Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Laura Palombi | -- | 2799 | 2023-06-21 12:09:59 | | | |
| 2 | Rita Xu | Meta information modification | 2799 | 2023-06-26 03:27:22 | | | | |
| 3 | Rita Xu | Meta information modification | 2799 | 2023-06-26 04:12:01 | | | | |
| 4 | Rita Xu | Meta information modification | 2799 | 2023-06-26 04:12:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Arcadi, A.; Morlacci, V.; Palombi, L. Sequential Reactions of α-Aminoalkynes with Carbonyls. Encyclopedia. Available online: https://encyclopedia.pub/entry/45930 (accessed on 24 July 2026).
Arcadi A, Morlacci V, Palombi L. Sequential Reactions of α-Aminoalkynes with Carbonyls. Encyclopedia. Available at: https://encyclopedia.pub/entry/45930. Accessed July 24, 2026.
Arcadi, Antonio, Valerio Morlacci, Laura Palombi. "Sequential Reactions of α-Aminoalkynes with Carbonyls" Encyclopedia, https://encyclopedia.pub/entry/45930 (accessed July 24, 2026).
Arcadi, A., Morlacci, V., & Palombi, L. (2023, June 21). Sequential Reactions of α-Aminoalkynes with Carbonyls. In Encyclopedia. https://encyclopedia.pub/entry/45930
Arcadi, Antonio, et al. "Sequential Reactions of α-Aminoalkynes with Carbonyls." Encyclopedia. Web. 21 June, 2023.
Copy Citation
Sequential reactions of aminoalkynes represent a powerful tool to easily assembly biologically important polyfunctionalized nitrogen heterocyclic scaffolds. Metal catalysis often plays a key role in terms of selectivity, efficiency, atom economy, and green chemistry of these sequential approaches.
sequential reactions
aminoalkynes
heterocycles
1. Introduction
Aminoalkynes are bifunctional derivatives that can undergo a diverse array of transformations. They offer sequential reactions with an electrophile and a nucleophile, and are ideal for cascade reactions. Sequential reactions represent a powerful tool to build up simple or more complex polyfunctionalized organic scaffolds from readily available reagents with high efficiency, selectivity, and atom economy [1][2][3]. Recently, applications of sequential reactions of aminoalkynes have been a very active research field in organic synthesis and medicinal chemistry. In particular, sequential reactions of β-, γ-, and δ-aminoalkynes to afford a variety of heterocyclic scaffolds were explored. Inactivated alkynes moieties are not very reactive toward nucleophiles. Their behavior changes by activation of the C-C triple bond by a metal catalyst. Various biologically important nitrogen heterocycles were directly synthesized in an easy way by means of intramolecular hydroamination of aminoalkynes in the presence of several transition metal as well as lanthanide catalysts [4][5]. The aptitude to form π- and σ-complexes can help in the choice of catalysts for the desired transformations when bi- or polyfunctional substrates are involved [6]. The reaction of γ- and δ-aminoalkynes with sulfonyl azides in the presence of Ru3(CO)12 catalyst efficiently afforded cyclic amidines of relevance in medicinal and coordination chemistry as well as in materials science [7]. The gold(I)-catalyzed tandem cyclization of γ-aminoalkynes with alkynes in water led to diversely substituted pyrrolo[1,2-a]quinolines [8]. Zhou et al. extended this reaction using less active terminal amidoalkynes in similar conditions [9]. The CuCl-catalyzed cascade transformation of internal β-aminoalkynes with alkynes under microwave irradiation gave diversely substituted tetrahydropyrrolo[1,2-a]quinolones [10]. An intramolecular gold-catalyzed hydroamination/aza-Diels–Alder tandem process of β-/γ-aminoalkynes with high regio- and diastereoselectivity and up to almost complete chemoselectivity showed great efficiency in a one-pot approach to the complex nitrogen heterocyclic derivatives of medicinal importance, such as the one-step synthesis of incargranine B aglycone and (±)-seneciobipyrrolidine (I) [11]. Faňanás, Rodríguez, and co-workers [12] described the preparation of complex pyrrolidines from readily available N-Boc-derived β-aminoalkynes and alkenes or alkynes through relay actions of PtII or Brønsted acids [13]. The reaction of α-aminoalkynes with carbon monoxide and selenium yielded 5-alkylideneselenazolin-2-ones stereoselectively via cycloaddition of in situ generated carbamoselenoates to a carbon–carbon triple bond. β-Aminoalkyne also afforded the corresponding six-membered selenium-containing heterocycle with the aid of CuI [14]. An operationally simple palladium-catalyzed intramolecular hydroaminocarbonylation of a variety of aminoalkynes directly provided a viable approach to a variety of valuable seven- and eight-membered lactams with high chemoselectivity and regioselectivity [15]. A sequential heterogeneous PtI2-catalyzed hydration of δ-aminoalkynes followed by intramolecular cyclization and intermolecular addition as well as ring-expansion cascade reaction with another electron-deficient alkynes was developed for the synthesis of various eight-membered nitrogen heterocycles with excellent yields under mild reaction conditions. The simple PtI2 could be easily recycled [16]. Moreover, a PtI2-catalyzed formal three-component cascade cycloaddition reactions between γ-aminoalkynes and electron-deficient alkynes gave highly functionalized cyclohexadiene-b-pyrrolidines with good yields [17]. Finally, among the domino and multicomponent processes that involve aminoalkynes, their cascade reactions with carbonyl derivatives stand out as a highly versatile tool to build up libraries of nitrogen-containing heterocyclic scaffolds with diversity and molecular complexity.
2. Sequential Reactions of α-Aminoalkynes (Propargylamines) with Carbonyls
Propargylic amine derivatives represent useful α-aminoalkynes building blocks for the construction of nitrogen-containing heterocyclic scaffolds through their sequential reactions with carbonyls. The gold-catalyzed reaction of propargylamine 1 with dialkyl acyclic/cyclic ketones, methyl, aryl/heteroaryl ketones and aldehydes bearing α-hydrogens 2 allowed a simple approach to pyridines 3 through a sequential amination–cyclization–aromatization cascade (Scheme 1) [18].
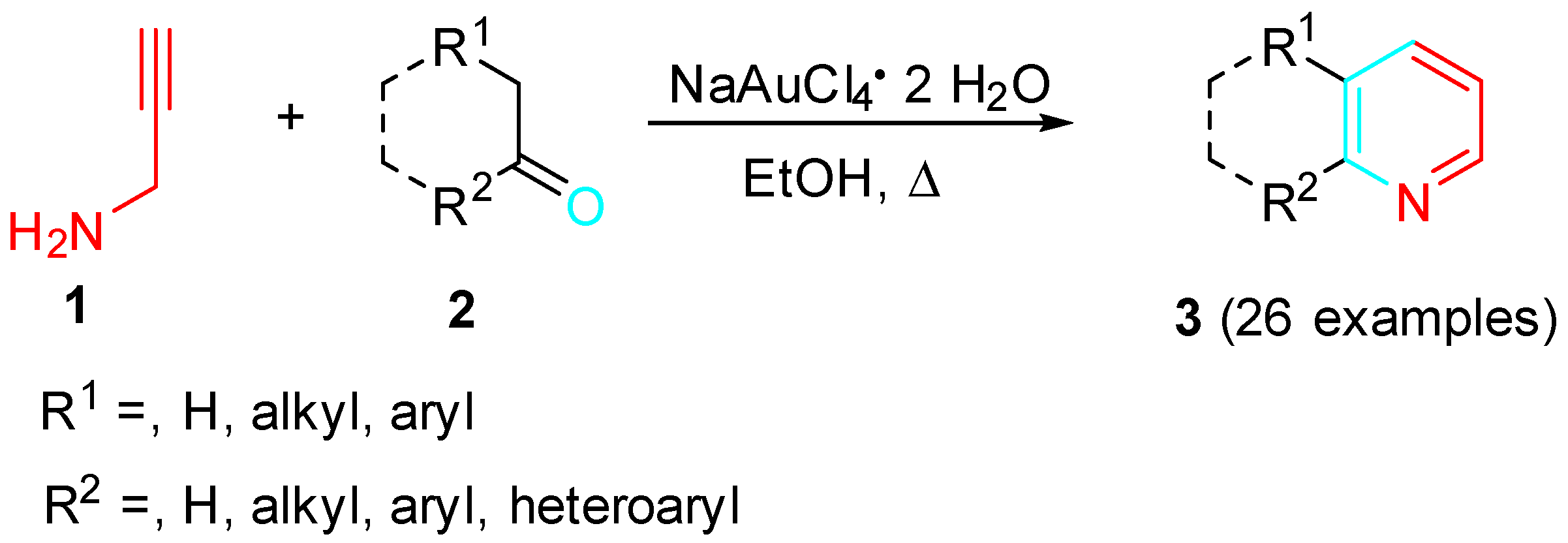
Scheme 1. Gold-catalyzed synthesis of pyridines from propargylamine and carbonyls.
The catalyst was envisaged to promote both the amination of carbonyl compounds 2 and the regioselective 6-endo-dig cyclization of the N-propargylenamine (N-propargyldienamine) intermediate 5 (Scheme 2).
