Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shane Handelsman | -- | 3790 | 2023-06-21 13:33:39 | | | |
| 2 | Lindsay Dong | + 4 word(s) | 3794 | 2023-06-25 03:04:14 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Handelsman, S.; Overbey, J.; Chen, K.; Lee, J.; Haj, D.; Li, Y. Role of PD-L1 in Hematopoietic and Organ Transplant. Encyclopedia. Available online: https://encyclopedia.pub/entry/45935 (accessed on 09 August 2026).
Handelsman S, Overbey J, Chen K, Lee J, Haj D, Li Y. Role of PD-L1 in Hematopoietic and Organ Transplant. Encyclopedia. Available at: https://encyclopedia.pub/entry/45935. Accessed August 09, 2026.
Handelsman, Shane, Juliana Overbey, Kevin Chen, Justin Lee, Delour Haj, Yong Li. "Role of PD-L1 in Hematopoietic and Organ Transplant" Encyclopedia, https://encyclopedia.pub/entry/45935 (accessed August 09, 2026).
Handelsman, S., Overbey, J., Chen, K., Lee, J., Haj, D., & Li, Y. (2023, June 21). Role of PD-L1 in Hematopoietic and Organ Transplant. In Encyclopedia. https://encyclopedia.pub/entry/45935
Handelsman, Shane, et al. "Role of PD-L1 in Hematopoietic and Organ Transplant." Encyclopedia. Web. 21 June, 2023.
Copy Citation
One major limitation shared by hematopoietic cellular therapies and solid organ transplantation is the occurrence of graft-versus-host disease (GVHD) and graft failure, respectively. In both cases, an overactive immune system targets and destroys functional tissue, similar to autoimmune diseases. Promising targets for future immunotherapy include the interaction of programmed Death-Ligand 1 (PD-L1) with PD-1 or CD-80, which play a role in regulating T cells. Studies have shown that overexpression of PD-L1 in GVHD models improves survival and reduces pro-inflammatory cytokine secretion. Similarly, stimulating PD-L1 during organ transplantation prolongs the graft’s lifespan and reduces rejection rates.
alloreactive
graft rejection
PD-L1
graft versus host disease (GVHD)
autoimmunity
1. Introduction
Self-tolerance is developed by T cells in the thymus. Thymocytes with T cell receptors (TCR) that have a strong affinity for self-peptides presented by the major histocompatibility complex (MHC), or the MHC complex itself, undergo negative selection, resulting in self-reactive thymocytes that undergo apoptosis and do not mature into T cells. However, in organ transplant cases where the tissue within a host is a foreign entity, alloreactivity can develop as central tolerance does not develop towards the non-self/allogenic peptides [1][2]. This occurs in the case of chronic graft rejection, as portions of donor HLA molecules are presented by antigen-presenting cells (APC) in the same manner as foreign bacterial antigens. In acute rejection, TCRs instead identify whole human leukocyte antigen (HLA) molecules presented by the donor cells as foreign. Due to these rejection processes, almost all organ transplants have limited life spans, requiring immunosuppressive therapy in recipients to prolong their graft’s viability [3][4].
HLA matching is crucial for successful hematopoietic stem cell transplantation and to prevent graft-versus-host disease (GVHD). When there is a mismatch between the donor and recipient HLA, donor T cells may mistake host self-peptides for infectious agents due to a process called molecular mimicry. This happens when a donor MHC class I or II molecule displays a common self-peptide that is not normally immunogenic but appears infectious due to variances in the host’s MHC structure. In contrast, HLA-matched donors present alloantigens or minor histocompatibility antigens (mHAs), which can cause GVHD when they are presented by host MHC. mHAs are polymorphic peptides that differ between donor and host cells, and they are positioned within the peptide-binding groove on host MHCs. The binding of mHAs with donor TCRs can lead to an alloreaction where donor T cells attack host tissues [5][6]. GVHD affects up to 80% of mismatched hematopoietic cell transplant (HCT) recipients, and even fully HLA-matched related donors can still result in GVHD in approximately 40% of recipients [7][8].
In both HCT and organ transplantation, alloreactive T cells are activated, often targeting the foreign organ or organs through their presentation of alloantigen [9]. Therefore, a major challenge in these procedures is preserving functional host/graft tissues from alloreactive T cells. This immune response is primarily coordinated by the recognition of MHC and its associated antigen by APCs, which induce subsequent T cell responses. Depending on the subsequent presentation of co-stimulatory and/or co-inhibitory molecules by the APC in association with MHC, co-modulatory molecules can either enhance or weaken an inflammatory response against a target antigen [10].
2. Current Challenges in Transplantation
2.1. Graft versus Host Disease
One of the most common complications of hematopoietic transplantation therapy is GVHD. This is characterized by transplanted immune cells’ recognition of their new host as foreign and is followed by an immune response against the host following a HCT. GVHD is scored to describe the disease’s severity and functional impact on specific organs. This alloreactive condition can be categorized as either acute or chronic [11][12]
Acute graft versus host disease (aGVHD) is a major cause of morbidity and mortality in HCT. Up to 40% of all cell transplant recipients develop aGVHD [13]. Clinical manifestations of aGVHD most commonly occur within 100 days of transplantation, but acute cases can occur past this time point. Stage 1 aGVHD has limited skin involvement and can present with maculopapular rash, erythema, and pruritus. More severe diseases, stages 2–4, tend to involve the liver and/or gastrointestinal tract. This can present as hyperbilirubinemia, jaundice, and cholestatic hepatitis with liver involvement, as well as severe nausea, vomiting, weight loss, and diarrhea in the gastrointestinal tract [11][12][13][14][15].
The pathophysiological mechanism of aGVHD first involves generalized tissue damage induced by radiation or chemotherapeutic agents used as a conditioning regimen before stem cell transplantation [16]. This damage to host tissues releases Damage Associated Molecular Patterns (DAMPs), stimulating the release of a variety of inflammatory cytokines such as IL-1, IL-6, and TNF-α from host non-hematopoietic and remaining host hematopoietic cells. These cytokines upregulate MHC presentation and cell surface adhesion molecules on host APCs, priming the host’s organs for targeting by a subset of T cells that are termed “alloreactive” [17][18].
Host and new donor APCs then activate alloreactive donor T cells still present in the hematopoietic transplant that aggregate in secondary lymphoid tissue [19][20]. This results in increased proliferation, trafficking, and activation of both CD4+ and CD8+ T cells, with T cell activation further inducing the release of IL-2 and IFN-γ [21][22][23][24][25]. Finally, alloreactive effector T cells (including helper T cells and cytotoxic T cells), along with natural killer (NK) cells, migrate to various organ systems where they begin to cause cytotoxic tissue damage. This eventually leads to the characteristic manifestations of aGVHD [26][27][28].
In contrast to aGVHD, chronic graft versus host disease (cGVHD) typically occurs 100 or more days after transplantation, affecting 30–70% of transplant recipients [29][30][31]. Clinical manifestations of chronic GVHD can appear in a variety of organ systems and can either be restricted to a single organ or widespread throughout the body. Dermatologic manifestations of chronic GVHD include lichen-sclerosis-like lesions, depigmentation, sweat impairment, and heat intolerance, along with the typical erythema and maculopapular rash.
