Peptide Conjugates for in Vitro and in Vivo Targeting and Extraction of Cancer Cells. J. Am. Chem. Soc. 2008, 130, 10258–10262.
- Salem, A.F.; Wang, S.; Billet, S.; Chen, J.F.; Udompholkul, P.; Gambini, L.; Baggio, C.; Tseng, H.R.; Posadas, E.M.; Bhowmick, N.A.; et al. Reduction of Circulating Cancer Cells and Metastases in Breast-Cancer Models by a Potent EphA2-Agonistic Peptide-Drug Conjugate. J. Med. Chem. 2018, 61, 2052–2061.
- Guo, Z.; He, B.; Yuan, L.; Dai, W.; Zhang, H.; Wang, X.; Wang, J.; Zhang, X.; Zhang, Q. Dual Targeting for Metastatic Breast Cancer and Tumor Neovasculature by EphA2-Mediated Nanocarriers. Int. J. Pharm. 2015, 493, 380–389.
- Jha, K.; Shukla, M.; Pandey, M. Survivin Expression and Targeting in Breast Cancer. Surg. Oncol. 2012, 21, 125–131.
- Altieri, D.C. Survivin, Cancer Networks and Pathway-Directed Drug Discovery. Nat. Rev. Cancer. 2008, 8, 61–70.
- Ryan, B.M.; Konecny, G.E.; Kahlert, S.; Wang, H.J.; Untch, M.; Meng, G.; Pegram, M.D.; Podratz, K.C.; Crown, J.; Slamon, D.J.; et al. Survivin Expression in Breast Cancer Predicts Clinical Outcome and Is Associated with HER2, VEGF, Urokinase Plasminogen Activator and PAI-1. Ann. Oncol. 2006, 17, 597–604.
- Tanaka, K.; Kanazawa, T.; Horiuchi, S.; Ando, T.; Sugawara, K.; Takashima, Y.; Seta, Y.; Okada, H. Cytoplasm-responsive nanocarriers conjugated with a functional cell-penetrating peptide for systemic siRNA delivery. Intl. J. Pharm. 2013, 455, 40–47.
- Rodel, F.; Sprenger, T.; Kaina, B.; Liersch, T.; Rodel, C.; Fulda, S.; Hehlgans, S. Survivin as a Prognostic/Predictive Marker and Molecular Target in Cancer Therapy. Curr. Med. Chem. 2012, 19, 3679–3688.
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A Unique Target for Tumor Therapy. Cancer Cell Int. 2016, 16, 49.
- Tsuda, N.; Murayama, K.; Ishida, H.; Matsunaga, K.; Komiya, S.; Itoh, K.; Yamada, A. Expression of a Newly Defined Tumor-Rejection Antigen SART3 in Musculoskeletal Tumors and Induction of HLA Class I-Restricted Cytotoxic T Lymphocytes by SART3-Derived Peptides. J. Orthop. Res. 2001, 19, 346–351.
- Miyagi, Y.; Sasatomi, T.; Mine, T.; Isomoto, H.; Shirouzu, K.; Yamana, H.; Imai, N.; Yamada, A.; Katagiri, K.; Muto, A.; et al. Induction of Cellular Immune Responses to Tumor Cells and Peptides in Colorectal Cancer Patients by Vaccination with SART3 Peptides. Clin. Cancer Res. 2001, 7, 3950–3962.
- Sherman, E.J.; Mitchell, D.C.; Garner, A.L. The RNA-Binding Protein SART3 Promotes MiR-34a Biogenesis and G1 Cell Cycle Arrest in Lung Cancer Cells. J. Biol. Chem. 2019, 294, 17188–17196.
- Timani, K.A.; Gyorffy, B.; Liu, Y.; Mohammad, K.S.; He, J.J. Tip110/SART3 Regulates IL-8 Expression and Predicts the Clinical Outcomes in Melanoma. Mol. Cancer 2018, 17, 124.
- Lee, J.H.; Lee, S.W. The Roles of Carcinoembryonic Antigen in Liver Metastasis and Therapeutic Approaches. Gastroenterol. Res. Pract. 2017, 2017, 7521987.
- Turriziani, M.; Fantini, M.; Benvenuto, M.; Izzi, V.; Masuelli, L.; Sacchetti, P.; Modesti, A.; Bei, R. Carcinoembryonic Antigen (CEA)-Based Cancer Vaccines: Recent Patents and Antitumor Effects from Experimental Models to Clinical Trials. Recent. Pat. Anticancer Drug Discov. 2012, 7, 265–296.
- Ojima, T.; Iwahashi, M.; Nakamura, M.; Matsuda, K.; Nakamori, M.; Ueda, K.; Naka, T.; Ishida, K.; James Primus, F.; Yamaue, H. Successful Cancer Vaccine Therapy for Carcinoembryonic Antigen (CEA)-Expressing Colon Cancer Using Genetically Modified Dendritic Cells That Express CEA and T Helper-Type 1 Cytokines in CEA Transgenic Mice. Int. J. Cancer 2007, 120, 585–593.
- Gulley, J.L.; Arlen, P.M.; Tsang, K.-Y.; Yokokawa, J.; Palena, C.; Poole, D.J.; Remondo, C.; Cereda, V.; Jones, J.L.; Pazdur, M.P.; et al. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin. Cancer Res. 2008, 14, 3060–3069.
- Liu, D.; Guo, P.; McCarthy, C.; Wang, B.; Tao, Y.; Auguste, D. Peptide Density Targets and Impedes Triple Negative Breast Cancer Metastasis. Nat. Commun. 2018, 9, 2612.
- Zhang, Q.; Bergman, J.; Wiman, K.G.; Bykov, V.J.N. Role of Thiol Reactivity for Targeting Mutant P53. Cell Chem. Biol. 2018, 25, 1219–1230.
- Bauer, M.R.; Joerger, A.C.; Fersht, A.R. 2-Sulfonylpyrimidines: Mild Alkylating Agents with Anticancer Activity toward P53-Compromised Cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5271–E5280.
- Synnott, N.C.; Bauer, M.R.; Madden, S.; Murray, A.; Klinger, R.; O’Donovan, N.; O’Connor, D.; Gallagher, W.M.; Crown, J.; Fersht, A.R.; et al. Mutant P53 as a Therapeutic Target for the Treatment of Triple-Negative Breast Cancer: Preclinical Investigation with the Anti-P53 Drug, PK11007. Cancer Lett. 2018, 414, 99–106.
- Nijman, H.W.; Vermeij, R.; Leffers, N.; Van Der Burg, S.H.; Melief, C.J.; Daemen, T. Immunological and Clinical Effects of Vaccines Targeting P53-Overexpressing Malignancies. J. Biomed. Biotechnol. 2011, 2011, 702146.
- Vijayan, V.; Mohapatra, A.; Uthaman, S.; Park, I.K. Recent Advances in Nanovaccines Using Biomimetic Immunomodulatory Materials. Pharmaceutics 2019, 11, 534.
- Chianese-Bullock, K.A.; Lewis, S.T.; Sherman, N.E.; Shannon, J.D.; Slingluff, C.L. Multi-Peptide Vaccines Vialed as Peptide Mixtures Can Be Stable Reagents for Use in Peptide-Based Immune Therapies. Vaccine 2009, 27, 1764–1770.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
531
Revisions:
2 times
(View History)
Update Date:
27 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No