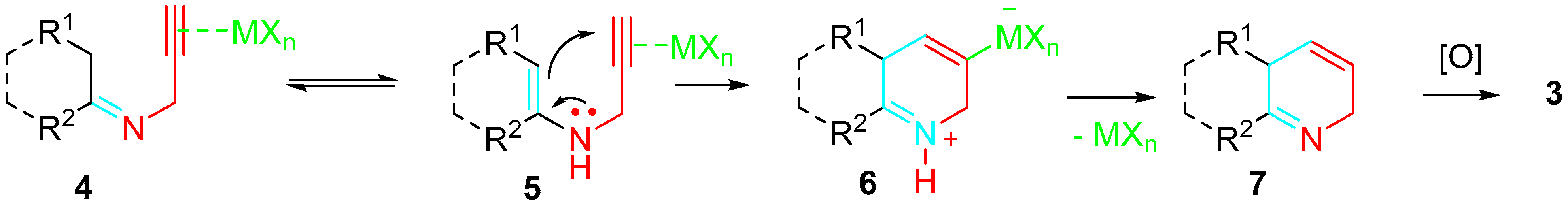
Scheme 2. Transition metal-catalyzed sequential amination–cyclization–aromatization reaction.
A variety of catalysts were tested in the reaction of 1 with 2. In particular, NaAuCl4·2H2O resulted in a highly efficient catalyst. Moreover, Au 8 was synthesized and applied as bifunctional catalyst. It was found that imidazolyl group acted as a Lewis base to catalyze the condensation of carbonyl compounds with propargylamine to form the imino intermediate, and the involved Au+-complex species activated the alkynyl moiety to give the dehydropyridine derivative, which underwent auto-oxidation reaction to afford the target pyridines (Scheme 3) [19].
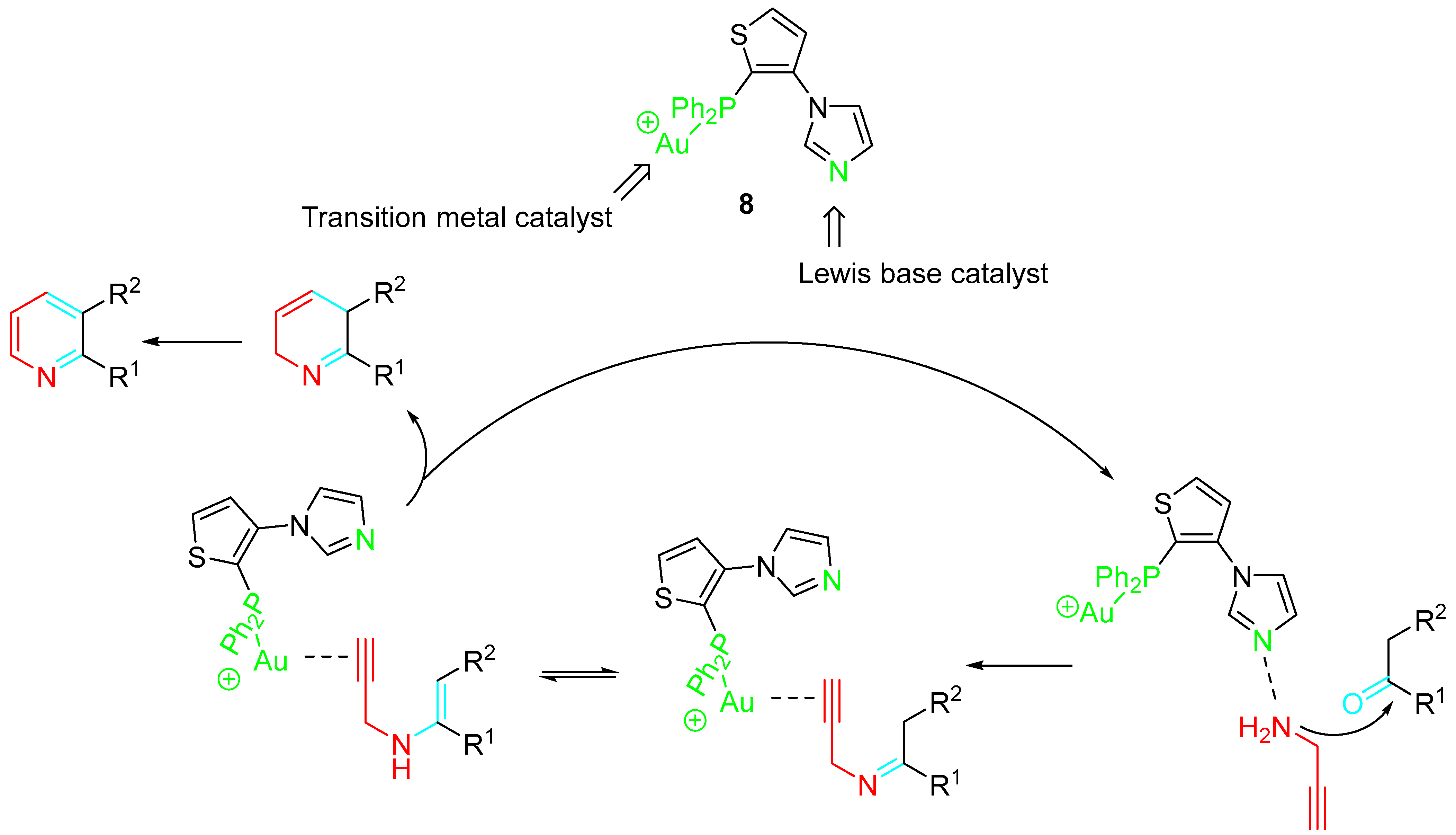
Scheme 3. Sequential reactions of carbonyl compounds and propargylamine catalyzed by Au-complex 8.
Copper salts were also effective catalysts in the reaction of cyclic ketones with propargylamine, and the highest product yields were observed in isopropanol (i-PrOH) in the presence of 5.0 mol% CuCl2 in air. Decreased yields among cyclic ketones were observed in the following order: six-membered >> eight-membered > five-membered ∼ seven-membered. However, the inexpensiveness of the catalyst and the tolerance to a wide number of functional groups (FG) in the ketone make the procedure very suitable for large-scale preparation of fused pyridines (Scheme 4) [20].
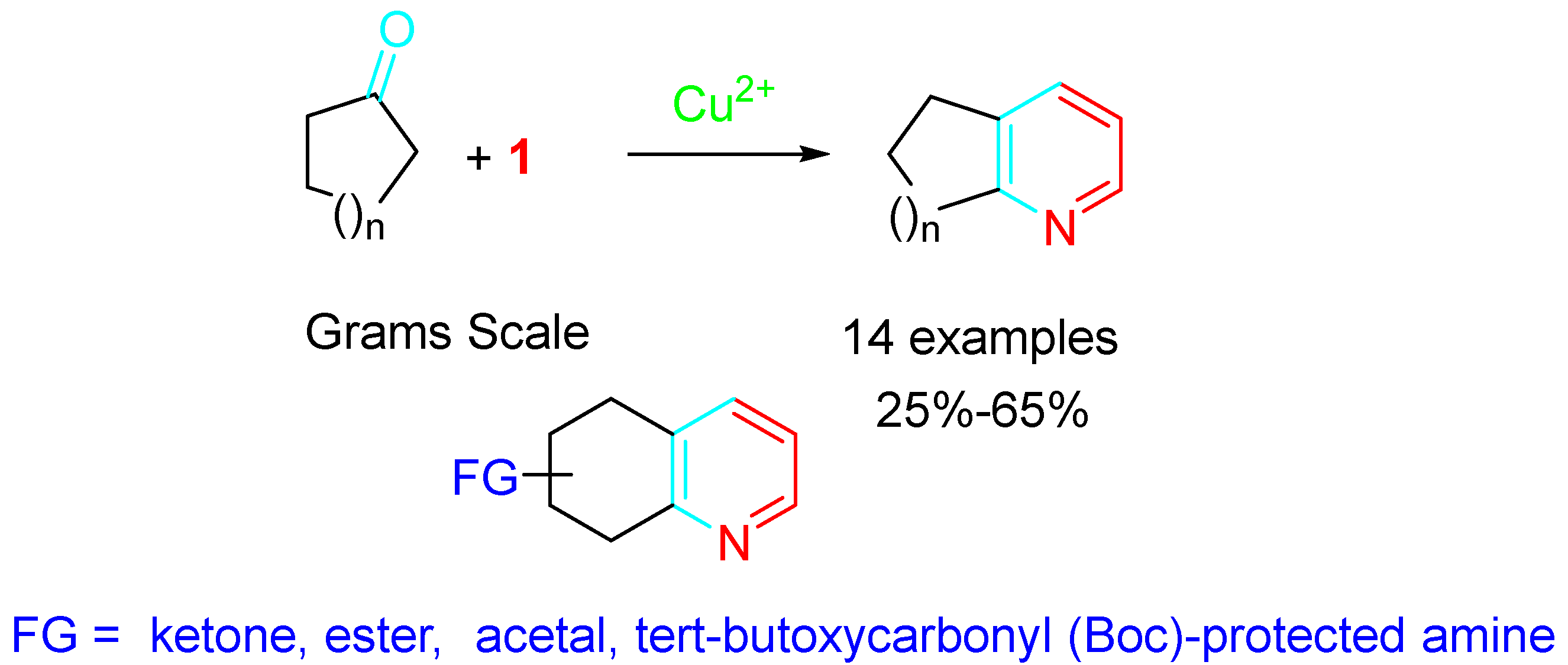
Scheme 4. Cu-catalyzed pyridine synthesis from cyclic ketones and propargylamine.