2.2. HCT Rejection
Graft failure is a severe complication following HCT that is divided into primary graft failure and secondary graft failure. In primary graft failure, there is a failure to achieve initial engraftment of the HCT. This is defined as an absolute neutrophil count (ANC) lower than 0.5 × 109/L by 28 days, platelet levels less than 20 × 109/L, or hemoglobin levels less than 80 g/L after transplantation. In secondary graft failure, there is a failure of the transplanted cells (ANC lower than 0.5 × 109/L) after initially successful engrafting [32].
The pathogenesis of graft failure in HCT is characterized by a complex, multifactorial, alloreactive immune response from host T cells and NK cells. Graft failure due to host NK cells most commonly occurs following MHC-mismatched HCT. However, even in HLA-matched HCT, host T cells have been shown to promote graft failure [33][34][35]. Host NK cells have inhibitory receptors that are unable to recognize the mismatched MHC class one molecules on the transplanted cells, resulting in NK-mediated cytotoxic rejection of the allogenic cells [36][37].
Interestingly, donor NK cells and donor T cells have been shown to promote engraftment by inhibiting the host cell response to the transplanted cells [38][39][40][41]. Both host and donor Treg cells have also been demonstrated to facilitate engraftment in HCT through the production of IL-10 and TGF-β, suppressing T cell and NK cell-mediated graft failure [42][43][44].
Even though graft rejection has an extremely poor prognosis, the condition is relatively rare among allogeneic HCT recipients, with an incidence of roughly 5% [45]. The current treatment for HCT graft failure mainly consists of G-CSF therapy and additional allogenic cells to increase the graft’s proliferative capacity and prevent its rejection [46].
2.3. The Pathophysiologic Process of Transplant Rejection
Graft failure remains a major limitation in other types of tissue transplantation beyond HCT. Kidney transplant is the most transplanted organ in the United States of America. In a review of United Network for Organ Sharing data on kidney transplantation from 2000 to 2014, 50,301 graft failures occurred, or about 3600 every year. Of this, 48% were due to chronic rejection, and 12% were due to acute rejection [47]. The immunological mechanism of transplant rejection occurs in three main forms: hyperacute, acute, and chronic. Hyperacute rejection (HAR) typically occurs within minutes to hours. Pre-formed circulating antibodies quickly bind to transplant tissue, typically resulting in irreversible graft thrombosis and ischemia. However, with the advent of screening technologies such as crossmatch testing and antibody screening over the last 40 years, HAR is now a rare occurrence [48]. Hyperacute rejection (HAR) is mainly a humoral immune response and is classified as a Type II hypersensitivity reaction. It occurs because pre-formed circulatory antibodies bind to the allogenic graft tissue.
In contrast, acute rejection (AR) occurs within days to weeks of transplantation. It is the result of the host immune system identifying the graft as foreign and destroying it, leading to graft failure. This type of rejection is largely controlled by immunosuppressive therapy, leading to a decreased incidence and better long-term outcomes. However, even with treatment, AR still occurs in about 7% of renal transplants every year [49]. Chronic rejection (CR) occurs months to years after transplantation and affects 100% of transplants to some degree. It is one of the main determinants of the longevity of solid organ transplants [50]. Its full etiology has yet to be understood, and proposed pathological mechanisms cite inflammatory, humoral, and cellular immune reactions.
Acute transplant rejection differs from HAR in that it involves both a humoral and cellular immune response. Additionally, in AR, the patient does not have pre-formed antibodies, but rather develops an activated immune response toward the graft tissue. This immune response arises because resident-donor antigen-presenting cells in the graft travel to sites of immune activation in the host, such as lymph nodes, and are recognized by a subset of T cells that are alloreactive. Alloreactive T cells are defined by the ability of their T cell receptors (TCRs) to bind non-self MHC molecules [51]. The mechanism of AR is the direct pathway of allorecognition, in that graft MHC molecules are directly presented to and recognized by host lymphocytes. Activation of alloreactive T cells leads to the typical host immune response to a foreign substance. This includes the activation of CD8+ T cells, Th1 T cells, Th2 T cells, and Follicular Helper T (TFH) cells. CD8+ T cells will migrate to the transplanted tissue and exert their direct cytotoxic effects. Activated Th1 cells will secrete IFN-γ resulting in the activation of macrophages and inflammation. TFH T cells serve as a link to humoral immunity, aiding B cell activation and the generation of donor-specific antibodies (DSAs) [52].
Whereas acute transplant rejection occurs via direct allorecognition, chronic transplant rejection occurs via the indirect pathway of allorecognition. The indirect pathway of allorecognition activates when graft T cells die in a manner that results in the release of cellular alloantigens, such as immune-mediated death or inflammasome-mediated pyroptosis. These cellular alloantigens, including peptide fragments of donor MHC molecules, are then phagocytosed by host APCs. Activated host immune cells travel to sites of immune activation and stimulate an immune response against the transplanted tissue, like the direct pathway of allorecognition [9]. While CR initially follows a similar cellular immune response activation as AR, there are several additional pathological changes that make CR a more complex and severe disease. One of the most distinctive features of chronic solid organ rejection is the activation of vascular smooth muscle cells and fibroblasts, resulting in intimal hyperplasia of blood vessels, and leading to ischemia of graft tissue.
