Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexandra Barbouti | -- | 4055 | 2023-06-26 15:18:46 | | | |
| 2 | Sirius Huang | Meta information modification | 4055 | 2023-06-28 03:09:45 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nousis, L.; Kanavaros, P.; Barbouti, A. Oxidative Stress-Induced Cellular Senescence. Encyclopedia. Available online: https://encyclopedia.pub/entry/46062 (accessed on 27 July 2026).
Nousis L, Kanavaros P, Barbouti A. Oxidative Stress-Induced Cellular Senescence. Encyclopedia. Available at: https://encyclopedia.pub/entry/46062. Accessed July 27, 2026.
Nousis, Lambros, Panagiotis Kanavaros, Alexandra Barbouti. "Oxidative Stress-Induced Cellular Senescence" Encyclopedia, https://encyclopedia.pub/entry/46062 (accessed July 27, 2026).
Nousis, L., Kanavaros, P., & Barbouti, A. (2023, June 26). Oxidative Stress-Induced Cellular Senescence. In Encyclopedia. https://encyclopedia.pub/entry/46062
Nousis, Lambros, et al. "Oxidative Stress-Induced Cellular Senescence." Encyclopedia. Web. 26 June, 2023.
Copy Citation
Cellular senescence, a cell state characterized by a generally irreversible cell cycle arrest, is implicated in various physiological processes and a wide range of age-related pathologies. Oxidative stress, a condition caused by an imbalance between the production and the elimination of reactive oxygen species (ROS) in cells and tissues, is a common driver of cellular senescence. ROS encompass free radicals and other molecules formed as byproducts of oxygen metabolism, which exhibit varying chemical reactivity.
oxidative stress
reactive oxygen species
labile iron
cellular senescence
telomeres
DNA-damage
lipofuscin
mitochondria
lysosomes
1. Cellular Senescence
1.1. A Brief Historical Overview and Some General Aspects of Cellular Senescence
Cellular senescence is a fundamental biological process, mainly characterized by a prolonged and generally irreversible arrest of cell proliferation. It was originally described six decades ago when Leonard Hayflick and Paul Sidney Moorhead showed in their seminal paper that normal diploid cells isolated from human fetuses gradually lost their ability to divide in vitro, despite the presence of ample space, growth factors, and nutrients in the culture medium [1]. Notably, the non-dividing cells remained alive and metabolically active over a long period until the eventual degeneration of the culture. This phenomenon was termed senescence, from the Latin word senex, which means “growing old”, as the authors assumed that it could play a causal role in organismal ageing. The underlying molecular explanation came a few decades later with the discovery that permanent cell cycle arrest is the result of telomere attrition [2]. Telomeres—from the Greek words telos (end) and meros (part) [3]—are the terminal chromatin structures that mask the natural ends of eukaryotic chromosomes and protect them from degradation and fusion [4][5]. However, the DNA at the very end of a linear chromosome (that is, the telomere) cannot be fully copied during replication. This phenomenon, commonly referred to as the “end-replication problem”, was observed in 1972 by James Watson [6]. At nearly the same time, Alexei Olovnikov first postulated that due to their incomplete replication, telomeres progressively becoming shorter with each cell doubling, and when this shortening reaches some threshold value, cells stop dividing and senesce [7]. For a long time, there was a controversy about whether telomere shortening and senescence had any relevance to organismal ageing or whether they were just a tissue culture phenomenon. This changed abruptly during the past few decades, as several studies in humans have shown that telomeres shorten with age, and shortening is associated with increased mortality risk [5][8][9][10]. Similarly, research in animal models revealed that short telomeres led to ageing-associated degenerative defects and loss of organismal viability [11][12][13], while telomere elongation delayed normal ageing [14].
Moreover, it became clear that apart from telomere attrition, several diverse signals can elicit senescence phenotypes. For instance, various stressors including oxidants, genotoxic agents, hypoxia, and aberrant activation of oncogenes trigger premature senescence, usually in the absence of telomere shortening [15][16][17]. Besides, the role of senescence has recently expanded beyond the contexts of telomere attrition or stressful insults, as cells with senescent features have been identified during embryogenesis, to particular transient anatomical structures, and during specific time windows of development in evolutionary distant organisms ranging from mammals to fish [16][18].
Conceivably, senescence as a stable growth-arrest program has evolved as a mechanism to prevent the propagation of unwanted cells and also to trigger their clearance by the immune system [16][19]. During development, senescence regulates embryonic growth and patterning [16][18], while in adulthood, senescence counteracts the uncontrolled growth of damaged cells and is a crucial barrier against cancer progression. Within this framework, senescence represents a beneficial response, essential for maintaining tissue homeostasis. Yet, senescent cells are not inert; they remain alive for prolonged periods and release factors that can harm neighboring healthy cells and the very cells that produce these factors. Accordingly, the accumulation of senescent cells within tissues and organs—when the immune system fails to efficiently remove them or when senescence persists—contributes to tissue dysfunction and gives rise to pathological manifestations, organ ageing, and age-related diseases [16][20][21][22][23]. Comprehensively, senescence is implicated in several physiological functions, but also in a wide variety of age-related pathologies, and displays beneficial effects as well as detrimental consequences on organismal health, depending on the context.
1.2. Features of Senescent Cells
A prominent feature of senescent cells is the irreversible growth arrest (Figure 1), which is largely mediated through either one or both the p53/p21 and the p16Ink4a/pRB signaling pathways [15][24]. Both pathways involve multiple upstream regulators and downstream effectors, and cross-regulate each other [25]. In addition, to some extent, they respond to different stimuli; DNA-damaging signals such as oxidants, and oncogenic or genotoxic stress, largely trigger senescence via the p53/p21 axis [24]. Upon entering senescence, cells display an abnormally enlarged and flat morphology with an increase in the cytoplasm-to-nucleus ratio. Moreover, they exhibit deregulated metabolism and accelerated accumulation of damage to DNA, proteins, and lipids (Figure 1) [26][27][28]. Last, but not least, senescent cells adopt a hyper-secretory phenotype, known as the senescence-associated secretory phenotype (SASP) (Figure 1) [15][24]. The exact composition remains elusive and varies significantly [29]; however, the SASP mainly includes proinflammatory cytokines, chemokines, angiogenic factors, matrix metalloproteinases, and ROS. Although SASP facilitates tissue homeostasis, when chronic, it mediates the pathophysiological effects of senescence; by acting in an autocrine or paracrine mode, the secreted factors reinforce and spread senescence, promote persistent chronic inflammation, stimulate tumorigenesis, impair the function of stem and progenitor cells, etc. [15][24]. This explains why senescent cells, although present in relatively low numbers in vivo, have such devastating consequences.