Selective aspects of the reaction of steroidal carbonyls with propargylamine were investigated. According to the results, the regioselective pyridine fusion to the cyclic skeleton was addressed by suitable choice between the substrate bearing a saturated or conjugated carbonyl group (Scheme 5) [18].
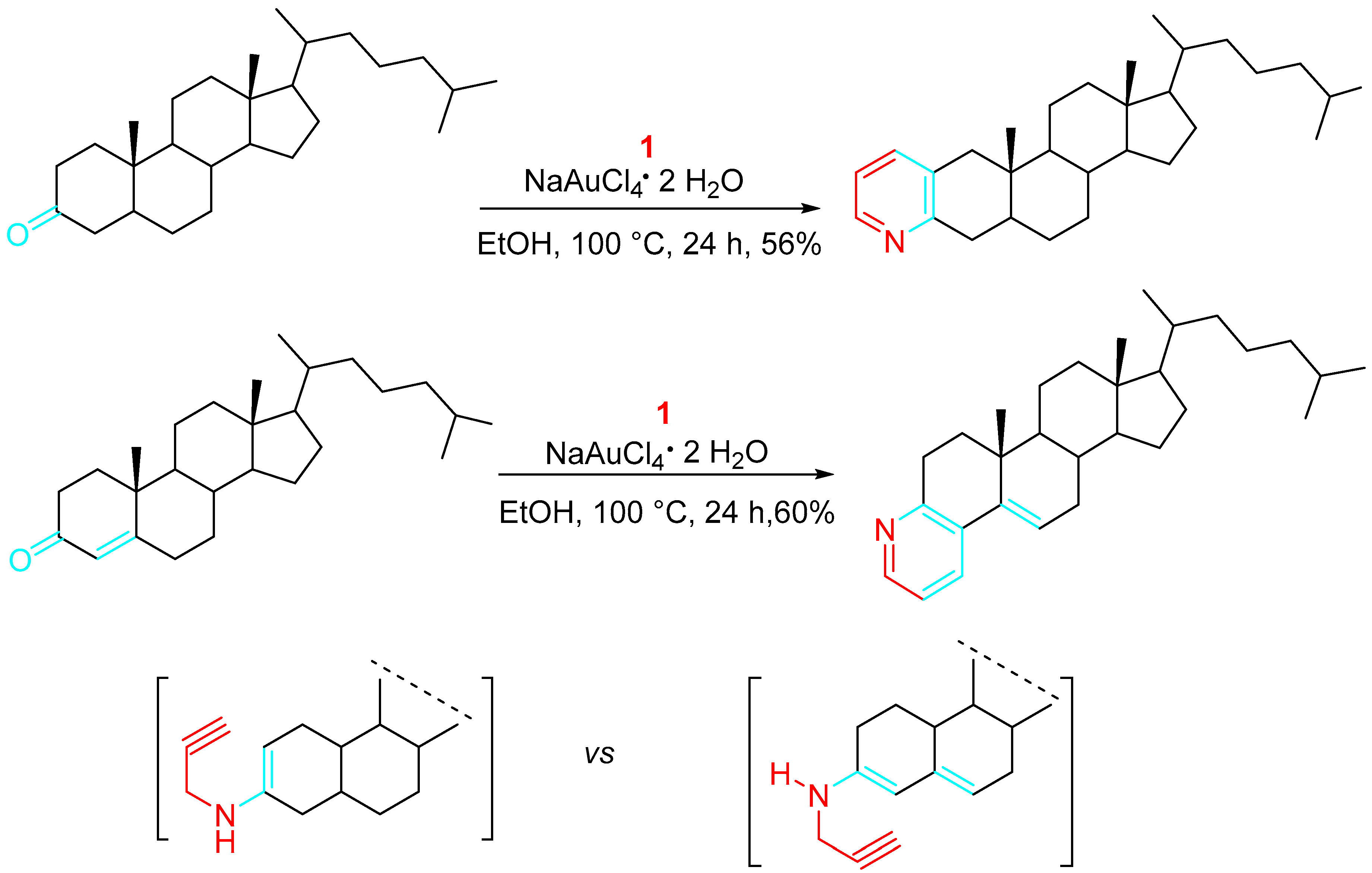
Scheme 5. Linear vs. angular steroidal pyridines.
Analogously, new A-ring pyridine fused androstanes in 17a-homo-17-oxa (D-homo lactone), 17α-picolyl or 17(E)-picolinylidene series were obtained by reacting 4-en-3-one or 4-ene-3,6-dione D-modified androstane derivatives with propargylamine under the presence of a Cu(II) catalyst, and evaluated for potential anticancer activity in vitro (Scheme 6) [21].
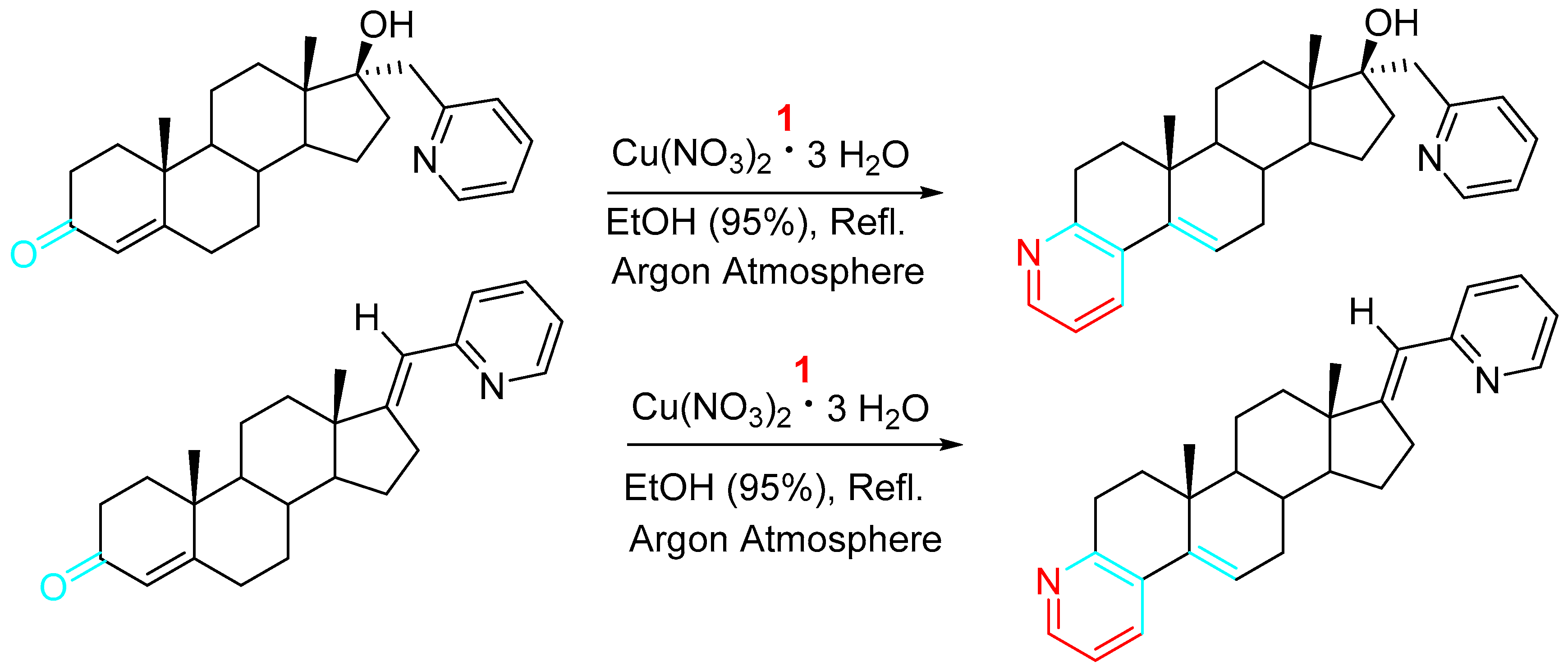
Scheme 6. Copper-catalyzed synthesis of A-ring fused pyridine D-modified androstane derivatives.
Similarly, the efficient synthesis of pyridine rings fused to the 3,4-positions of the steroid nucleus was described via the Cu(II)-catalyzed reaction of propargylamine with 17β-hydroxyandrost-4-en-3-one, 17α-methyl-17β-hydroxyandrost-4-en-3-one, or 17β-hydroxyestr-4-en-3-one [22]. The procedure was also applied to the synthesis of heterocyclic betulin derivatives (Scheme 7) [23][24].