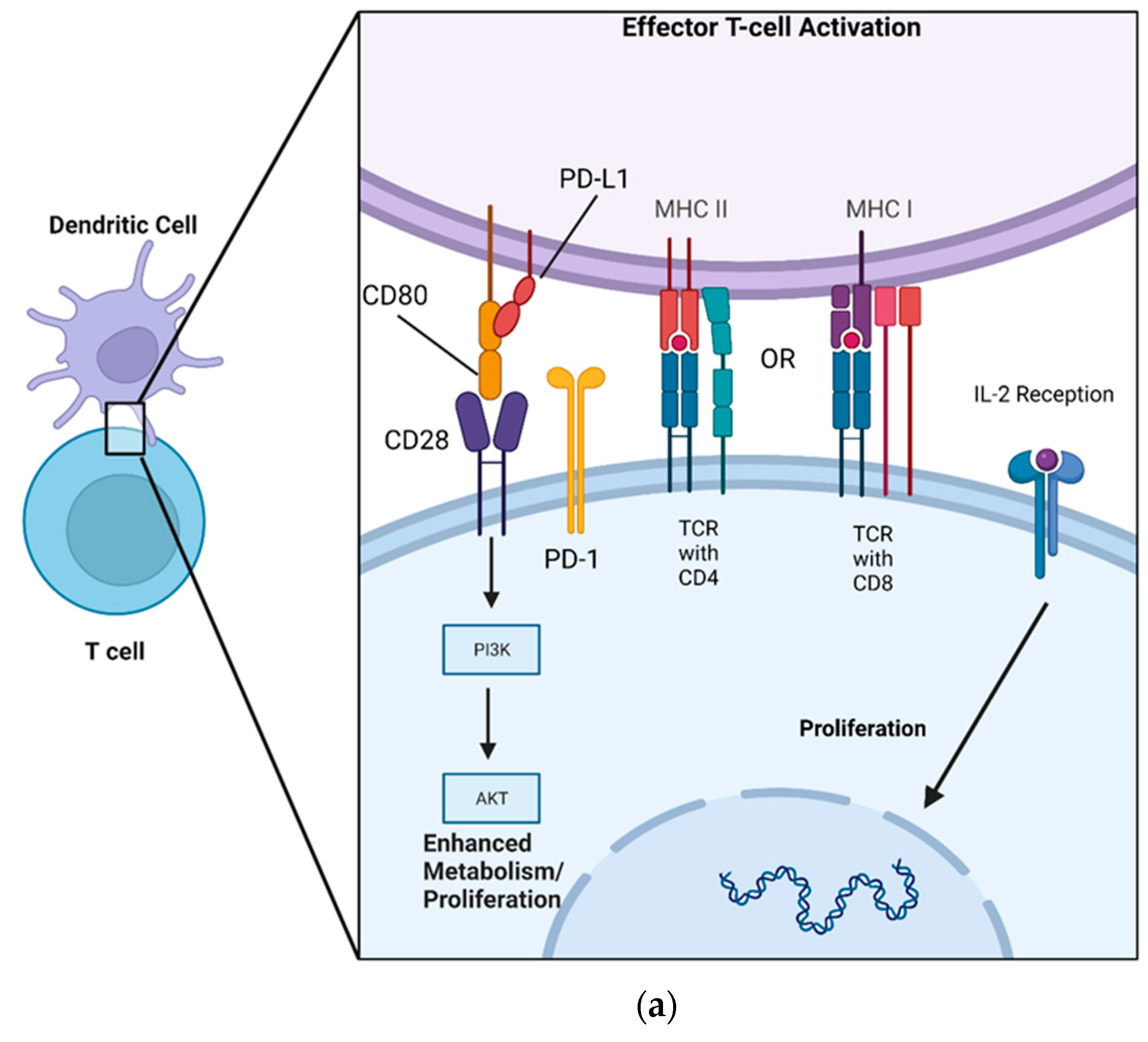
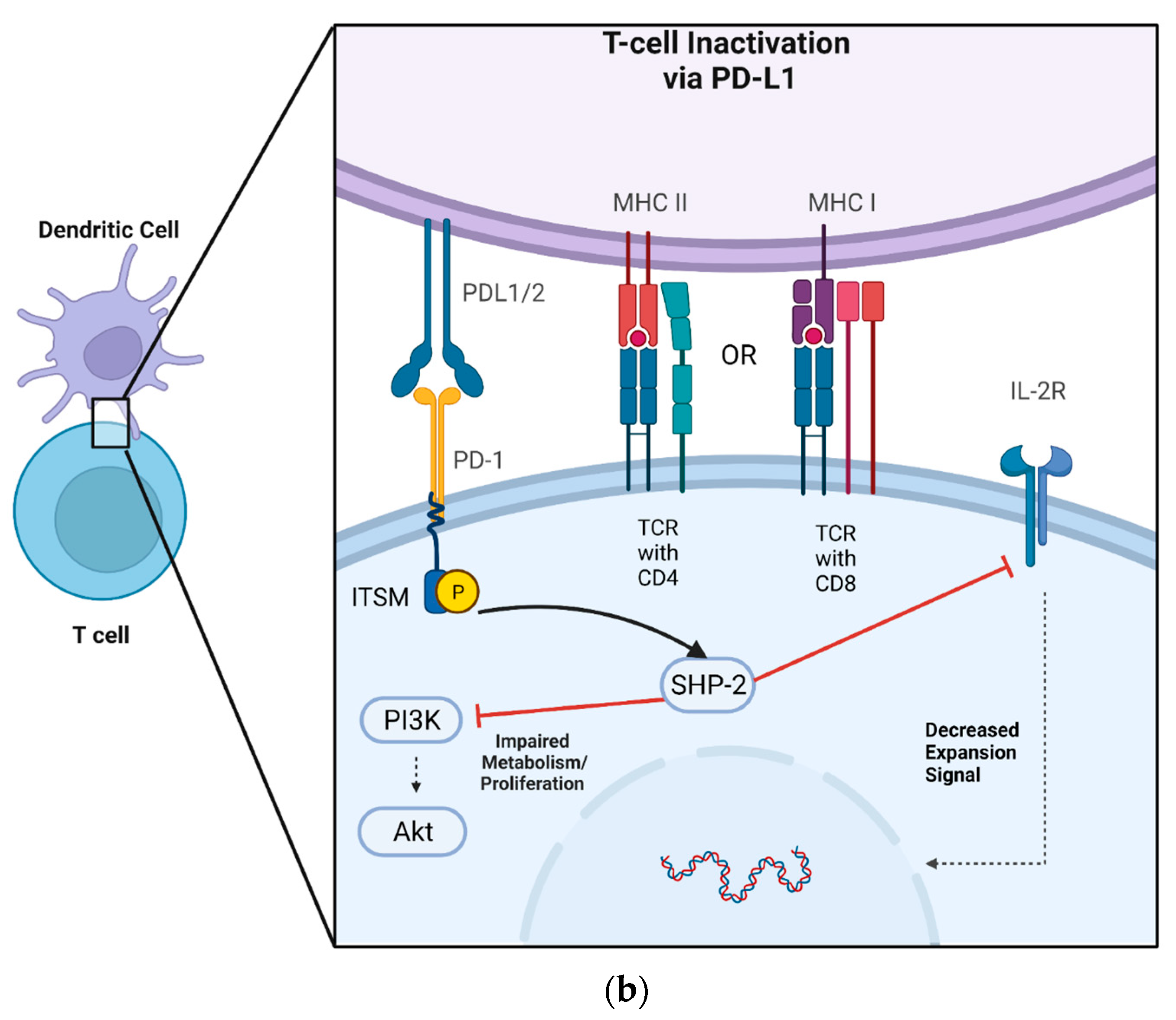
3. Programmed Death-Ligand 1 (PD-L1) Mechanism of Action
PD-L1 (also known as B7-H1) and its corresponding receptor, PD-1, are both transmembrane proteins that are a part of the immunoglobulin superfamily. PD-L1 is constitutively present on the surface of hematopoietic cells such as monocytes and T cells. However, monocytes in peripheral blood do not tend to express PD-L1. Certain immune-privileged sites, such as the placenta and cornea, have also been shown to maintain expression of PD-L1. Additionally, the ligand’s expression is upregulated during active immune responses. This upregulation has been linked to the presence of pro-inflammatory cytokines such as IFN-γ or exogenous LPS on the surface of hematopoietic cells and at the surface of their epithelial and endothelial cells [25][53][54]. PD-L1 mRNA has been found in human organs, including the heart, kidney, and lung, but not in the colon or small intestine [55].
Although soluble versions of PD-L1 and PD-1 exist, conventionally, the ligand and its receptor are transmembrane-bound proteins that are displayed on the cell’s surface. More specifically, PD-1 has an extracellular domain that allows for interaction with PD-L1, a localizing transmembrane portion, and an intracellular domain that allows for signal transduction [56]. The receptor’s production is upregulated via TCR recognition of MHC, thereby preventing the overactivation of T cells and limiting immune-mediated damage to native tissue [57][58]. PD-L1 has been shown to decrease the expansion of CD4+ T cells and NK cells, limit the cytotoxic effects of CD8+ T cells, and simultaneously increase the differentiation of CD4+ T cells into Tregs [59][60][61]. PD-1 can be stimulated following TCR activation in a two-step sequence whereby TCR engagement with MHC is followed by co-inhibition by PD-L1 [62]. Once PD-L1 is recognized by PD-1 in peripheral (non-immune) tissues, it initiates a co-inhibitory signal, limiting T cell proliferation (Figure 1b). This immunomodulatory effect is driven by the downregulation of a variety of intracellular pathways responsible for cellular metabolism and proliferation.