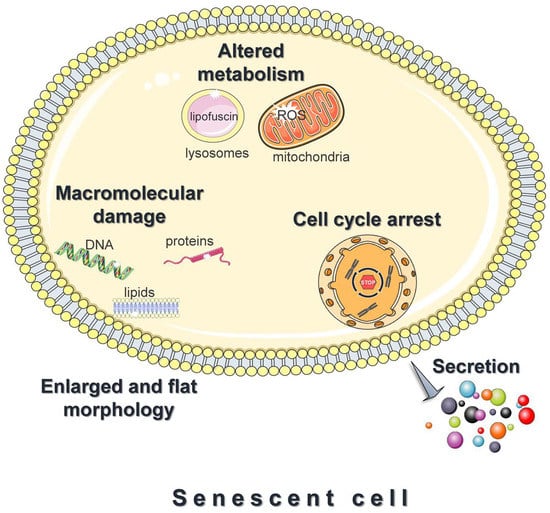
Figure 1. Features of senescent cells. Cellular senescence is the essentially irreversible growth arrest that occurs when cells experience stressful insults or certain physiological processes. Common features of senescent cells are an abnormally enlarged and flat morphology, altered metabolism, and the accelerated accumulation of damage to DNA, proteins, and lipids. Moreover, senescent cells adopt a hyper-secretory phenotype, known as the senescence-associated secretory phenotype (SASP). The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
2. Intracellular Damage in Oxidative Stress-Induced Cellular Senescence: Is Iron Involved?
A well-known feature of cellular senescence is its intimate association with macromolecular damage: various damaging insults signal cellular senescence, while the accelerated accumulation of macromolecular damage is an almost universal hallmark of senescent cells [15]. The senescent state is also characterized by an altered metabolic profile. Lysosomes exhibit progressively deteriorated function and accumulate lipofuscin, a non-degradable aggregate of intracellular catabolism [19]. Lysosomal dysfunction attenuates mitophagy (a mitochondrial quality control mechanism that degrades damaged mitochondria) leading to the accumulation of damaged mitochondria which often produce elevated ROS levels [30]. This, in turn, targets lysosomes and enhances macromolecular damage, aggravating senescence phenotypes [30].
ROS are common drivers of cellular senescence and trigger all the aforementioned senescence traits. The term ROS encompasses species with varying chemical reactivity. For instance, O2•− and H2O2 are not reactive enough to directly oxidize cellular macromolecules and induce damage. However, in the presence of labile iron, peroxides can be converted to extremely reactive free radicals such as HO• and RO•. These free radicals are short-lived and highly reactive; thus, they attack and oxidize their targets close to the site of their formation (diffusion-controlled reactivity). Consequently, when peroxides are elevated (oxidative stress conditions), labile iron can trigger single- or double-strand breakage and oxidative modifications to DNA bases, oxidative damage to proteins, and peroxidation of lipids [31]; all these effects can elicit cellular senescence. Moreover, labile iron is not uniformly distributed within cellular compartments. Lysosomes and mitochondria, organelles that undergo the most remarkable alterations during senescence, contain higher amounts of labile iron compared with other cellular compartments [32][33]. The high iron content makes these organelles extra sensitive in oxidative stress conditions [34][35][36][37]. Considering the above, the implication of labile iron in the mechanisms of oxidative stress-induced cellular senescence is a quite reasonable assumption.
2.1. DNA Damage
2.1.1. Activation of the DNA Damage Response Pathway
Nuclear DNA, although bearing hereditary information, is particularly susceptible to oxidative stress. ROS cause various lesions to DNA, such as single- and double-strand breakage, and oxidation to purines and pyrimidines [38]. Eukaryotic cells are equipped with a complex network known as the DNA damage response (DDR), which protects the genome from detrimental insults [39]. The DDR machinery can detect DNA lesions and set the cell fate depending on the cell type and the severity of the damage [40][41][42]. When genotoxic agents such as ROS trigger DNA damage, the DDR machinery temporarily arrests cell proliferation and gives time for efficient DNA repair. After the damage is repaired, the cell exits from the arrest phase and resumes cell cycle progression. However, if the damage is severe and irreparable, the prolonged DDR activation may cause a permanent cell cycle arrest and the senescence phenotype, or otherwise may initiate cell death programs (apoptosis) [40][41][42].
The DDR initiates with the recognition of the DNA damage by sensor proteins and the activation of signaling protein kinases such as ataxia-telangiectasia-mutated (ATM) and ATM- and Rad3-related (ATR) kinases. ATM and ATR phosphorylate histone γ-H2A.X, which regulates various downstream mediators (such as 53BP1) to coordinate the DDR machinery, and the downstream kinases CHK1 and CHK2, which ultimately transmit the damaging signal to p53. The p53 transcription factor regulates the expression of the cyclin-dependent kinase (CDK) inhibitor p21, leading to cell growth arrest and senescence [43].
Telomere shortening due to cellular replication also results in DDR activation and induction of cellular senescence [40][41]. Interestingly, as it turns out, oxidative stress interferes with telomere homeostasis, and ROS-induced telomere dysfunction may signal persistent DDR activation and senescence, which can be either dependent on or independent of telomere length [5][44][45][46].
2.1.2. Telomeres and how they signal senescence
Telomeres are specialized structures at the ends of eukaryotic chromosomes essential for maintaining genomic stability. They consist of tandem nucleotide repeats coated with a six-member protein complex known as the shelterin [5]. Human telomeres consist of roughly 5–15 kilobases that terminate in a 3′ single-stranded overhang of approximately 50–200 nucleotides. In humans and mammals, their basic DNA repeat is the hexanucleotide sequence 5’-TTAGGG-3’ in the strand that contains the 3’-end [47]. The telomeric end folds back onto itself, forming a lariat-like structure (t-loop), while the single-stranded overhang invades the telomeric double-stranded region [48]. This highly organized structure, which is arranged and stabilized via the shelterin proteins, safeguards the single-stranded terminus from being recognized as a potential breakpoint and the resulting erroneous activation of the DNA damage response (DDR) machinery [49].