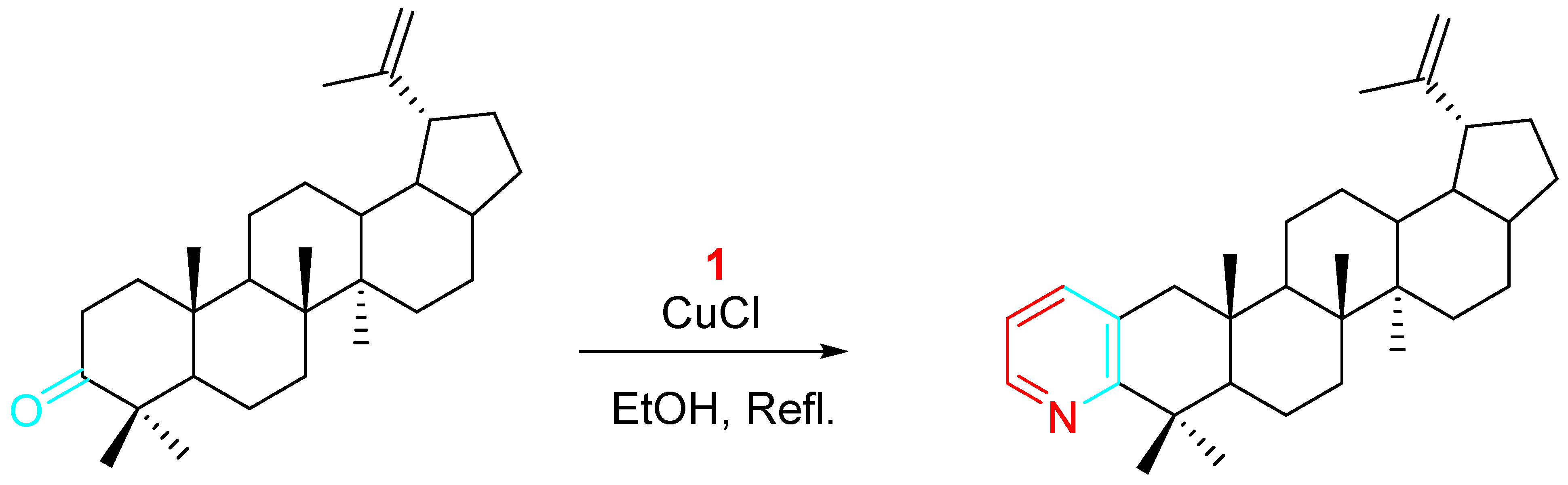
Scheme 7. Copper-catalyzed synthesis of betulin derivatives.
Optimization of the synthesis of steroidal pyridines was tried by prolonging the reaction time and varying the catalyst loading. In some cases, the use of NaAuCl4·2H2O instead of CuCl and the addition of activated molecular sieves (MS) to the reaction mixture led to significant improvement (Scheme 8) [25].
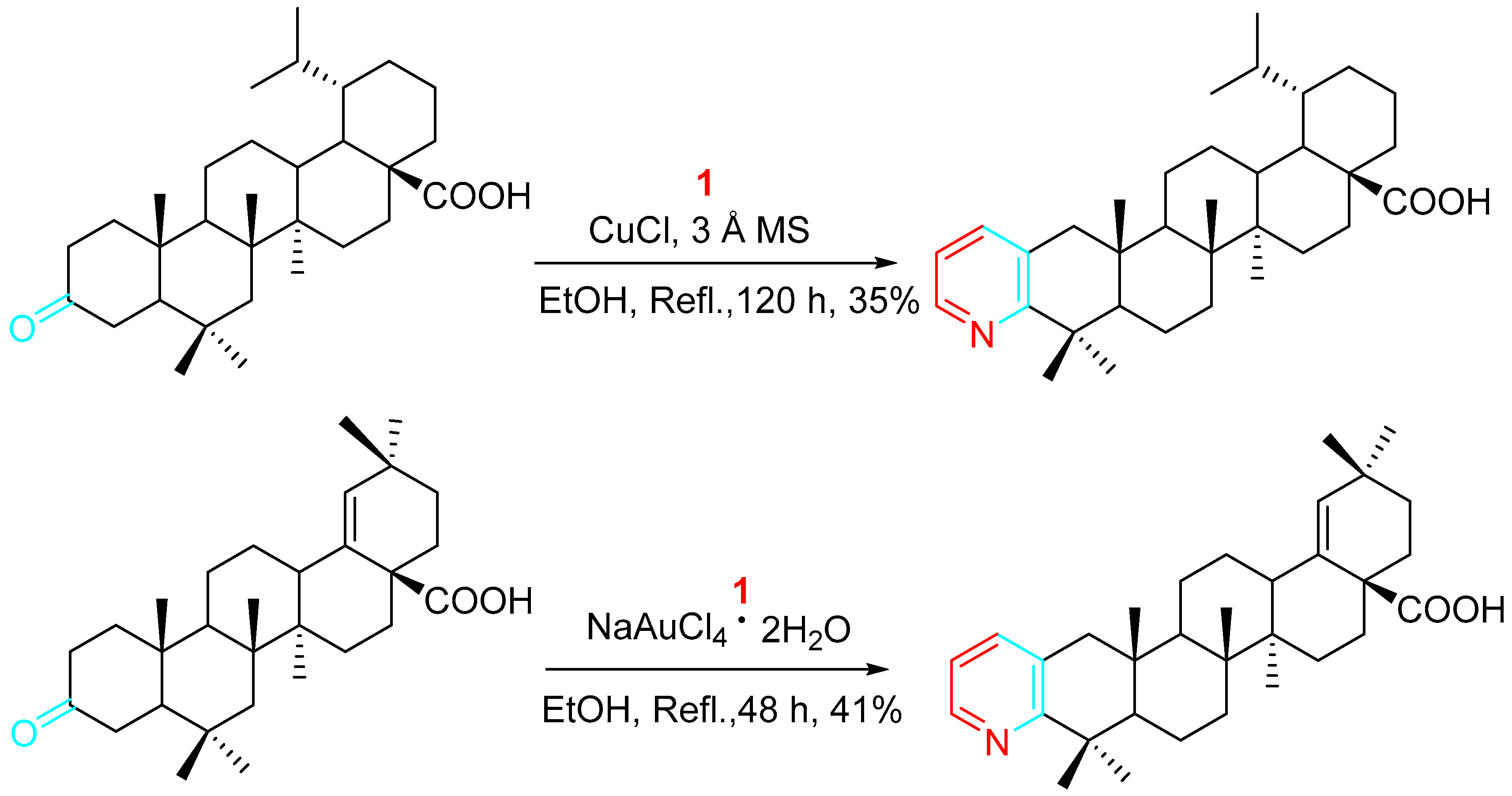
Scheme 8. Optimization of the synthesis of relevant steroidal pyridines.
Several other applications of the methodology accomplished the preparation of significant scaffolds. Indeed, the chemical synthesis of highly potent and acid-stable inhibitors of hedgehog signaling carbacyclopamine analogue 11 was reported. The gold-catalyzed amination–annulation–aromatization sequence applied to the inseparable mixture of the isomers 9 and 10 regioselectively furnished, after removal of the tert-butyldimethylsilyl ether (tetrabutylammonium fluoride, THF, 25 °C), carbacyclopamine analogue 11 with 36% overall yield for the two steps (Scheme 9) [26].
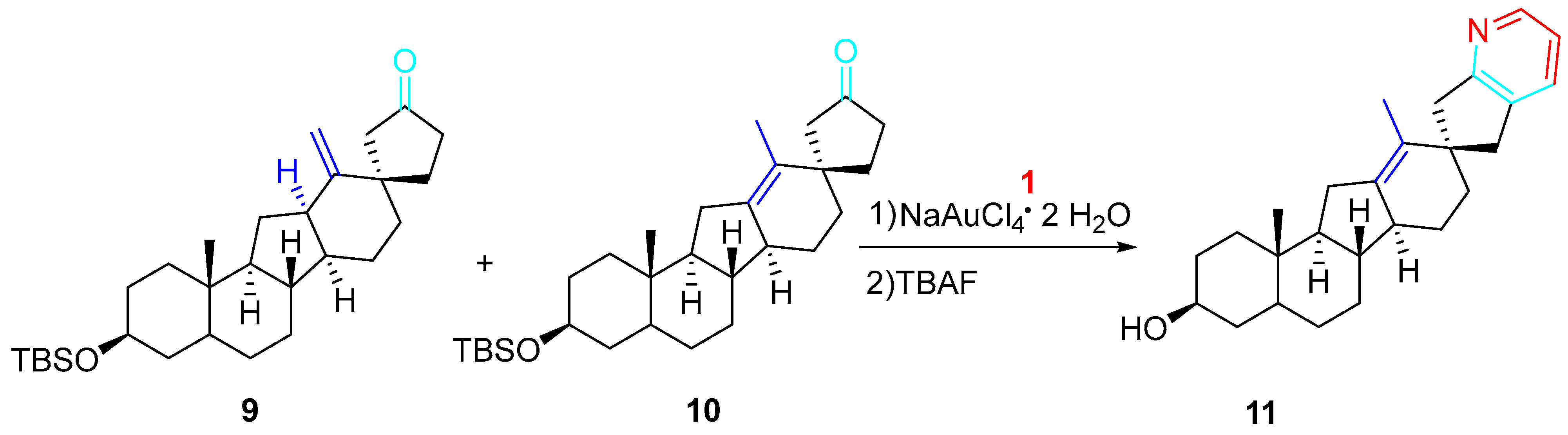
Scheme 9. Gold-catalyzed synthesis of the carbacycloamine analogue 11.