Figure 1. A DC acts as an APC for both CD8+ and CD4+ T cells via MHC I and MHCII, respectively, allowing for co-stimulatory/inhibitory signaling to occur. (a) MHC presentation of a complimentary peptide to the TCR allows for CD80’s activation of CD28 on the T cell, which subsequently enhances T cell metabolism and proliferation via the PI3K/AKT pathway. PD-L1 is sequestered by CD80, forming a heterodimer. CD80 simultaneously blocks the activation of PD-1 while activating CD28. (b) MHC presentation of a complimentary peptide to the TCR, followed by PD-L1 recognition by PD-1, induces phosphorylation of ITSM on the intracellular portion of PD-1. Phosphorylated ITSM then interacts with SHP-2, inhibiting the PIK3/AKT signaling cascade and limiting T cell production of IL-2.
Crucial to this intracellular regulation are intracellular signaling domains on the PD-1 molecule known as immunoreceptor tyrosine-based switch motifs (ITSM). Following TRC activation and PD-1/PD-L1 binding, these ITSMs are phosphorylated, allowing for their interaction with a cytoplasmic tyrosine phosphatase called Src homology 2 domain-containing phosphatase 2 (SHP-2). SHP-2 contains an enzymatic tail portion that allows for downstream inhibition of phosphatidylinositol-3-kinase (PI3K) and Akt as well as a reduction in IL-2 production [63][64][65][66]. PD-L1’s inhibition of PI3K/AKT thereby directly counteracts the effects exerted by CD80 (also known as B7-1), a costimulatory molecule present on APCs [67][68] (Figure 1a).
4. PD-L1 Amelioration of GVHD, Autoimmunity, and Graft Rejection
4.1. GVHD
A major goal of therapeutics following hematopoietic transplantation is to prevent the development of GVHD. PD-L1 appears to have the potential to promote immune tolerance following transplant and minimize the potential for developing adverse reactions caused by stem cell therapy. Tang et al. demonstrated how in vivo hydrodynamic gene transfer, which induces overexpression of PD-L1 in murine aGVHD models, results in lower lethality due to aGVHD. This overexpression of PD-L1 was found to result in the inhibition of donor T cells and reduce effector memory status. Moreover, PD-L1 overexpression resulted in murine models exhibiting lower levels of pro-inflammatory cytokines secreted from effector T cells, less proliferation, and increases in the apoptotic activity of these effector T cells [69].
It is important to note that the development of GVHD is dependent on which cells express PD-L1. While expression of the ligand on donor T cells induces GVHD, expression on parenchymal tissue such as hepatocytes results in protection from the development of GVHD [70][71].
4.2. Autoimmunity
The mechanism by which PD-L1 plays a role in autoimmune disease can be translated thematically to the expression of PD-L1 during cell transplantation. A study by Hu et al., using DBA/1J mouse models (a strain of mice bred to develop rheumatoid arthritis), studied the impact of PD-L1 overexpression on these mice. Researchers administered a PD-L1 lentiviral vector to transfect mouse bone marrow mesenchymal cells, which induced PD-L1 overexpression in the cells. DBA/1J mice that were treated with mesenchymal stem cells that had received the PD-L1 vector exhibited less joint damage, less activation of pro-inflammatory cytokines, and an inhibition of T and B cell activation. Moreover, there was limited cartilage damage, synovial hyperplasia, inflammatory cell infiltration, and bone erosion in the mice treated with the PD-L1 overexpression vector. These transplanted stem cells decreased the DCs found in mouse joints and increased the Tregs in the tissue, promoting a tolerant and non-inflammatory environment [72].
4.3. Solid Organ Tolerance
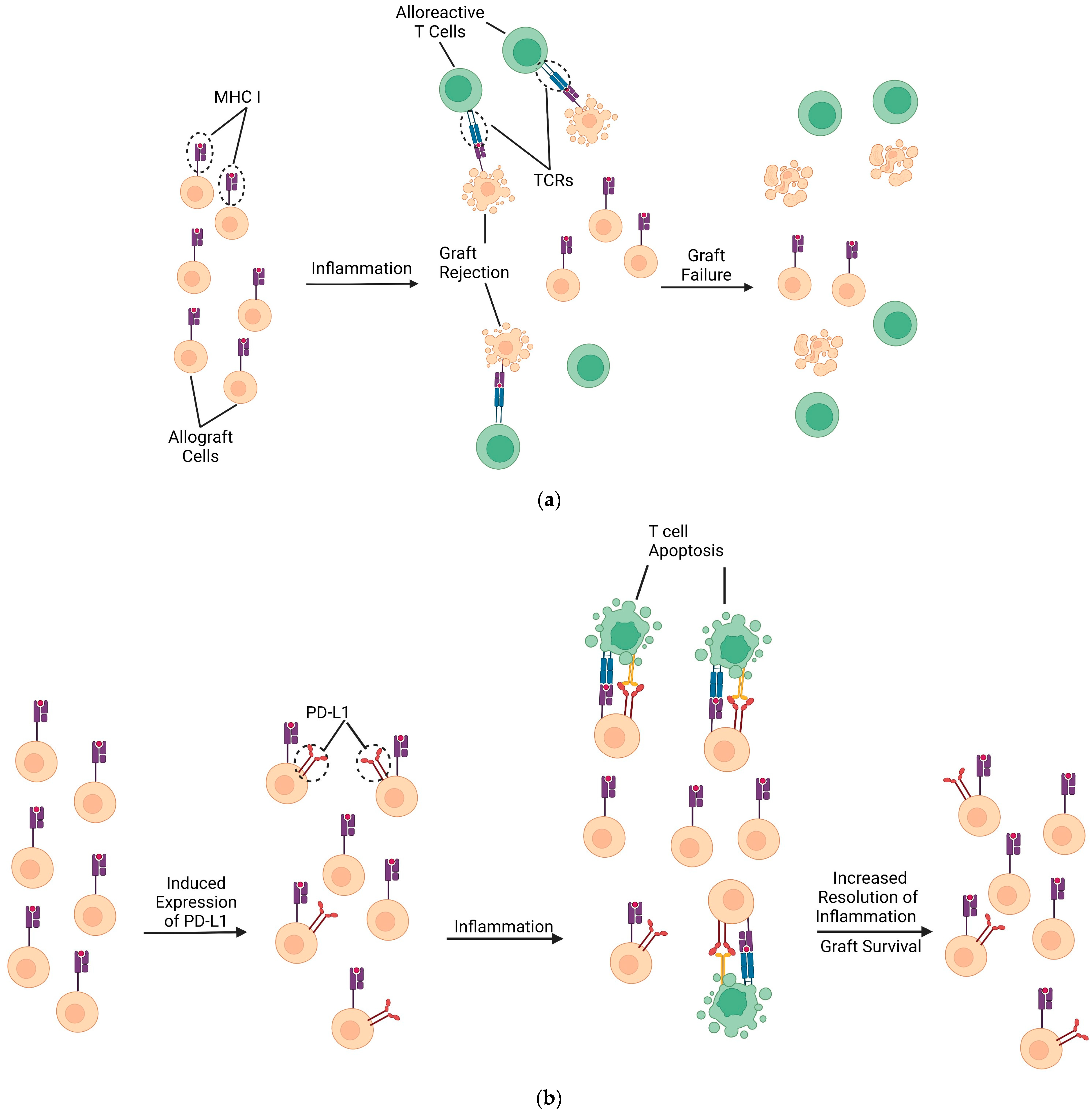
In addition to PD-L1’s role in GVHD prevention, the role this ligand holds in organ transplantation has also been explored. Tanaka et al. studied the effects of PD-L1’s presence in mice that received a mismatched cardiac allograft that underwent concurrent immunosuppression with CTLA4Ig. Their study showed that blocking PD-L1 resulted in significantly reduced numbers of Tregs and significantly increased portions of alloreactive cytotoxic and IFN-γ-producing T cells within the spleen of allograft recipient mice. Regarding allograft function, Tanaka et al. demonstrated that PD-L1 blocking antibodies induced graft rejection within this same population of transplanted mice (Figure 2a,b). Moreover, PD-L1-deficient mice receiving heart transplants also had a significant reduction in graft survival. Histologically, it seems a possible cause of these effects in PD-L1 knockout mice was the increased vasculopathy within the cardiac tissue of mice [73].