Telomeres have been intimately linked with the onset of senescence [5]. Their length decreases with each cellular replication in almost all somatic cells of adult organisms, due to an inability of the replicative polymerases to complete the synthesis at the ends of linear chromosomes. This phenomenon can be counteracted by telomerase, the specialized enzyme that compensates for telomere loss by elongating chromosomal ends. However, since telomerase is repressed in the majority of human somatic cells, telomeres become progressively shorter as cells divide, and when tthey get below a crucial length, they cannot sufficiently bind the shelterin and the telomeric loop destabilizes [44]. This results in the exposure of their terminus region, which is sensed as a double-strand break and accumulates proteins involved in DDR machinery. The DDR signaling through the induction of cell cycle inhibitors promotes the stable arrest of the cell cycle, also known as replicative senescence [40][50].
2.1.3. New Insights into Oxidative Stress-Induced Telomere Shortening and/or Dysfunction
The initial hypothesis concerning the link between telomeres and senescence was that the former are merely a “biological clock” that measures mitotic time and stops proliferation after a more or less fixed number of cell divisions (when telomere length reaches a certain cutoff point) [5]. This simplistic concept has been switched to a more complicated perception, as it has become apparent that telomere homeostasis strongly correlates with oxidative stress, and it was proposed that oxidative stress may contribute to telomere shortening more than the end-replication problem alone [51][52]. During the past few decades, several in vitro studies in human cultured cells have revealed that oxidative stress accelerates telomere shortening and inhibits cell proliferation [46][51][53][54][55], while treatment with antioxidants prevents oxidative stress-induced telomere shortening and extends the proliferative lifespan [51][56]. Moreover, evidence from animal models and human studies supports the negative correlation between oxidative stress and telomere length [57]. Remarkably, telomere loss is not solely a stochastic event, since telomeric DNA is more vulnerable to oxidative damage compared with the bulk of the genome, and this is due to many reasons.
First of all, the damage that occurs within telomeric regions is less efficiently repaired compared with elsewhere in the chromosome [41][58][59]. This is mainly because the shelterin protein-complex coats telomeres, rendering their damage unrecognizable by the DNA repair enzymes [41][58][60][61]. The unrepaired, and therefore persistent, telomeric damage causes a prolonged DDR signaling, ultimately leading to permanent cell cycle arrest and the senescence phenotype [41][58][62].
Apart from being hard to repair, telomeres are also more sensitive to oxidative stress compared with the bulk of the genome, a feature recently called “TelOxidation” [63]. One main reason for telomeres’ susceptibility to oxidative stress is their high content in guanine, the DNA base most vulnerable to oxidation. Ιn vitro studies have shown that under oxidative stress conditions, 8-oxo deoxyguanine (8-oxoG)—the primary product of deoxyguanine oxidation—is more predominant in telomeric sequences compared with non-telomeric sequences [55][64]. A recent study in immortalized cells by Fouquerel et al. has shown that the persistent 8-oxoG induction exclusively within telomeric regions hastened telomere shortening, impaired cell growth, and triggered replication stress, even though telomeres are only a minor proportion of the chromosomes [65]. The researchers proposed that the persistent telomeric 8-oxoG induction interferes with telomere replication, aggravating their shortening.
While telomeric damage can occur through the direct attack of oxidants on DNA, it may also arise by oxidative modifications of the cellular nucleotide pool [66]. In this case, the insertion of oxidized nucleotides—particularly the oxidized form of the guanine nucleotide—inhibits telomerase, preventing further telomere extension [47][67][68][69]. In this way, the oxidation of free nucleotides hastens telomere shortening and loss.
An additional threat for telomeres under oxidative stress conditions is the dissociation of shelterin proteins. 8-oxoG lesions or intermediates of base excision repair in telomeric DNA reportedly disrupt the binding of the TRF1 and TRF2 (telomere repeat binding factors 1 and 2) proteins, which are essential for telomere stability [70].
Notably, although the majority of studies so far supports that oxidative stress hastens telomere shortening and the onset of senescence, recent reports suggest otherwise. Barnes et al., have shown that in non-diseased cells, telomeric 8-oxoG formation induced multiple markers of p53-dependent premature senescence, but remarkably, this was not accompanied by telomere shortening. Yet, their results suggest that telomeric 8-oxoG lesions in non-diseased cells activate DDR signaling and enforce p53-dependent premature senescence by disrupting DNA replication and increasing telomere fragility [45]. Moreover, in vivo experiments confirmed that infiltrating neutrophils in the liver induces ROS-mediated telomere dysfunction and senescence in neighboring hepatocytes, which was not accompanied by telomere shortening [46].
2.1.4. The Putative Role of Labile Iron in Oxidative Stress-Mediated DNA Damage and Cellular Senescence
ROS accumulation can damage DNA and activate the DDR pathways, causing various responses, including cellular senescence. Evidence in the literature is clear in highlighting that the intracellular availability of labile iron represents a prerequisite for ROS-induced adverse effects [71][72][73]. This is because iron catalyzes the conversion of relatively unreactive peroxides to extremely reactive free radicals that can instantly attack and oxidize all essential cellular macromolecules, impairing cellular function. Previous in vitro and in vivo studies revealed that the diminution of intracellular iron protects against oxidative stress-induced DNA damage and cell death [73][74][75][76][77]. Given that iron is essential for the generation of extremely reactive free radicals that can damage DNA (telomeric or non-telomeric), it may well contribute to oxidative stress-mediated cellular senescence. Recent studies have indicated that excess intracellular iron accelerates cellular senescence by damaging the DNA via Fenton chemistry [78]. This process was defined by Sfera et al. as “ferrosenescence” [78]. Moreover, iron levels have been associated with specular changes in p53 activity in mouse hepatocytes and rat liver [79].
2.2. Protein Oxidation
Apart from DNA damage, impaired proteostasis is another prominent feature of senescent cells [80]. A well-known cause of proteotoxicity is ROS, which provoke oxidative modifications leading to protein misfolding and aggregation. ROS may directly attack and cleave protein backbones, generating protein fragments, or may oxidize protein amino acids, especially the aromatic and sulfur-containing ones [81]. Oxidative modifications may also be induced via indirect reactions with reactive intermediates originating from lipid or carbohydrate oxidation [81][82].
Notably, some types of protein oxidation—for example, certain oxidations of thiol groups—can be reversed enzymatically. Such oxidation may regulate signaling mechanisms and are relevant in physiological processes (this is referred to as redox signaling) [27][71][83]. Nevertheless, most types of oxidative damage in proteins cannot be reversed and, since their accumulation compromises cell function, their elimination is extremely important. The maintenance of proteostasis is attained through precisely coordinated networks that rapidly reverse or degrade oxidized proteins [27]. To some extent, non-reversibly oxidized proteins are degraded by the proteasomal or autophagy–lysosomal systems. However, when the rate of their formation overwhelms the rate of their degradation, oxidized proteins accumulate, raising the risk of the formation of insoluble protein aggregates which may further impair the activity of proteasomal and lysosomal degradation systems [84].