Furthermore, the gold-catalyzed reaction of 1-benzylpiperidin-4-one with propargylamine efficiently afforded the potassium channel modulator 6-benzyl-5,6,7,8-tetrahydro-1,6-naphthirine 12 (Scheme 10) [27].
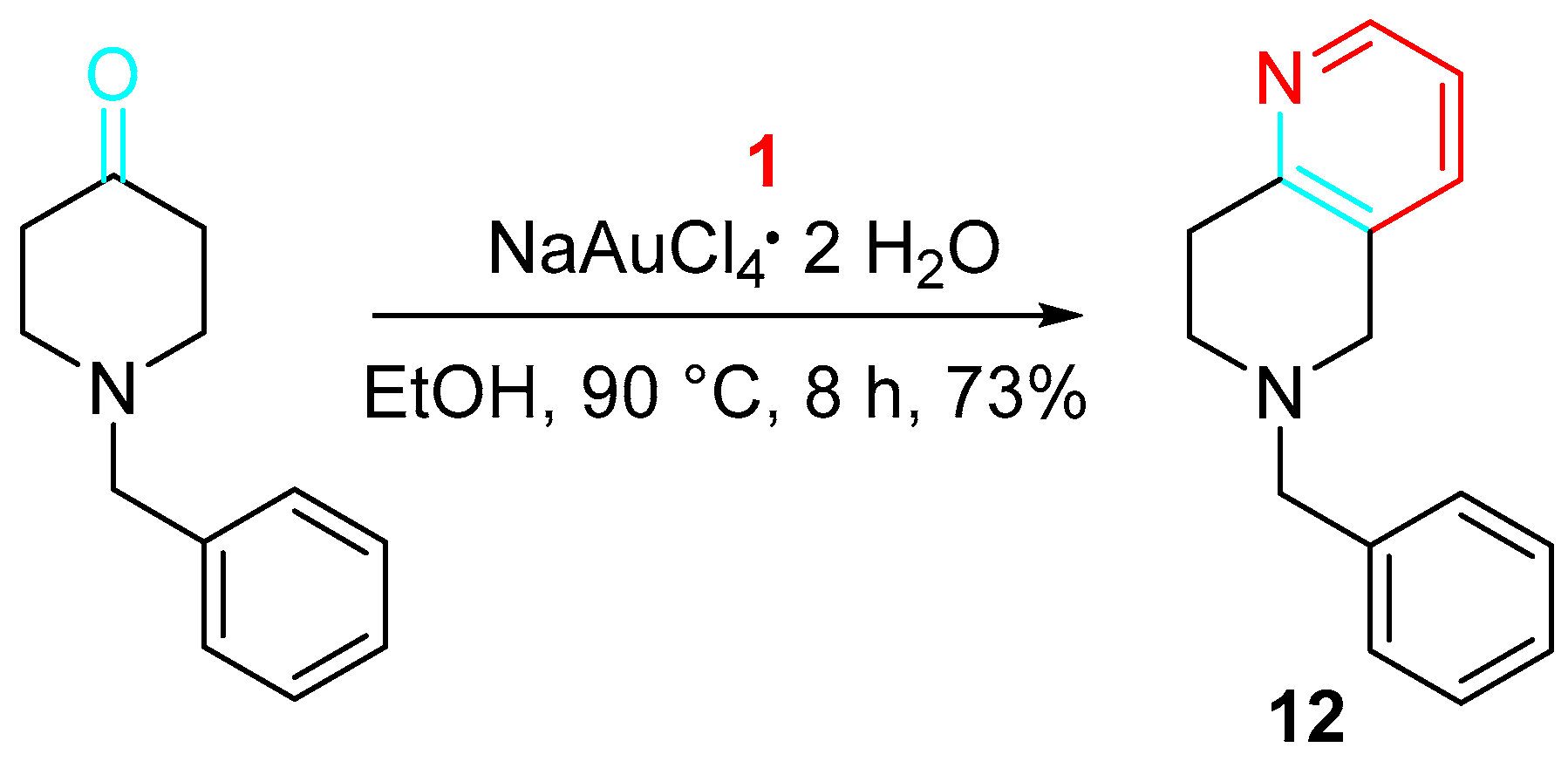
Scheme 10. Gold-catalyzed synthesis of the potassium channel modulator 12.
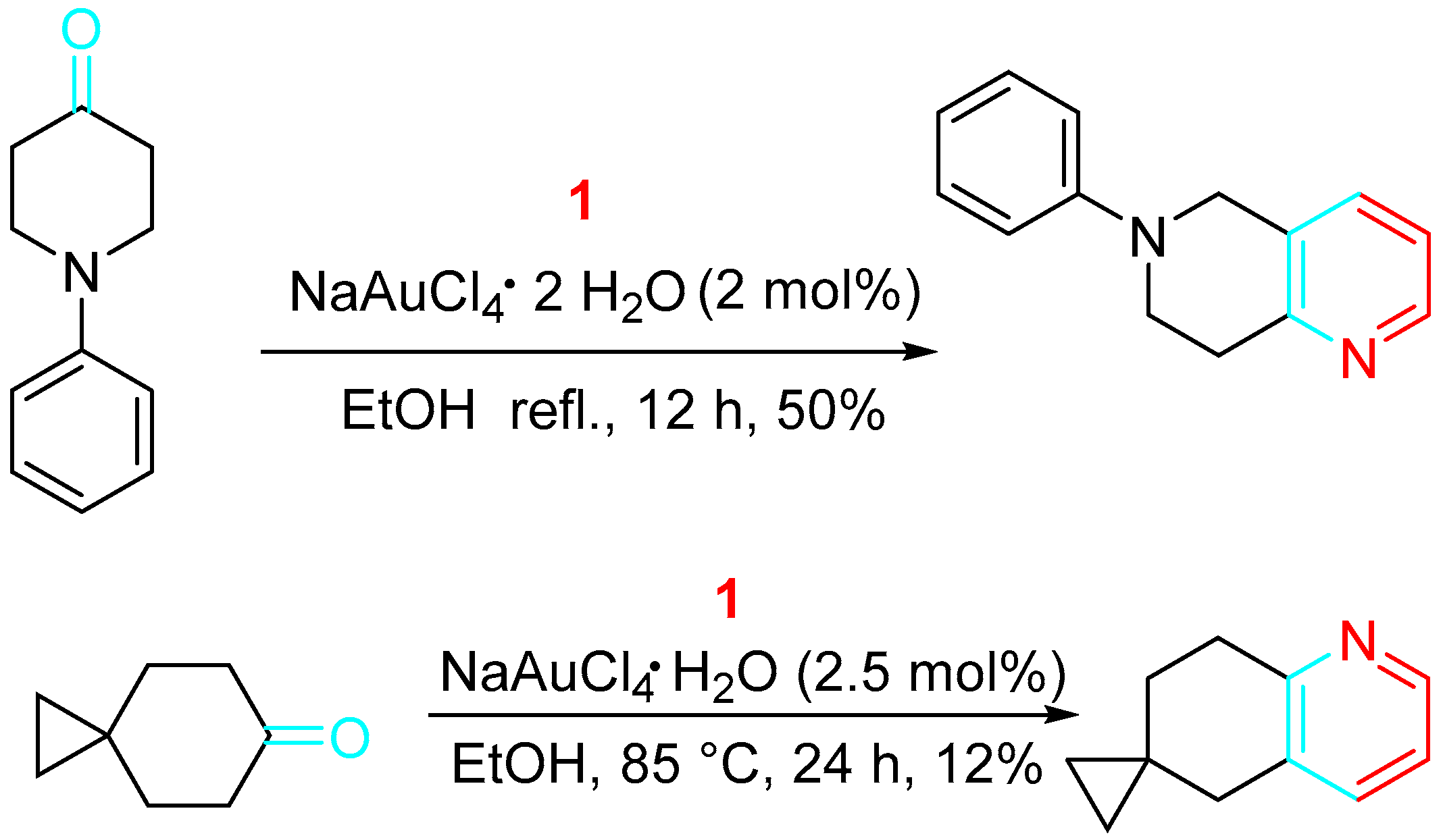
Scheme 11. Synthesis of Wnt signal path inhibitors.
The gold-catalyzed synthesis of BAY-298, a nanomolar small molecule (SMOL) hLH-R antagonist, reducing sex hormone levels in vivo has been described (Scheme 12) [30].