Figure 2. Proposed protective effects of PD-L1 during transplantation. (a) Limited PD-L1 expression by transplanted cells provides less protection from cytotoxic T cell infiltration (and less protection from helper T cell infiltration). Inflammation progresses to acute rejection and eventual graft failure as the tissue loses viable graft cells. (b) PD-L1 may be upregulated by methods such as IFN-γ administration or hydrodynamic gene transfer onto transplanted DCs and certain graft tissues such as hepatocytes [55][69][70][74]. If PD-L1 upregulation is sufficient to overcome CD80 sequestration/signaling, a reduction in alloreactive T cells would prevent graft cell induced apoptosis. Ideally, this technique would preserve graft function by preventing T cell infiltration.
4.4. PD-L1 Viral Reactivation
The potential of PD-L1 to prevent transplant rejection and graft survival must be weighed against its immunosuppressive properties, especially in the case of viral reactivation. The expression of PD-L1 on CD8+ T cells decreases the proliferative capacity and cytotoxic function of these cells. Given these effects, it is not surprising that many viruses induce PD-L1 overexpression, thereby decreasing the host cytotoxic T cell response and contributing to viral immune escape. Recently implicated viruses include Hepatitis B virus (HBV), HIV, Epstein–Barr virus, hantaviruses, and even coronaviruses [75][76][77][78][79]. Consequently, any therapy involving the upregulation of PD-L1 represents a clear pathway for the reactivation of underlying viral infections.
However, the effect of any immunotherapy interacting with PD-L1 is not so simple. While one would expect that any immunotherapy downregulating PD-L1 would lead to increased immune function and, therefore, decrease viral reactivations, there is clinical evidence of increased rates of HBV reactivation in cancer patients being treated with anti-PD-L1 monoclonal antibodies [80]. The exact mechanism by which this occurs is still unclear. Potential theories include the role of PD-1 in preventing immune-associated hepatocyte damage or the role of PD-1 in Treg apoptosis [81][82][83]. Overall, there is much to be discovered about the effects of PD-L1 immunotherapies in vivo.
Additionally, the potential adverse effects of PD-L1-induced immunosuppression must be viewed in the context of the current gold standard of post-transplant and GVHD immunosuppressive therapies, such as calcineurin inhibitors and corticosteroids. The high rates of infection in post-transplant patients due to immunosuppression are already well understood [4][84]. This is especially evident in the occurrence of viral reactivation. Traditional immunosuppressive therapies after solid organ transplant have been strongly implicated in the reactivation of Epstein–Barr virus, HBV, BK polyomavirus, and many more [84][85][86][87]. Overall, the potential immunosuppressive effects of PD-L1 must be further investigated, as it represents a promising alternative to current traditional immunosuppressive therapies.
5. Current State of PD-L1/PD-1 Targeting Therapies
Pharmaceutical targeting of PD-L1 has primarily focused on the inhibition of excess PD-L1 produced by cancer cells. Several monoclonal antibodies have been approved to target the PD-L1/PD-1 interaction, including pembrolizumab, nivolumab, and cemiplibab. Atezolizumab, avelumab, and durvalumab are all approved to target and inhibit PD-L1 [75]. Each of these therapeutics seeks to inhibit the immunosuppression seen in tumor cells that overexpress PD-L1, preventing immune tolerance in cancerous growth. However, these treatments have also been found to lead to the development of self-reactive T cells and have been linked to the development of autoimmune and inflammatory diseases [76].
In the absence of PD-L1/PD-1-induced anergy, patients receiving treatment that inhibits this pathway are at increased risk for developing inflammatory arthritis as well as hematologic diseases, including immune thrombocytopenia and autoimmune hemolytic anemia [77][78]. To circumvent some of the adverse effects noted in patients undergoing treatment with monoclonal antibodies, some researchers are pursuing combination therapies with small molecules such as osimertinib, an EGFR inhibitor, that mutually function to limit the over-activation of PD-L1. These combination therapies are in development for the treatment of lymphomas, NSCLC, colon cancer, and melanoma [79].
Other researchers have considered targeting non-membranous PD-L1 expression with small molecules that inhibit the expression of PD-L1 in its nuclear, cytoplasmic, or extra-cellular vesicle expression to ultimately reduce signaling sequelae [80][82]. Another avenue for investigation into PD-L1/PD-1 inhibition is more indirect by targeting its expression via known regulators of the PD-L1/PD-1 pathway, such as epigenetic modification and transcriptional and post-transcriptional regulators [83]. These emerging immunotherapies offer a variety of methods to enhance the body’s ability to target and kill cancer cells in a more precise manner. Researchers’ focus on PD-L1 and its role in the protection of cancer through anergy has resulted in prolonged lives for many. However, up until this point in time, researchers’ goal with PD-L1 targeting therapy has been to increase immune response via inhibition of the ligand. Therapeutic induction of PD-L1 expression seeks the opposite goal from most current pharmacologic agents in development—to promote local immunosuppression.
6. Conclusions
One major limitation shared by hematopoietic cellular therapies and solid organ transplantation is the occurrence of graft-versus-host disease (GVHD) and graft failure, respectively. In both cases, an overactive immune system targets and destroys functional tissue, similar to autoimmune diseases.
Promising targets for future immunotherapy include the interaction of PD-L1 with PD-1 or CD-80, which play a role in regulating T cells. Studies have shown that overexpression of PD-L1 in GVHD models improves survival and reduces pro-inflammatory cytokine secretion. Similarly, stimulating PD-L1 during organ transplantation prolongs the graft’s lifespan and reduces rejection rates. Additionally, in autoimmune disease models, increased PD-L1 expression leads to slower disease progression and limited tissue damage.
Although PD-L1 has primarily been studied in the context of cancer treatment, it has the potential to be a valuable tool in the management of allo- and auto-immunity. While most research has focused on blocking PD-L1 to inhibit immunosuppressive processes, enhancing PD-L1 activity could be a promising therapeutic approach as it dampens a destructive immune reaction. Further investigation is needed to understand how increased PD-L1 expression and availability in the context of GVHD, autoimmunity, and organ transplantation can promote immune tolerance.