Oxidized proteins are elevated in oxidative stress-related pathologies, aged organisms, and senescent cells as well [27][31][80][81][85][86][87]. Although not necessarily indicative of senescent cells, the accumulation of oxidized proteins and protein aggregates is tightly related to senescence [31][87]. In agreement with that, protein degradation by proteasomal and lysosomal systems is affected in senescent cells [31]. Moreover, oxidized proteins form lipofuscin, a non-degradable aggregate that represents a universal hallmark of senescence cells, as will be discussed below.
Although the interactions of labile iron with cellular proteins are ill-defined, there is evidence that some ROS-mediated signaling pathways are dependent on iron availability [71].
2.3. Lipid Oxidation
A large number of existing studies support that senescent cells alter lipid metabolism, although our knowledge concerning the specific lipid metabolite composition or its contribution to the senescent phenotype is sparse [88][89]. Elevated ROS levels fuel lipid peroxidation and breakage of lipids, altering the permeability and fluidity of the membrane lipid bilayer and therefore cell integrity [71]. Increased availability of labile iron favors the generation of highly reactive lipid radicals (RO•), which can drive detrimental cellular processes, including ferroptosis, an iron-dependent form of cell death [71]. RO• (like HO•) are extremely reactive and have some very local effects. Moreover, the breakdown byproducts of lipid peroxidation, mostly reactive aldehydes such as malonaldehyde and 4-hydroxynonenal, are highly electrophile compounds, and they easily modify intracellular macromolecules (mainly proteins) by forming covalent electrophilic addition products. Such highly damaging lipid peroxidation products have been found in increased levels in cells and biological fluids in ageing and age-related diseases [26]. Concerning senescence, it has been reported that excess ROS generated from dysfunctional mitochondria induce lipid oxidative damage and lipid deposits [90][91]. Apart from lipid damage, products of lipid peroxidation are also elevated in senescent cells [89]. Finally, like proteins, oxidized lipids are also components of lipofuscin.
2.4. Lipofuscin Formation and Accumulation
2.4.1. Lipofuscin: A Non-Degradable Product That Accumulates in Postmitotic and Senescent Cells
Lipofuscin is a pigmented, non-degradable biological “garbage” of intracellular catabolism, mainly found within lysosomes but also, in lesser amounts, in the cytosol [92][93]. It can be conventionally generated in virtually any cell type; however, its levels are nearly undetectable in normal proliferative cells, probably because its concentration is diluted with constant divisions. Still, lipofuscin progressively accumulates over time in postmitotic cells which no longer divide, and in cells undergoing senescence [15][93][94][95][96][97].
Despite its name, which implies a lipid structure (lipos is the Greek word for “fat”), lipofuscin is a heterogenous complex mixture consisting mainly of highly oxidized proteins, a lesser amount of lipids, and a few carbohydrates, ribonucleic acids, and metals [84][98], particularly iron ions [36]. The heavy cross-linking of the macromolecules within lipofuscin makes it difficult to identify its exact composition. However, there is strong evidence for the partial mitochondrial origin of lipofuscin [28][30][84][99].
An issue of debate is whether lipofuscin accumulation contributes to the initiation of senescence or whether it is just a mere consequence of this process. However, the finding that synthetic lipofuscin can by itself induce cellular senescence in human fibroblasts argues for a causal role in the process [100]. In any case, lipofuscin accumulation inhibits both proteasomal and lysosomal degradation systems, deteriorates cellular functionality, and is inversely correlated with longevity [101][102].
2.4.2. Mechanisms of Lipofuscin Formation: The Role of Intracellular Iron Homeostasis and Oxidative Stress
Lipofuscin originates via oxidation and polymerization reactions of various cellular macromolecules and structures [93][98]. A requirement for the initiation of such devastating reactions is the generation of highly reactive oxidants. Intensive and/or prolonged oxidative stress leads to the formation of over-oxidized non-degradable materials [27][84] which finally aggregate, polymerize, and accumulate inside the cells, aggravating the rate of lipofuscin accumulation [28].
Lipofuscin resides primarily within lysosomes, acidic organelles with high-level degenerative potential that recycle non-essential, or eliminate harmful, cytoplasmic macromolecules and organelles delivered to them via the autophagy, phagocytosis, and endocytosis processes [103]. Since many of these materials contain iron, lysosomes are rich in this potentially harmful transition metal. The presence of labile iron and low pH make lysosomes ideal for Fenton-type reactions [104]. The generated highly reactive free radicals induce chain oxidation of lysosomal components leading to lipofuscin formation. This is supported by the observation that iron status regulates lipofuscin accumulation in rat heart myocytes cultured under oxidative stress conditions [105].
2.5. Alterations in Mitochondria
Numerous studies highlight that senescent cells show remarkable alterations in mitochondrial function, structure, and dynamics [106]. In different models of cellular senescence, including oxidative stress-induced senescence, mitochondria become hyperfused and elongated, while their mass is increased [53][107][108][109][110]. Mitochondria are also less efficient in producing ATP and show increased proton leak and decreased mitochondrial membrane potential [53][107][111][112][113]. In addition, the autophagic degradation of dysfunctional or superfluous mitochondria within lysosomes (mitophagy) is impaired in senescent cells [114]. As a result, dysfunctional mitochondria which produce excessive ROS accumulate. It has been reported that mitochondrially derived ROS aggravate senescence by inducing genomic damage, particularly at telomeric regions, while interventions that diminish mitochondrial ROS hamper telomere shortening and extend the replicative lifespan [53][56][90]. Moreover, mitochondrial ROS are essential for the establishment of the senescent phenotype [53][107]. Particularly, excess ROS production from dysregulated mitochondria potentiates telomeric DNA damage and induces a prolonged DDR activation which is both necessary and sufficient for the establishment of stable cell growth arrest and senescence [107]. Mitochondrially derived ROS in senescence do not influence solely the genome but induce damage to other intracellular macromolecules as well. Recently, oxidatively modified proteins have been observed in the mitochondria of senescent cells [115], while ROS derived from dysfunctional mitochondria have been reported to induce lipid oxidative damage and lipid deposits [90][91]. Finally, mitochondrially derived ROS from senescent cells have been reported to spread senescence to neighboring healthy cells, through the induction of a DDR response [116][117].