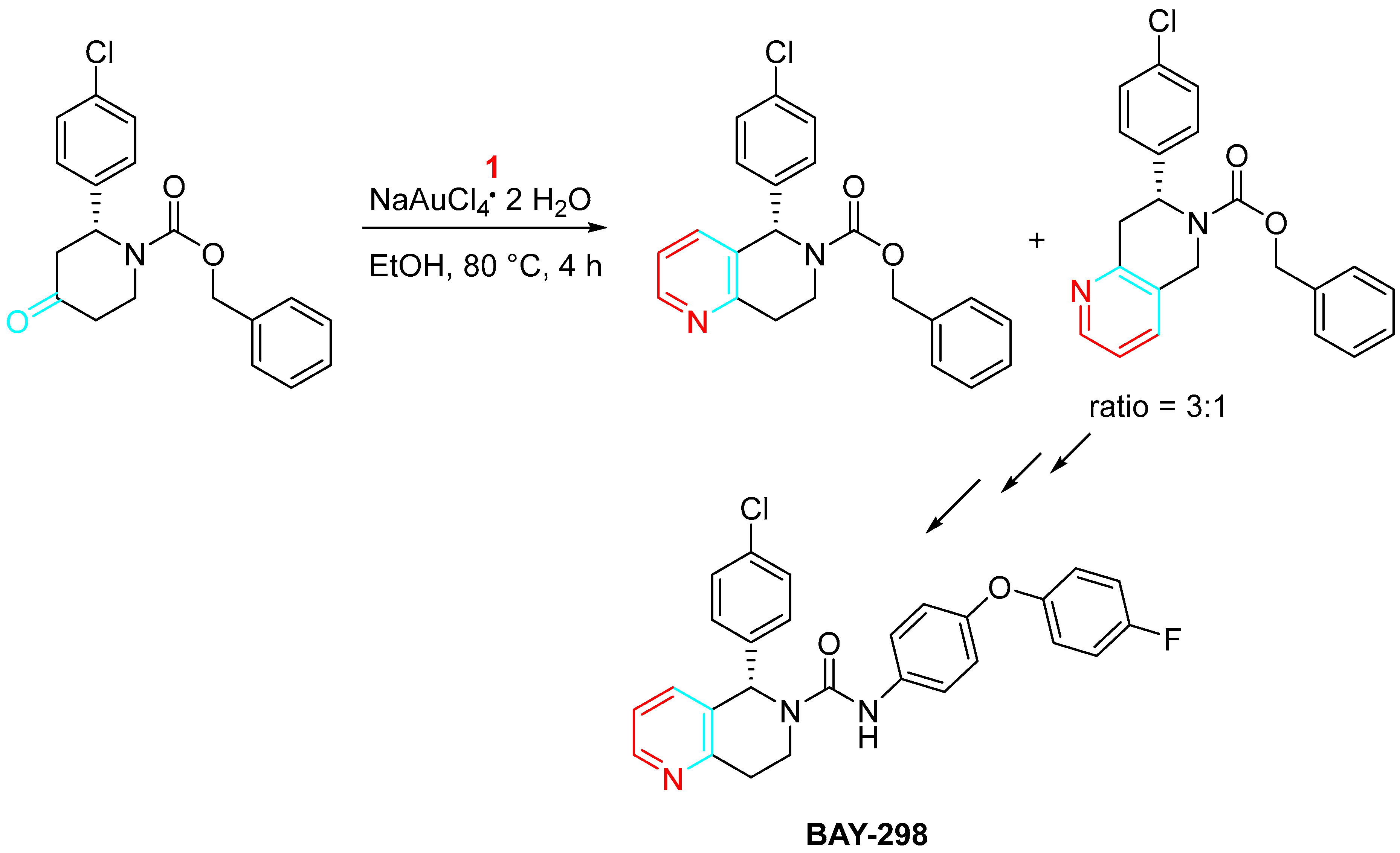
Scheme 12. Gold-catalyzed synthesis of BAY-298.
Moreover, the gold-catalyzed sequential condensation–cyclization–aromatization procedure was extended as the key step for the preparation of BMS-846372, a potent and orally active human CGRP receptor antagonist employed for migraine therapy (Scheme 13) [31].
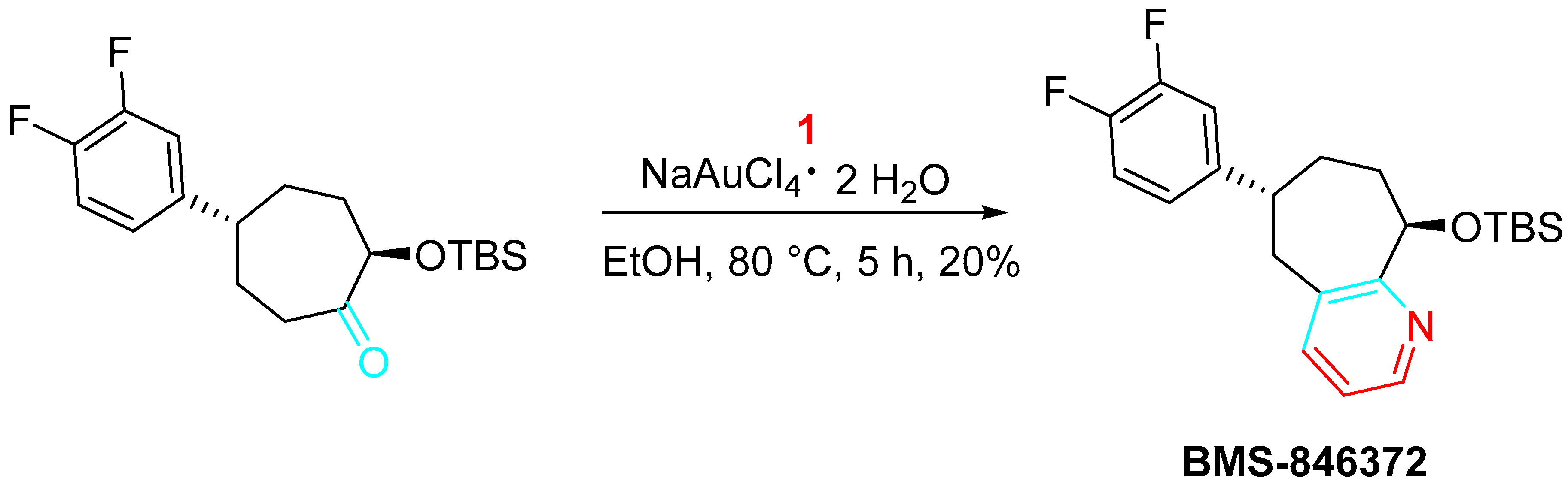
Scheme 13. Synthesis of BMS-846372.
The methodology also resulted in a viable tool for the preparation of aryl and heteroaryl derivatives of benzomorphanes 13, pharmacologically active as inhibitors of 11ß-hydroxysteroid dehydrogenase (HSD1) (Scheme 14) [32].
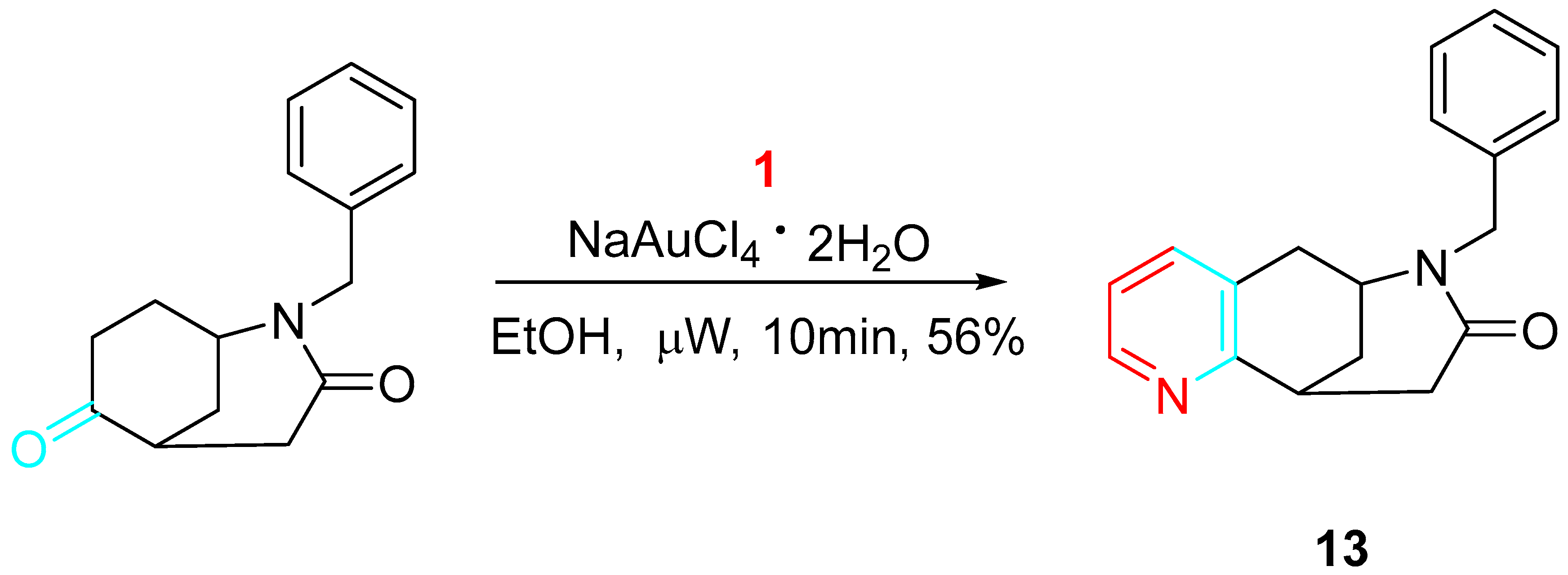
Scheme 14. Gold-catalyzed synthesis of the benzomorphane derivative 13.