References
- Xing, Y.; Hogquist, K.A. T-cell tolerance: Central and peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4, a006957.
- Felix, N.J.; Allen, P.M. Specificity of T-cell alloreactivity. Nat. Rev. Immunol. 2007, 7, 942–953.
- García, M.A.A.; Yebra, B.G.; Flores, A.L.L.; Guerra, E.G. The major histocompatibility complex in transplantation. J. Transpl. 2012, 2012, 842141.
- Nelson, J.; Alvey, N.; Bowman, L.; Schulte, J.; Segovia, M.C.; McDermott, J.; Te, H.S.; Kapila, N.; Levine, D.J.; Gottlieb, R.L.; et al. Consensus recommendations for use of maintenance immunosuppression in solid organ transplantation: Endorsed by the American College of Clinical Pharmacy, American Society of Transplantation, and International Society for Heart and Lung Transplantation: An executive summary. Pharmacotherapy 2022, 42, 594–598.
- Spierings, E. Minor histocompatibility antigens: Past, present, and future. Tissue Antigens 2014, 84, 360.
- Koyama, M.; Hill, G.R. Alloantigen presentation and graft-versus-host disease: Fuel for the fire. Blood 2016, 127, 2963–2970.
- Ferrara, J.L.M.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 373, 1550–1561.
- Spellman, S.R. Hematology 2022-what is complete HLA match in 2022? Hematol. Am. Soc. Hematol. Educ. Program 2022, 2022, 83–89.
- Siu, J.H.Y.; Surendrakumar, V.; Richards, J.A.; Pettigrew, G.J. T cell Allorecognition Pathways in Solid Organ Transplantation. Front. Immunol. 2018, 9, 2548.
- Sharpe, A.H. Mechanisms of costimulation. Immunol. Rev. 2009, 229, 5–11.
- Filipovich, A.H.; Weisdorf, D.; Pavletic, S.; Socie, G.; Wingard, J.R.; Lee, S.J.; Martin, P.; Chien, J.; Przepiorka, D.; Couriel, D.; et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol. Blood Marrow Transpl. 2005, 11, 945–956.
- Jagasia, M.H.; Greinix, H.T.; Arora, M.; Williams, K.M.; Wolff, D.; Cowen, E.W.; Palmer, J.; Weisdorf, D.; Treister, N.S.; Cheng, G.-S.; et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol. Blood Marrow Transpl. 2015, 21, 389–401.e1.
- Weisdorf, D.; Haake, R.; Blazar, B.; Miller, W.; McGlave, P.; Ramsay, N.; Kersey, J.; Filipovich, A. Treatment of moderate/severe acute graft-versus-host disease after allogeneic bone marrow transplantation: An analysis of clinical risk features and outcome. Blood 1990, 75, 1024–1030.
- Harris, A.C.; Young, R.; Devine, S.; Hogan, W.J.; Ayuk, F.; Bunworasate, U.; Chanswangphuwana, C.; Efebera, Y.A.; Holler, E.; Litzow, M.; et al. International, Multicenter Standardization of Acute Graft-versus-Host Disease Clinical Data Collection: A Report from the Mount Sinai Acute GVHD International Consortium. Biol. Blood Marrow Transpl. 2016, 22, 4–10.
- Zeiser, R.; Blazar, B.R. Acute Graft-versus-Host Disease—Biologic Process, Prevention, and Therapy. N. Engl. J. Med. 2017, 377, 2167–2179.
- Flinn, A.M.; Gennery, A.R. Treatment of Pediatric Acute Graft-versus-Host Disease-Lessons from Primary Immunodeficiency? Front. Immunol. 2017, 8, 328.
- Alegre, M.L.; Lakkis, F.G.; Morelli, A.E. Antigen Presentation in Transplantation. Trends Immunol. 2016, 37, 831–843.
- Hill, G.R.; Koyama, M. Cytokines and costimulation in acute graft-versus-host disease. Blood 2020, 136, 418–428.
- Duffner, U.A.; Maeda, Y.; Cooke, K.R.; Reddy, P.; Ordemann, R.; Liu, C.; Ferrara, J.L.M.; Teshima, T. Host dendritic cells alone are sufficient to initiate acute graft-versus-host disease. J. Immunol. 2004, 172, 7393–7398.
- Gao, E.K.; Kosaka, H.; Surh, C.D.; Sprent, J.T. Cell contact with Ia antigens on nonhemopoietic cells in vivo can lead to immunity rather than tolerance. J. Exp. Med. 1991, 174, 435–446.
- Chakraverty, R.; Sykes, M. The role of antigen-presenting cells in triggering graft-versus-host disease and graft-versus-leukemia. Blood 2007, 110, 9–17.
- Mohty, M.; Blaise, D.; Faucher, C.; Vey, N.; Bouabdallah, R.; Stoppa, A.-M.; Viret, F.; Gravis, G.; Olive, D.; Gaugler, B. Inflammatory cytokines and acute graft-versus-host disease after reduced-intensity conditioning allogeneic stem cell transplantation. Blood 2005, 106, 4407–4411.
- Nimer, S.D.; Giorgi, J.; Gajewski, J.L.; Ku, N.; Schiller, G.J.; Lee, K.; Territo, M.; Ho, W.; Feio, S.; Selch, M.; et al. Selective depletion of CD8+ cells for prevention of graft-versus-host disease after bone marrow transplantation. A randomized controlled trial. Transplantation 1994, 57, 82–87.
- Via, C.S.; Finkelman, F.D. Critical role of interleukin-2 in the development of acute graft-versus-host disease. Int. Immunol. 1993, 5, 565–572.
- Shustov, A.; Nguyen, P.; Finkelman, F.; Elkon, K.B.; Via, C.S. Related Content Fas Expression on Antigen-Speciic T Cells Has Costimulatory, Helper, and Down-Regulatory Functions In Vivo for Cytotoxic T Cell Responses but Not for T Cell-Dependent B Cell Responses. J. Immunol. 1998, 161, 2848–2855.
- Koyama, M.; Cheong, M.; Markey, K.A.; Gartlan, K.; Kuns, R.D.; Locke, K.R.; Lineburg, K.E.; Teal, B.E.; Mouttie, L.L.-E.; Bunting, M.D.; et al. Donor colonic CD103+ dendritic cells determine the severity of acute graft-versus-host disease. J. Exp. Med. 2015, 212, 1303–1321.
- Ghayur, T.; Seemayer, T.A.; Kongshavn, P.A.; Gartner, J.G.; Lapp, W.S. Graft-versus-host reactions in the beige mouse. An investigation of the role of host and donor natural killer cells in the pathogenesis of graft-versus-host disease. Transplantation 1987, 44, 261–267.
- Ruggeri, L.; Aversa, F.; Martelli, M.F.; Velardi, A. Allogeneic hematopoietic transplantation and natural killer cell recognition of missing self. Immunol. Rev. 2006, 214, 202–218.
- Ball, L.M.; Egeler, R.M. Acute GvHD: Pathogenesis and classification. Bone Marrow Transplant. 2008, 41, S58–S64.