Mitochondria contain high amounts of iron, up to 20–50% of the total cellular iron in some cell types [118]. Within the mitochondria, iron is primarily utilized for the biosynthesis of heme and Fe-S clusters; excess iron can be stored in the organelle-specific form of ferritin [119]. Iron accumulates in mitochondria with age, leading to mitochondrial dysfunction, increased oxidative damage, and cell death [120]. Similarly, mitochondrial iron overload correlates with oxidative stress and apoptotic cell death. However, evidence for the potential role of mitochondrial iron in senescence is sparse. It has been reported that frataxin deficiency, which is known to induce mitochondrial iron accumulation, provokes mitochondrial dysfunction, oxidative stress, and cellular senescence [121]. Moreover, a recent study in endothelial cells revealed that loss of neuropilin-1, a protein widely known for its role in angiogenesis, induces mitochondrial iron accumulation and iron-dependent oxidative stress, which results in mitochondrial dysfunction and senescence [122]. Additionally, treatment with mitoTEMO, a mitochondria-targeted antioxidant compound, or the iron-chelating drug deferoxamine (DFO), inhibited mitochondrial ROS production and protected against cellular senescence [122].
2.6. Alterations in Lysosomes
Another common trait of senescent cells is alterations in lysosomal morphology and function. Lysosomes increase in number and size in senescence [30][34][123]. Moreover, lysosomes contain high amounts of iron, which favors Fenton reactions [30][32][124] and makes these organelles ideal sites for the generation of lipofuscin [101][104]. In addition, lipofuscin seems to be another major intracellular source of ROS (together with mitochondria) in senescent cells, as the presence of iron within its structure catalyzes the formation of highly reactive free radicals [36][37]. Thus, lipofuscin itself can further oxidize cellular macromolecules, which has been reported to be a common trait of senescent cells [36]. Remarkably, as mentioned above, lysosomal dysfunction results in decreased mitophagy and the accumulation of damaged and dysfunctional mitochondria which produce elevated levels of ROS [30]. Mitochondrially derived ROS in senescent cells target intracellular macromolecules (DNA, protein, lipids) and organelles (including lysosomes), forming feedback loops that potentiate macromolecular and organelle damage and establish senescence [34][37].
References
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621.
- Campisi, J. The Biology of Replicative Senescence. Eur. J. Cancer 1997, 33, 703–709.
- Muller, H.J. The Remaking of Chromosomes. Collect. Net 1938, 8, 182–198.
- d’Adda di Fagagna, F.; Teo, S.-H.; Jackson, S.P. Functional Links between Telomeres and Proteins of the DNA-Damage Response. Genes Dev. 2004, 18, 1781–1799.
- Shay, J.W.; Wright, W.E. Telomeres and Telomerase: Three Decades of Progress. Nat. Rev. Genet. 2019, 20, 299–309.
- Watson, J.D. Origin of Concatemeric T7DNA. Nature. New Biol. 1972, 239, 197–201.
- Olovnikov, A.M. A Theory of Marginotomy. J. Theor. Biol. 1973, 41, 181–190.
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres Shorten during Ageing of Human Fibroblasts. Nature 1990, 345, 458–460.
- Boonekamp, J.J.; Simons, M.J.P.; Hemerik, L.; Verhulst, S. Telomere Length Behaves as Biomarker of Somatic Redundancy Rather than Biological Age. Aging Cell 2013, 12, 330–332.
- Blasco, M.A. Telomere Length, Stem Cells and Aging. Nat. Chem. Biol. 2007, 3, 640–649.
- Armanios, M.; Alder, J.K.; Parry, E.M.; Karim, B.; Strong, M.A.; Greider, C.W. Short Telomeres Are Sufficient to Cause the Degenerative Defects Associated with Aging. Am. J. Hum. Genet. 2009, 85, 823–832.
- Herrera, E.; Samper, E.; Martín-Caballero, J.; Flores, J.M.; Lee, H.W.; Blasco, M.A. Disease States Associated with Telomerase Deficiency Appear Earlier in Mice with Short Telomeres. EMBO J. 1999, 18, 2950–2960.
- Rudolph, K.L.; Chang, S.; Lee, H.-W.; Blasco, M.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, Stress Response, and Cancer in Aging Telomerase-Deficient Mice. Cell 1999, 96, 701–712.
- Bernardes de Jesus, B.; Vera, E.; Schneeberger, K.; Tejera, A.M.; Ayuso, E.; Bosch, F.; Blasco, M.A. Telomerase Gene Therapy in Adult and Old Mice Delays Aging and Increases Longevity without Increasing Cancer. EMBO Mol. Med. 2012, 4, 691–704.
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827.
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496.
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular Senescence and Its Effector Programs. Genes Dev. 2014, 28, 99–114.
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism That Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217.
- Rodier, F.; Campisi, J. Four Faces of Cellular Senescence. J. Cell Biol. 2011, 192, 547–556.
- Hudgins, A.D.; Tazearslan, C.; Tare, A.; Zhu, Y.; Huffman, D.; Suh, Y. Age- and Tissue-Specific Expression of Senescence Biomarkers in Mice. Front. Genet. 2018, 9, 59.
- Barbouti, A.; Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.G.; Papoudou-Bai, A.; Gorgoulis, V.G.; Kanavaros, P. Implications of Oxidative Stress and Cellular Senescence in Age-Related Thymus Involution. Oxid. Med. Cell. Longev. 2020, 2020, 7986071.
- Barbouti, A.; Evangelou, K.; Pateras, I.S.; Papoudou-Bai, A.; Patereli, A.; Stefanaki, K.; Rontogianni, D.; Muñoz-Espín, D.; Kanavaros, P.; Gorgoulis, V.G. In Situ Evidence of Cellular Senescence in Thymic Epithelial Cells (TECs) during Human Thymic Involution. Mech. Ageing Dev. 2019, 177, 88–90.
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593.
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705.
- Hamsanathan, S.; Gurkar, A.U. Lipids as Regulators of Cellular Senescence. Front. Physiol. 2022, 13, 796850.
- Höhn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; König, J.; Grune, T.; Castro, J.P. Happily (n)Ever after: Aging in the Context of Oxidative Stress, Proteostasis Loss and Cellular Senescence. Redox Biol. 2017, 11, 482–501.
- Höhn, A.; Grune, T. Lipofuscin: Formation, Effects and Role of Macroautophagy. Redox Biol. 2013, 1, 140–144.
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular Senescence in Aging and Age-Related Disease: From Mechanisms to Therapy. Nat. Med. 2015, 21, 1424–1435.