The synthetic approach has yielded a number of scaffolds suitable for the design of performance-diverse screening libraries (Scheme 15) [33].
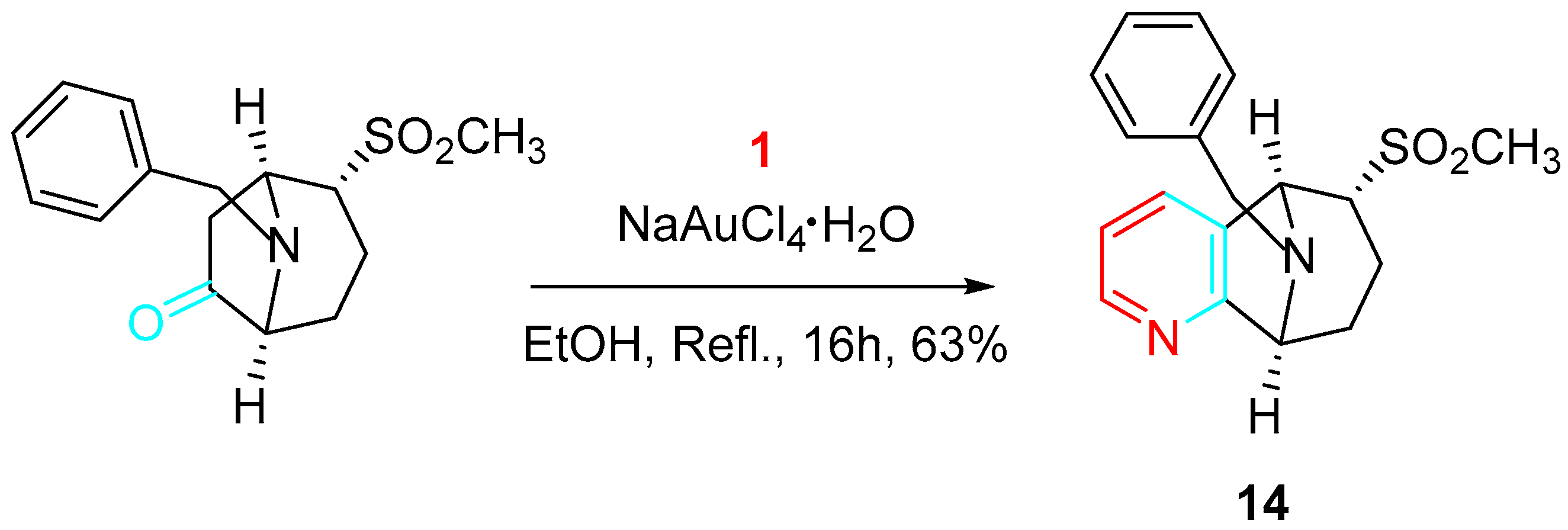
Scheme 15. Gold-catalyzed synthesis of the tropane-related scaffold 14.
The reaction of 2-tetralones and propargylamine in the presence of complexes of gold or copper, preferably NaAuCl4 and CuC1, was employed to synthesize octahydrobenzoquinoline derivatives 16 as inhibitors of 11β-hydroxysteroid dehydrogenase for the treatment of metabolic disorders, such as metabolic syndrome, diabetes, obesity, and dyslipidemia. The reaction is usually run in alcohols at temperatures ranging from 20 to 120 °C through conventional heating or microwave irradiation. The resulting pyridine was reduced to the corresponding piperidine (Scheme 16) [34].
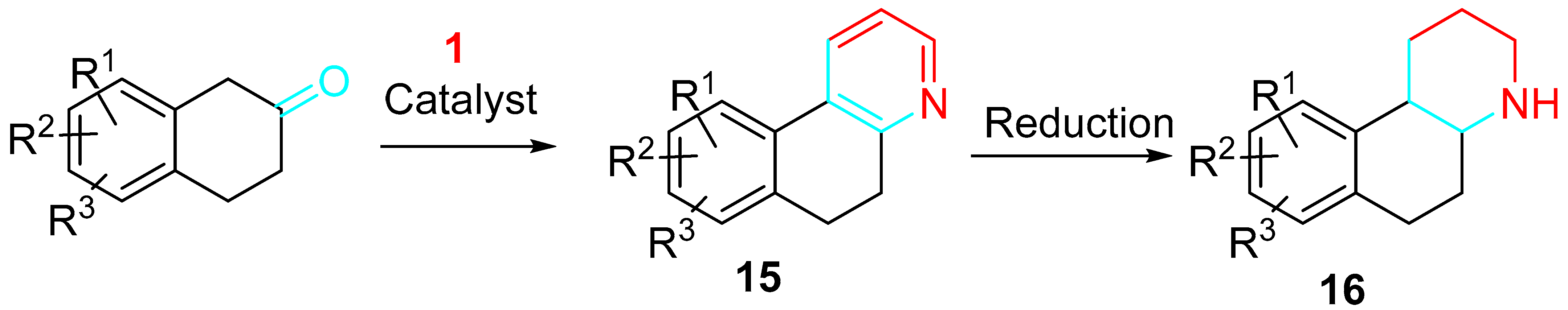
Scheme 16. Synthesis of 11β-hydroxysteroid dehydrogenase inhibitors.
The sequential gold-catalyzed condensation/annulation reaction of the 1,3-dihyrdo-2H-inden-2-one with the propargylamine provided the corresponding 9H-indeno [2,1-b]pyridine 17 as the ligand for the synthesis of an olefin polymerization catalyst (Scheme 17) [35].
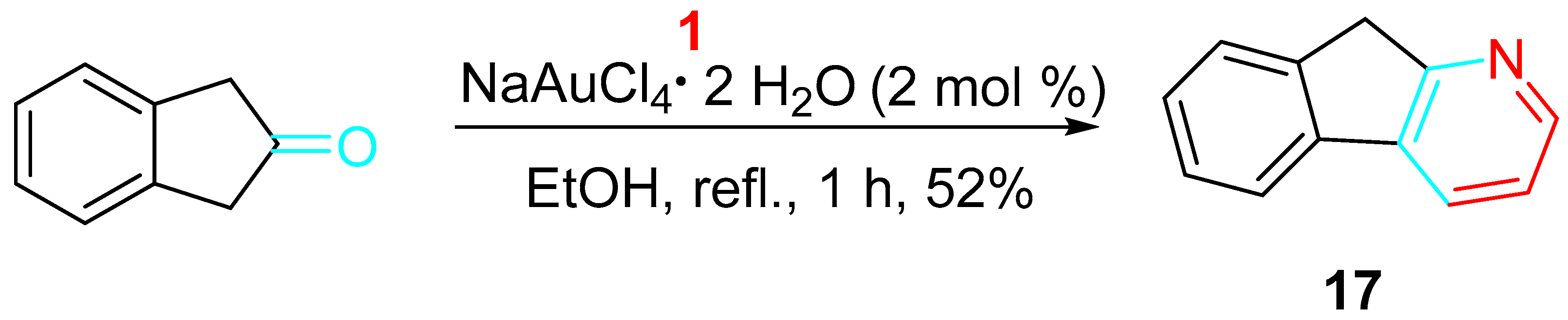
Scheme 17. Gold-catalyzed synthesis of the ligand 17.
The gold(III)-catalyzed reaction of simple β-ketoesters with propargylamines achieved the synthesis of potentially bioactive 2,5-dihydropyridines 18 with satisfactory yields. The best results were observed using 5 mol% of the cheaper NaAuCl4 in MeOH as solvent. The dichloro(2-pyridinecarboxilato)gold (pic)AuCl2) resulted in a less effective catalyst, and the reaction failed to occur in the presence of (Ph3P)AuCl/AgOTf catalytic system or by using platinum(II) and platinum(IV) catalysts. Recovery of the starting materials when triflic acid (TfOH) was used instead of NaAuCl4 ruled out the formation of the product by Brønsted acid catalysis. Propargylamines unsubstituted at the triple bond (R4=H) or with an aromatic ring at this position gave higher yields than propargylamines bearing an aliphatic chain at the same position (Scheme 18) [36].