- Arai, S.; Arora, M.; Wang, T.; Spellman, S.R.; He, W.; Couriel, D.R.; Urbano-Ispizua, A.; Cutler, C.S.; Bacigalupo, A.A.; Battiwalla, M.; et al. Increasing Incidence of Chronic Graft-versus-Host Disease inAllogeneic Transplantation: A Report from the Center for International Blood and Marrow Transplant Research. Biol. Blood Marrow Transplant. 2015, 21, 266–274.
- Flowers, M.E.D.; Parker, P.M.; Johnston, L.J.; Matos, A.V.B.; Storer, B.; Bensinger, W.I.; Storb, R.; Appelbaum, F.R.; Forman, S.J.; Blume, K.G.; et al. Comparison of chronic graft-versus-host disease after transplantation of peripheral blood stem cells versus bone marrow in allogeneic recipients: Long-term follow-up of a randomized trial. Blood 2002, 100, 415–419.
- Chaturvedi, S.; George, B.; Savani, B.N. Bleeding and thrombotic complications. In The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies; Springer: Berlin/Heidelberg, Germany, 2018; pp. 301–306.
- Shatry, A.M.; Roopenian, D.C.; Levy, R.B. Survival and function of MiHA epitope-specific host CD8 TM cells following ablative conditioning and HCT. Biol. Blood Marrow Transpl. 2007, 13, 293–298.
- Marijt, W.A.; Kernan, N.A.; Diaz-Barrientos, T.; Veenhof, W.F.; O’Reilly, R.J.; Willemze, R.; Falkenburg, J.H. Multiple minor histocompatibility antigen-specific cytotoxic T lymphocyte clones can be generated during graft rejection after HLA-identical bone marrow transplantation. Bone Marrow Transpl. 1995, 16, 125–132.
- Marijt, W.A.; Veenhof, W.F.; Brand, A.; Goulmy, E.; Fibbe, W.E.; Willemze, R.; van Rood, J.J.; Falkenburg, J.H. Minor histocompatibility antigen-specific cytotoxic T Cell lines, capable of lysing human hematopoietic progenitor cells, can be generated in vitro by stimulation with HLA-identical bone marrow cells. J. Exp. Med. 1991, 173, 101–109.
- Taylor, M.A.; Ward, B.; Schatzle, J.D.; Bennett, M. Perforin- and Fas-dependent mechanisms of natural killer cell-mediated rejection of incompatible bone marrow cell grafts. Eur. J. Immunol. 2002, 32, 793–799.
- Bennett, M.; Taylor, P.A.; Austin, M.; Baker, M.B.; Schook, L.; Rutherford, M.S.; Kumar, V.; Podack, E.R.; Mohler, K.M.; Levy, R.B.; et al. Cytokine and cytotoxic pathways of NK cell rejection of class I-deficient bone marrow grafts: Influence of mouse colony environment. Int. Immunol. 1998, 10, 785–790.
- Lapidot, T.; Faktorowich, Y.; Lubin, I.; Reisner, Y. Enhancement of T-cell-depleted bone marrow allografts in the absence of graft-versus-host disease is mediated by CD8+ CD4− and not by CD8- CD4+ thymocytes. Blood 1992, 80, 2406–2411.
- Martin, P.J. Prevention of allogeneic marrow graft rejection by donor T Cells that do not recognize recipient alloantigens: Potential role of a veto mechanism. Blood 1996, 88, 962–969.
- Martin, P.J. Donor CD8 cells prevent allogeneic marrow graft rejection in mice: Potential implications for marrow transplantation in humans. J. Exp. Med. 1993, 178, 703–712.
- Murphy, W.J.; Bennett, M.; Kumar, V.; Longo, D.L. Donor-type activated natural killer cells promote marrow engraftment and B cell development during allogeneic bone marrow transplantation. J. Immunol. 1992, 148, 2953–2960.
- Fujisaki, J.; Wu, J.; Carlson, A.L.; Silberstein, L.; Putheti, P.; Larocca, R.; Gao, W.; Saito, T.I.; Celso, C.L.; Tsuyuzaki, H.; et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 2011, 474, 216–219.
- Joffre, O.; Santolaria, T.; Calise, D.; Al Saati, T.; Hudrisier, D.; Romagnoli, P.; Van Meerwijk, J.P.M. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat. Med. 2008, 14, 88–92.
- Barao, I.; Hanash, A.M.; Hallett, W.; Welniak, L.A.; Sun, K.; Redelman, D.; Blazar, B.R.; Levy, R.B.; Murphy, W.J. Suppression of natural killer cell-mediated bone marrow cell rejection by CD4+CD25+ regulatory T Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 5460–5465.
- Park, J.H.; Lee, J.-H.; Lee, J.-H.; Park, H.-S.; Choi, E.-J.; Kang, Y.-A.; Kang, H.; Woo, J.M.; Lee, Y.-S.; Jeon, M.; et al. Incidence, Management, and Prognosis of Graft Failure and Autologous Reconstitution after Allogeneic Hematopoietic Stem Cell Transplantation. J. Korean Med. Sci. 2021, 36, e151.
- Servais, S.; Beguin, Y.; Baron, F. Emerging drugs for prevention of graft failure after allogeneic hematopoietic stem cell transplantation. Expert Opin. Emerg. Drugs 2013, 18, 173–192.
- Astor, B.C.; Melamed, M.L.; Mandelbrot, D.A.; Djamali, A. Seasonality of mortality and graft failure among kidney transplant recipients in the US—A retrospective study. Transpl. Int. 2018, 31, 293–301.
- Tinckam, K. Histocompatibility methods. Transpl. Rev. 2009, 23, 80–93.
- Lentine, K.L.; Smith, J.; Hart, A.; Miller, J.; Skeans, M.; Larkin, L.; Robinson, A.; Gauntt, K.; Israni, A.; Hirose, R.; et al. OPTN/SRTR 2020 Annual Data Report: Kidney. Am. J. Transpl. 2022, 22 (Suppl. 2), 21–136.
- Kloc, M.; Ghobrial, R.M. Chronic allograft rejection: A significant hurdle to transplant success. Burn. Trauma 2014, 2, 3–10.
- Bestard, O.; Nickel, P.; Cruzado, J.M.; Schoenemann, C.; Boenisch, O.; Sefrin, A.; Grinyó, J.M.; Volk, H.-D.; Reinke, P. Circulating Alloreactive T Cells Correlate with Graft Function in Longstanding Renal Transplant Recipients. J. Am. Soc. Nephrol. 2008, 19, 1419–1429.
- Kishimoto, K.; Sandner, S.; Imitola, J.; Sho, M.; Li, Y.; Langmuir, P.B.; Rothstein, D.M.; Strom, T.B.; Turka, L.A.; Sayegh, M.H. Th1 cytokines, programmed cell death, and alloreactive T Cell clone size in transplant tolerance. J. Clin. Investig. 2002, 109, 1471–1479.
- Kythreotou, A.; Siddique, A.; Mauri, F.A.; Bower, M.; Pinato, D.J. PD-L1. J. Clin. Pathol. 2018, 71, 189–194.
- Liang, S.C.; Latchman, Y.E.; Buhlmann, J.E.; Tomczak, M.F.; Horwitz, B.H.; Freeman, G.J.; Sharpe, A.H. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur. J. Immunol. 2003, 33, 2706–2716.
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369.
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992, 11, 3887–3895.