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of Age-Related Accumulation and Influence on Cell Function. Free Radic. Biol. Med. 2002, 33, 611–619.
- Ogrodnik, M.; Salmonowicz, H.; Gladyshev, V.N. Integrating Cellular Senescence with the Concept of Damage Accumulation in Aging: Relevance for Clearance of Senescent Cells. Aging Cell 2019, 18, e12841.
- Petrat, F.; Rauen, U.; de Groot, H. Determination of the Chelatable Iron Pool of Isolated Rat Hepatocytes by Digital Fluorescence Microscopy Using the Fluorescent Probe, Phen Green SK. Hepatology 1999, 29, 1171–1179.
- Ma, Y.; de Groot, H.; Liu, Z.; Hider, R.C.; Petrat, F. Chelation and Determination of Labile Iron in Primary Hepatocytes by Pyridinone Fluorescent Probes. Biochem. J. 2006, 395, 49–55.
- Park, J.T.; Lee, Y.-S.; Cho, K.A.; Park, S.C. Adjustment of the Lysosomal-Mitochondrial Axis for Control of Cellular Senescence. Ageing Res. Rev. 2018, 47, 176–182.
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial Turnover and Aging of Long-Lived Postmitotic Cells: The Mitochondrial–Lysosomal Axis Theory of Aging. Antioxid. Redox Signal. 2010, 12, 503–535.
- Höhn, A.; Jung, T.; Grimm, S.; Grune, T. Lipofuscin-Bound Iron Is a Major Intracellular Source of Oxidants: Role in Senescent Cells. Free Radic. Biol. Med. 2010, 48, 1100–1108.
- Brunk, U.T.; Terman, A. The Mitochondrial-Lysosomal Axis Theory of Aging: Accumulation of Damaged Mitochondria as a Result of Imperfect Autophagocytosis. Eur. J. Biochem. 2002, 269, 1996–2002.
- Cadet, J.; Davies, K.J.A. Oxidative DNA Damage & Repair: An Introduction. Free Radic. Biol. Med. 2017, 107, 2–12.
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA Damage Response and Immune Signaling Alliance: Is It Good or Bad? Nature Decides When and Where. Pharmacol. Ther. 2015, 154, 36–56.
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA Damage Checkpoint Response in Telomere-Initiated Senescence. Nature 2003, 426, 194–198.
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere Dysfunction in Ageing and Age-Related Diseases. Nat. Cell Biol. 2022, 24, 135–147.
- Davalli, P.; Marverti, G.; Lauriola, A.; D’Arca, D. Targeting Oxidatively Induced DNA Damage Response in Cancer: Opportunities for Novel Cancer Therapies. Oxid. Med. Cell. Longev. 2018, 2018, 2389523.
- Engeland, K. Cell Cycle Regulation: P53-P21-RB Signaling. Cell Death Differ. 2022, 29, 946–960.
- de Lange, T. How Telomeres Solve the End-Protection Problem. Science 2009, 326, 948–952.
- Barnes, R.P.; de Rosa, M.; Thosar, S.A.; Detwiler, A.C.; Roginskaya, V.; Van Houten, B.; Bruchez, M.P.; Stewart-Ornstein, J.; Opresko, P.L. Telomeric 8-Oxo-Guanine Drives Rapid Premature Senescence in the Absence of Telomere Shortening. Nat. Struct. Mol. Biol. 2022, 29, 639–652.
- Lagnado, A.; Leslie, J.; Ruchaud-Sparagano, M.; Victorelli, S.; Hirsova, P.; Ogrodnik, M.; Collins, A.L.; Vizioli, M.G.; Habiballa, L.; Saretzki, G.; et al. Neutrophils Induce Paracrine Telomere Dysfunction and Senescence in ROS-dependent Manner. EMBO J. 2021, 40, e106048.
- Ahmed, W.; Lingner, J. Impact of Oxidative Stress on Telomere Biology. Differentiation 2018, 99, 21–27.
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian Telomeres End in a Large Duplex Loop. Cell 1999, 97, 503–514.
- de Lange, T. Shelterin: The Protein Complex That Shapes and Safeguards Human Telomeres. Genes Dev. 2005, 19, 2100–2110.
- Herbig, U.; Jobling, W.A.; Chen, B.P.C.; Chen, D.J.; Sedivy, J.M. Telomere Shortening Triggers Senescence of Human Cells through a Pathway Involving ATM, P53, and P21CIP1, but Not P16INK4a. Mol. Cell 2004, 14, 501–513.
- von Zglinicki, T. Oxidative Stress Shortens Telomeres. Trends Biochem. Sci. 2002, 27, 339–344.
- von Zglinicki, T.; Pilger, R.; Sitte, N. Accumulation of Single-Strand Breaks Is the Major Cause of Telomere Shortening in Human Fibroblasts. Free Radic. Biol. Med. 2000, 28, 64–74.
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial Dysfunction Accounts for the Stochastic Heterogeneity in Telomere-Dependent Senescence. PLoS Biol. 2007, 5, e110.
- von Zglinicki, T.; Saretzki, G.; Döcke, W.; Lotze, C. Mild Hyperoxia Shortens Telomeres and Inhibits Proliferation of Fibroblasts: A Model for Senescence? Exp. Cell Res. 1995, 220, 186–193.
- Kawanishi, S.; Oikawa, S. Mechanism of Telomere Shortening by Oxidative Stress. Ann. N. Y. Acad. Sci. 2004, 1019, 278–284.
- Saretzki, G.; Murphy, M.P.; von Zglinicki, T. MitoQ Counteracts Telomere Shortening and Elongates Lifespan of Fibroblasts under Mild Oxidative Stress. Aging Cell 2003, 2, 141–143.
- Reichert, S.; Stier, A. Does Oxidative Stress Shorten Telomeres in Vivo? A Review. Biol. Lett. 2017, 13, 20170463.
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA Damage Is Irreparable and Causes Persistent DNA-Damage-Response Activation. Nat. Cell Biol. 2012, 14, 355–365.
- Kruk, P.A.; Rampino, N.J.; Bohr, V.A. DNA Damage and Repair in Telomeres: Relation to Aging. Proc. Natl. Acad. Sci. USA 1995, 92, 258–262.
- Bombarde, O.; Boby, C.; Gomez, D.; Frit, P.; Giraud-Panis, M.-J.; Gilson, E.; Salles, B.; Calsou, P. TRF2/RAP1 and DNA–PK Mediate a Double Protection against Joining at Telomeric Ends. EMBO J. 2010, 29, 1573–1584.