- Chen, L.; Flies, D.B. Molecular mechanisms of T Cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242.
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242.
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346.e3.
- Zhulai, G.; Oleinik, E. Targeting regulatory T cells in anti-PD-1/PD-L1 cancer immunotherapy. Scan. J. Immunol. 2022, 95, e13129.
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T Cells. J. Exp. Med. 2009, 206, 3015–3029.
- Bretscher, P.A. A two-step, two-signal model for the primary activation of precursor helper T Cells. Proc. Natl. Acad. Sci. USA 1999, 96, 185–190.
- Patsoukis, N.; Duke-Cohan, J.S.; Chaudhri, A.; Aksoylar, H.-I.; Wang, Q.; Council, A.; Berg, A.; Freeman, G.J.; Boussiotis, V.A. Interaction of SHP-2 SH2 domains with PD-1 ITSM induces PD-1 dimerization and SHP-2 activation. Commun. Biol. 2020, 3, 128.
- Chikuma, S.; Terawaki, S.; Hayashi, T.; Nabeshima, R.; Yoshida, T.; Shibayama, S.; Okazaki, T.; Honjo, T. PD-1-mediated suppression of IL-2 production induces CD8+ T Cell anergy in vivo. J. Immunol. 2009, 182, 6682–6689.
- Boussiotis, V.A.; Chatterjee, P.; Li, L. Biochemical signaling of PD-1 on T Cells and its functional implications. Cancer J. 2014, 20, 265–271.
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T Cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217.
- Appleman, L.J.; van Puijenbroek, A.A.F.L.; Shu, K.M.; Nadler, L.M.; Boussiotis, V.A. CD28 costimulation mediates down-regulation of p27kip1 and cell cycle progression by activation of the PI3K/PKB signaling pathway in primary human T Cells. J. Immunol. 2002, 168, 2729–2736.
- Riley, J.L. PD-1 signaling in primary T Cells. Immunol. Rev. 2009, 229, 114–125.
- Tang, L.; Ma, S.; Gong, H.; Wang, J.; Xu, Y.; Wu, D.; Sun, A. PD-L1 Ameliorates Murine Acute Graft-versus-Host Disease by Suppressing Effector but Not Regulatory T Cells Function. Arch. Immunol. Ther. Exp. 2019, 67, 179–187.
- Li, X.; Deng, R.; He, W.; Liu, C.; Wang, M.; Young, J.; Meng, Z.; Du, C.; Huang, W.; Chen, L.; et al. Loss of B7-H1 expression by recipient parenchymal cells leads to expansion of infiltrating donor CD8+ T Cells and persistence of graft-versus-host disease. J. Immunol. 2012, 188, 724–734.
- Saha, A.; O’connor, R.S.; Thangavelu, G.; Lovitch, S.; Dandamudi, D.B.; Wilson, C.B.; Vincent, B.G.; Tkachev, V.; Pawlicki, J.M.; Furlan, S.; et al. Programmed death ligand-1 expression on donor T Cells drives graft-versus-host disease lethality. J. Clin. Investig. 2016, 126, 2642–2660.
- Hu, Q.-Y.; Yuan, Y.; Li, Y.-C.; Yang, L.-Y.; Zhou, X.-Y.; Xiong, D.-Q.; Zhao, Z.-Y. Programmed Cell Death Ligand 1-Transfected Mouse Bone Marrow Mesenchymal Stem Cells as Targeted Therapy for Rheumatoid Arthritis. BioMed Res. Int. 2021, 2021, 5574282.
- Tanaka, K.; Albin, M.J.; Yuan, X.; Yamaura, K.; Habicht, A.; Murayama, T.; Grimm, M.; Waaga, A.M.; Ueno, T.; Padera, R.F.; et al. PDL1 is required for peripheral transplantation tolerance and protection from chronic allograft rejection. J. Immunol. 2007, 179, 5204–5210.
- Cha, J.-H.; Chan, L.-C.; Li, C.-W.; Hsu, J.L.; Hung, M.-C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370.
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 Pathway: Signaling, Cancer, and Beyond. Adv. Exp. Med. Biol. 2020, 1248, 33–59.
- Hall, K.H.; Liu, Y.; Jiang, C.; Harvey, R.D. New and Worsening Long-term Immune-Related Adverse Events with PD-1/PD-L1 Pathway Agents in Patients with Cancer. Pharmacotherapy 2020, 40, 133–141.
- Delanoy, N.; Michot, J.-M.; Comont, T.; Kramkimel, N.; Lazarovici, J.; Dupont, R.; Champiat, S.; Chahine, C.; Robert, C.; Herbaux, C.; et al. Haematological immune-related adverse events induced by anti-PD-1 or anti-PD-L1 immunotherapy: A descriptive observational study. Lancet Haematol 2019, 6, e48–e57.
- Canavan, M.; Floudas, A.; Veale, D.J.; Fearon, U. The PD-1:PD-L1 axis in Inflammatory Arthritis. BMC Rheumatol. 2021, 5, 1.
- Wu, Q.; Jiang, L.; Li, S.-C.; He, Q.-J.; Yang, B.; Cao, J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol. Sin. 2021, 42, 1–9.
- Xiong, W.; Gao, Y.; Wei, W.; Zhang, J. Extracellular and nuclear PD-L1 in modulating cancer immunotherapy. Trends Cancer 2021, 7, 837–846.
- Wang, Y.; Huang, L.; Huang, T.; Geng, S.; Chen, X.; Huang, X.; Lai, P.; Du, X.; Weng, J. The Gut Bacteria Dysbiosis Contributes to Chronic Graft-versus-Host Disease Associated with a Treg/Th1 Ratio Imbalance. Front. Microbiol. 2022, 13, 813576.
- Ying, H.; Zhang, X.; Duan, Y.; Lao, M.; Xu, J.; Yang, H.; Liang, T.; Bai, X. Non-cytomembrane PD-L1: An atypical target for cancer. Pharmacol. Res. 2021, 170, 105741.
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 2021, 14, 10.
- Elalouf, A. Infections after organ transplantation and immune response. Transpl. Immunol. 2023, 77, 101798.
- Vietzen, H.; Furlano, P.L.; Cornelissen, J.J.; Böhmig, G.A.; Jaksch, P.; Puchhammer-Stöckl, E. HLA-E-restricted immune responses are crucial for the control of EBV infections and the prevention of PTLD. Blood 2023, 141, 1560–1573.
- Park, J.-I.; Song, G.-W.; Ryu, J.H.; Choi, S.-T.; Choi, N.-G.; Jung, B.-H.; Chu, C.W.; Kim, K.-K.; Jung, D.-H.; Ha, T.-Y.; et al. A Multicenter, Randomized, Open-Label Study to Compare the Efficacy and Safety of Tacrolimus and Corticosteroids in Combination with or without Mycophenolate Mofetil in Liver Transplantation Recipients Infected with Hepatitis B Virus. Transplant Proc. 2023, 55, 387–395.
- Burek Kamenaric, M.; Ivkovic, V.; Kovacevic Vojtusek, I.; Zunec, R. The Role of HLA and KIR Immunogenetics in BK Virus Infection after Kidney Transplantation. Viruses 2020, 12, 1417.
More
Information
Subjects:
Allergy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
680
Revisions:
2 times
(View History)
Update Date:
25 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No