- Hewitt, G.; Jurk, D.; Marques, F.D.M.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres Are Favoured Targets of a Persistent DNA Damage Response in Ageing and Stress-Induced Senescence. Nat. Commun. 2012, 3, 708.
- Fumagalli, M.; Rossiello, F.; Mondello, C.; d’Adda di Fagagna, F. Stable Cellular Senescence Is Associated with Persistent DDR Activation. PLoS ONE 2014, 9, e110969.
- Jacome Burbano, M.S.; Cherfils-Vicini, J.; Gilson, E. Neutrophils: Mediating TelOxidation and Senescence. EMBO J. 2021, 40, e108164.
- Oikawa, S.; Kawanishi, S. Site-Specific DNA Damage at GGG Sequence by Oxidative Stress May Accelerate Telomere Shortening. FEBS Lett. 1999, 453, 365–368.
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 2019, 75, 117–130.e6.
- Sekiguchi, M.; Tsuzuki, T. Oxidative Nucleotide Damage: Consequences and Prevention. Oncogene 2002, 21, 8895–8904.
- Fouquerel, E.; Lormand, J.; Bose, A.; Lee, H.-T.; Kim, G.S.; Li, J.; Sobol, R.W.; Freudenthal, B.D.; Myong, S.; Opresko, P.L. Oxidative Guanine Base Damage Regulates Human Telomerase Activity. Nat. Struct. Mol. Biol. 2016, 23, 1092–1100.
- Aeby, E.; Ahmed, W.; Redon, S.; Simanis, V.; Lingner, J. Peroxiredoxin 1 Protects Telomeres from Oxidative Damage and Preserves Telomeric DNA for Extension by Telomerase. Cell Rep. 2016, 17, 3107–3114.
- Smith, S. Telomerase Can’t Handle the Stress. Genes Dev. 2018, 32, 597–599.
- Opresko, P.L.; Fan, J.; Danzy, S.; Wilson, D.M.; Bohr, V.A. Oxidative Damage in Telomeric DNA Disrupts Recognition by TRF1 and TRF2. Nucleic Acids Res. 2005, 33, 1230–1239.
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron Homeostasis and Oxidative Stress: An Intimate Relationship. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2019, 1866, 118535.
- Galaris, D.; Pantopoulos, K. Oxidative Stress and Iron Homeostasis: Mechanistic and Health Aspects. Crit. Rev. Clin. Lab. Sci. 2008, 45, 1–23.
- Barbouti, A.; Doulias, P.T.; Zhu, B.Z.; Frei, B.; Galaris, D. Intracellular Iron, but Not Copper, Plays a Critical Role in Hydrogen Peroxide-Induced DNA Damage. Free Radic. Biol. Med. 2001, 31, 490–498.
- Barbouti, A.; Amorgianiotis, C.; Kolettas, E.; Kanavaros, P.; Galaris, D. Hydrogen Peroxide Inhibits Caspase-Dependent Apoptosis by Inactivating Procaspase-9 in an Iron-Dependent Manner. Free Radic. Biol. Med. 2007, 43, 1377–1387.
- Doulias, P.-T.; Christoforidis, S.; Brunk, U.T.; Galaris, D. Endosomal and Lysosomal Effects of Desferrioxamine: Protection of HeLa Cells from Hydrogen Peroxide-Induced DNA Damage and Induction of Cell-Cycle Arrest. Free Radic. Biol. Med. 2003, 35, 719–728.
- Tenopoulou, M.; Doulias, P.-T.; Barbouti, A.; Brunk, U.; Galaris, D. Role of Compartmentalized Redox-Active Iron in Hydrogen Peroxide-Induced DNA Damage and Apoptosis. Biochem. J. 2005, 387, 703–710.
- Mantelou, A.G.; Barbouti, A.; Goussia, A.; Zacharioudaki, A.; Papoudou-Bai, A.; Vlachou, C.; Kokkoris, S.; Papalois, A.; Galaris, D.; Glantzounis, G.K. Combined Administration of Membrane-Permeable and Impermeable Iron-Chelating Drugs Attenuates Ischemia/Reperfusion-Induced Hepatic Injury. Free Radic. Biol. Med. 2022, 193, 227–237.
- Sfera, A.; Bullock, K.; Price, A.; Inderias, L.; Osorio, C. Ferrosenescence: The Iron Age of Neurodegeneration? Mech. Ageing Dev. 2018, 174, 63–75.
- Dongiovanni, P.; Fracanzani, A.L.; Cairo, G.; Megazzini, C.P.; Gatti, S.; Rametta, R.; Fargion, S.; Valenti, L. Iron-Dependent Regulation of MDM2 Influences P53 Activity and Hepatic Carcinogenesis. Am. J. Pathol. 2010, 176, 1006–1017.
- Kaushik, S.; Cuervo, A.M. Proteostasis and Aging. Nat. Med. 2015, 21, 1406–1415.
- Kehm, R.; Baldensperger, T.; Raupbach, J.; Höhn, A. Protein Oxidation—Formation Mechanisms, Detection and Relevance as Biomarkers in Human Diseases. Redox Biol. 2021, 42, 101901.
- Castro, J.P.; Jung, T.; Grune, T.; Siems, W. 4-Hydroxynonenal (HNE) Modified Proteins in Metabolic Diseases. Free Radic. Biol. Med. 2017, 111, 309–315.
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462.
- Grune, T. Oxidized Protein Aggregates: Formation and Biological Effects. Free Radic. Biol. Med. 2020, 150, 120–124.
- Labbadia, J.; Morimoto, R.I. The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 2015, 84, 435–464.
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of Cellular Proteostasis in Aging and Disease. J. Cell Biol. 2018, 217, 51–63.
- Cavinato, M.; Madreiter-Sokolowski, C.T.; Büttner, S.; Schosserer, M.; Zwerschke, W.; Wedel, S.; Grillari, J.; Graier, W.F.; Jansen-Dürr, P. Targeting Cellular Senescence Based on Interorganelle Communication, Multilevel Proteostasis, and Metabolic Control. FEBS J. 2021, 288, 3834–3854.
- Wiley, C.D.; Campisi, J. The Metabolic Roots of Senescence: Mechanisms and Opportunities for Intervention. Nat. Metab. 2021, 3, 1290–1301.
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (per) Oxidation in Mitochondria: An Emerging Target in the Ageing Process? Biogerontology 2017, 18, 859–879.
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria Are Required for Pro-ageing Features of the Senescent Phenotype. EMBO J. 2016, 35, 724–742.
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular Senescence Drives Age-Dependent Hepatic Steatosis. Nat. Commun. 2017, 8, 15691.
- Kurz, T.; Terman, A.; Brunk, U.T. Autophagy, Ageing and Apoptosis: The Role of Oxidative Stress and Lysosomal Iron. Arch. Biochem. Biophys. 2007, 462, 220–230.
- Terman, A.; Brunk, U.T. Oxidative Stress, Accumulation of Biological “Garbage”, and Aging. Antioxid. Redox Signal. 2006, 8, 197–204.
- Salmonowicz, H.; Passos, J.F. Detecting Senescence: A New Method for an Old Pigment. Aging Cell 2017, 16, 432–434.
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Muñoz-Espín, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, Universal Biomarker Assay to Detect Senescent Cells in Biological Specimens. Aging Cell 2017, 16, 192–197.
- Sitte, N.; Merker, K.; Grune, T.; von Zglinicki, T. Lipofuscin Accumulation in Proliferating Fibroblasts in Vitro: An Indicator of Oxidative Stress. Exp. Gerontol. 2001, 36, 475–486.
- Jung, T.; Bader, N.; Grune, T. Lipofuscin: Formation, Distribution, and Metabolic Consequences. Ann. N. Y. Acad. Sci. 2007, 1119, 97–111.
- Barbouti, A.; Lagopati, N.; Veroutis, D.; Goulas, V.; Evangelou, K.; Kanavaros, P.; Gorgoulis, V.G.; Galaris, D. Implication of Dietary Iron-Chelating Bioactive Compounds in Molecular Mechanisms of Oxidative Stress-Induced Cell Ageing. Antioxidants 2021, 10, 491.
- König, J.; Ott, C.; Hugo, M.; Jung, T.; Bulteau, A.-L.; Grune, T.; Höhn, A. Mitochondrial Contribution to Lipofuscin Formation. Redox Biol. 2017, 11, 673–681.
- Von Zglinicki, T.; Nilsson, E.; Döcke, W.D.; Brunk, U.T. Lipofuscin Accumulation and Ageing of Fibroblasts. Gerontology 1995, 41, 95–108.
- Terman, A.; Brunk, U.T. Lipofuscin. Int. J. Biochem. Cell Biol. 2004, 36, 1400–1404.
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 464.
- Yim, W.W.-Y.; Mizushima, N. Lysosome Biology in Autophagy. Cell Discov. 2020, 6, 6.
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes in Iron Metabolism, Ageing and Apoptosis. Histochem. Cell Biol. 2008, 129, 389–406.
- Marzabadi, M.R.; Sohal, R.S.; Brunk, U.T. Effect of Ferric Iron and Desferrioxamine on Lipofuscin Accumulation in Cultured Rat Heart Myocytes. Mech. Ageing Dev. 1988, 46, 145–157.
- Martini, H.; Passos, J.F. Cellular Senescence: All Roads Lead to Mitochondria. FEBS J. 2022, 290, 1186–1202.
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between P21 and Reactive Oxygen Production Is Necessary for Cell Senescence. Mol. Syst. Biol. 2010, 6, 347.
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial Dysfunction and Cell Senescence: Deciphering a Complex Relationship. FEBS Lett. 2019, 593, 1566–1579.
- Lee, H.-C.; Yin, P.-H.; Chi, C.-W.; Wei, Y.-H. Increase in Mitochondrial Mass in Human Fibroblasts under Oxidative Stress and during Replicative Cell Senescence. J. Biomed. Sci. 2002, 9, 517–526.
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy Impairment with Lysosomal and Mitochondrial Dysfunction Is an Important Characteristic of Oxidative Stress-Induced Senescence. Autophagy 2017, 13, 99–113.
- Hutter, E.; Renner, K.; Pfister, G.; Stöckl, P.; Jansen-Dürr, P.; Gnaiger, E. Senescence-Associated Changes in Respiration and Oxidative Phosphorylation in Primary Human Fibroblasts. Biochem. J. 2004, 380, 919–928.
- Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschênes-Simard, X.; Ferbeyre, G. Mitochondrial Dysfunction Contributes to Oncogene-Induced Senescence. Mol. Cell. Biol. 2009, 29, 4495–4507.
- Stöckl, P.; Hütter, E.; Zwerschke, W.; Jansen-Dürr, P. Sustained Inhibition of Oxidative Phosphorylation Impairs Cell Proliferation and Induces Premature Senescence in Human Fibroblasts. Exp. Gerontol. 2006, 41, 674–682.
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? eBioMedicine 2017, 21, 7–13.
- Hamon, M.; Ahmed, E.K.; Baraibar, M.A.; Friguet, B. Proteome Oxidative Modifications and Impairment of Specific Metabolic Pathways During Cellular Senescence and Aging. Proteomics 2020, 20, 1800421.
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A Senescent Cell Bystander Effect: Senescence-induced Senescence. Aging Cell 2012, 11, 345–349.
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barrett, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent Human Melanocytes Drive Skin Ageing via Paracrine Telomere Dysfunction. EMBO J. 2019, 38, e101982.
- Ward, D.M.; Cloonan, S.M. Mitochondrial Iron in Human Health and Disease. Annu. Rev. Physiol. 2019, 81, 453–482.
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361.
- Seo, A.Y.; Xu, J.; Servais, S.; Hofer, T.; Marzetti, E.; Wohlgemuth, S.E.; Knutson, M.D.; Chung, H.Y.; Leeuwenburgh, C. Mitochondrial Iron Accumulation with Age and Functional Consequences. Aging Cell 2008, 7, 706–716.
- Bolinches-AmorÃ3s, A.; Mollá, B.; Pla-MartÃn, D.; Palau, F.; González-Cabo, P. Mitochondrial Dysfunction Induced by Frataxin Deficiency Is Associated with Cellular Senescence and Abnormal Calcium Metabolism. Front. Cell. Neurosci. 2014, 8, 124.
- Issitt, T.; Bosseboeuf, E.; De Winter, N.; Dufton, N.; Gestri, G.; Senatore, V.; Chikh, A.; Randi, A.M.; Raimondi, C. Neuropilin-1 Controls Endothelial Homeostasis by Regulating Mitochondrial Function and Iron-Dependent Oxidative Stress. iScience 2019, 11, 205–223.
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453.
- Ma, Y.; Abbate, V.; Hider, R.C. Iron-Sensitive Fluorescent Probes: Monitoring Intracellular Iron Pools. Met. Integr. Biometal Sci. 2015, 7, 212–222.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
951
Revisions:
2 times
(View History)
Update Date:
28 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No