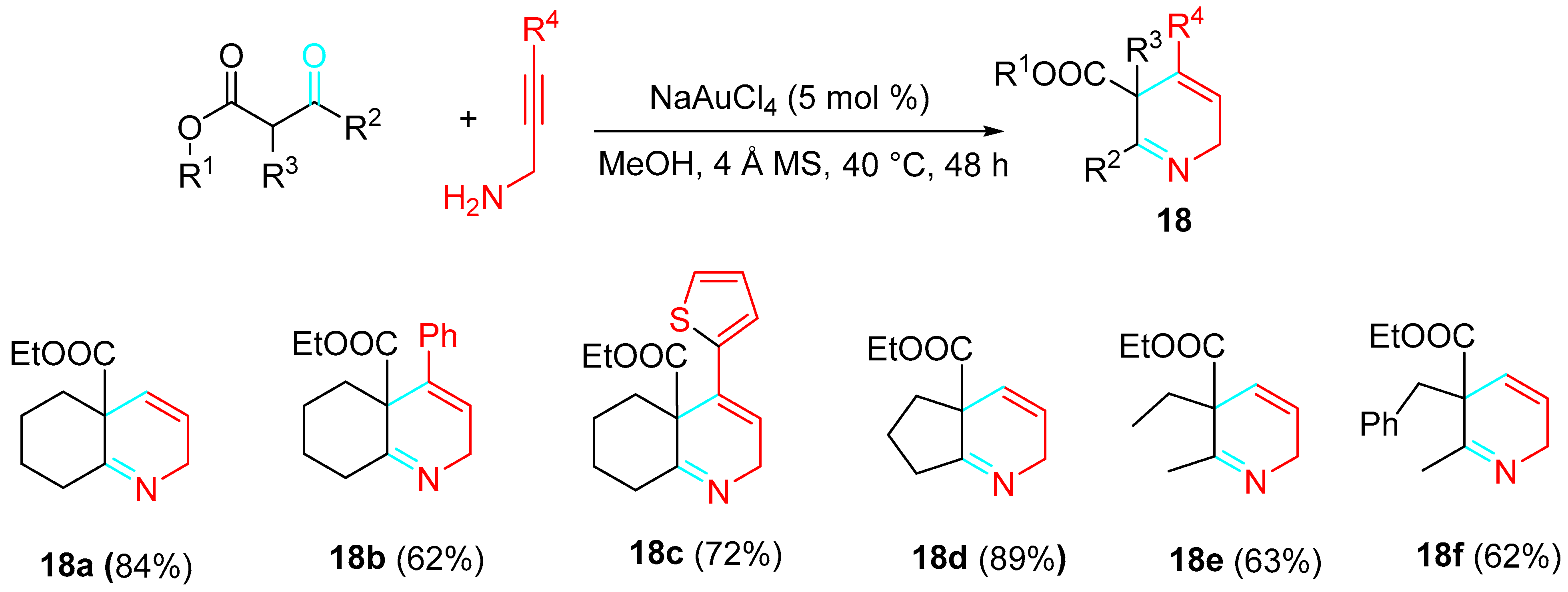
Scheme 18. Gold-catalyzed synthesis of 2,5-dihydropyridines 18.
Substituted pyridinium salts 19 were obtained under mild conditions by a condensation reaction between carbonyls and propargylamine under the presence of an Ag2CO3/HNTf2 synergistically acting catalyst system. The one-pot transformation should proceed via sequential 6-endo-dig cyclization of the in situ generated propargylenamine/protonolysis of the resulting vinyl–silver intermediate. The silver(I)-catalyzed cyclization reaction was exclusively selective for the formation of six-membered rings. Only 6-endo-dig cyclized pyridinium products were obtained, even with substrates bearing an electron-withdrawing group at the acetylenic position, which underwent unusual inversion of the reactivity usually observed in Michael-type reactions. CH3CN was the solvent of choice in this one-pot transformation (Scheme 19) [37].
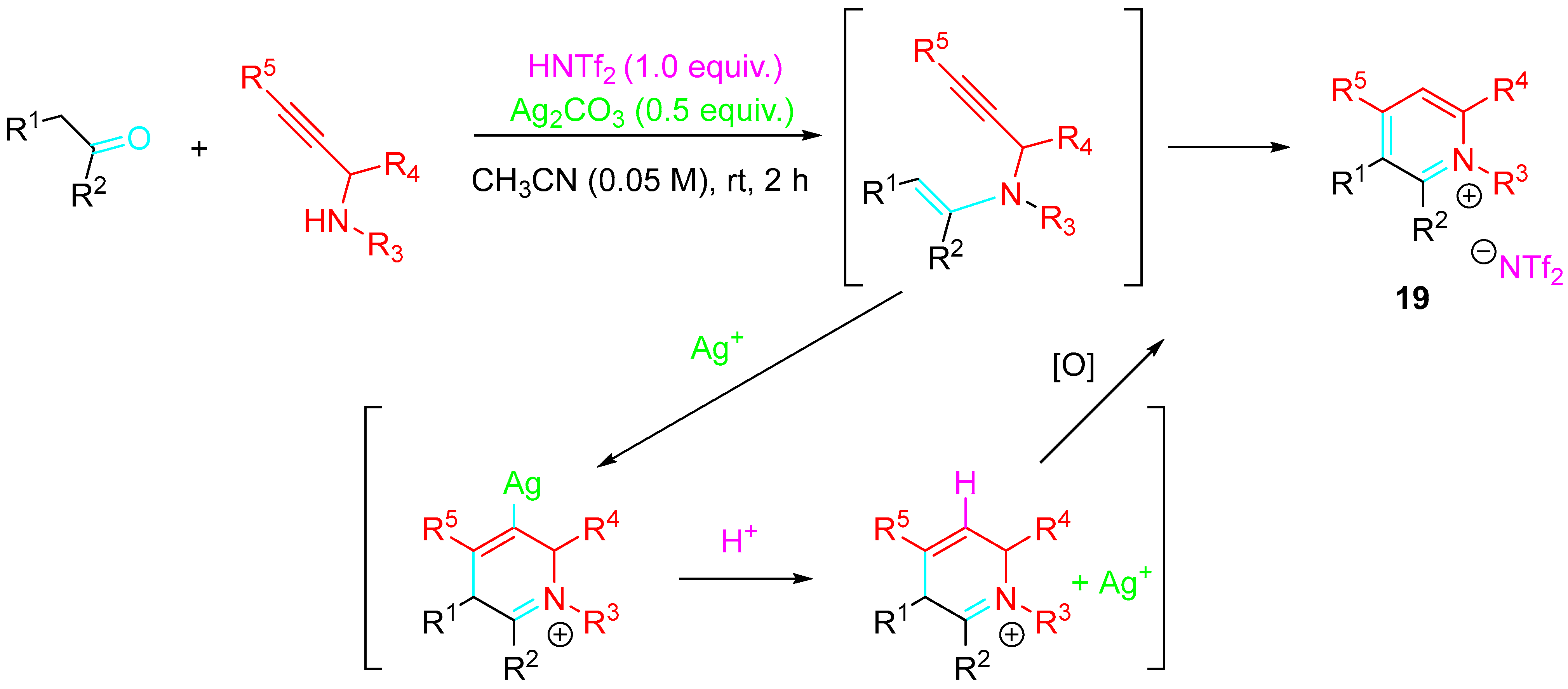
Scheme 19. Synthesis of pyridinium salts.
Interestingly, hetero-anthracene derivatives such as 20, used in the preparation of organic light-emitting devices, were practically obtained under metal-free conditions (Scheme 20) [38].
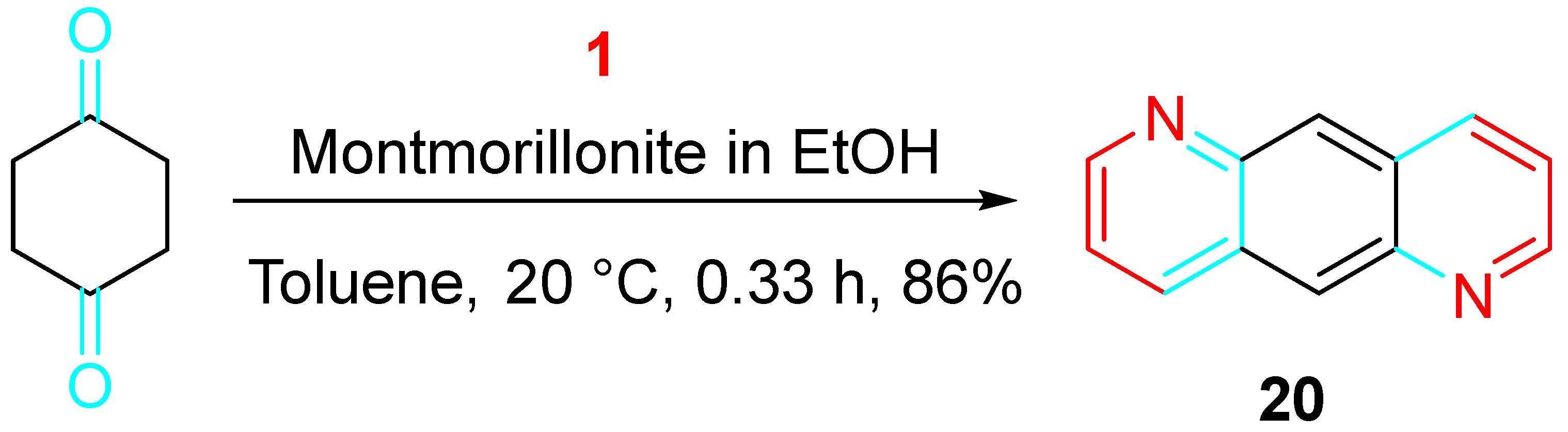
Scheme 20. Metal-free synthesis of the hetero-anthracene derivative 20.
Moreover, substituted dihydrophenanthrolines 21 were easily obtained from 2-substituted 6,7-dihydroquinoline-8(5H)-ketones and propargylamine in alcohol at 70–130 °C. This metal-free method has the advantages of safety, cleanness and wide substrate applicability. The product can be efficiently isolated by adjusting the temperature or prolonging the reaction time (Scheme 21) [39].
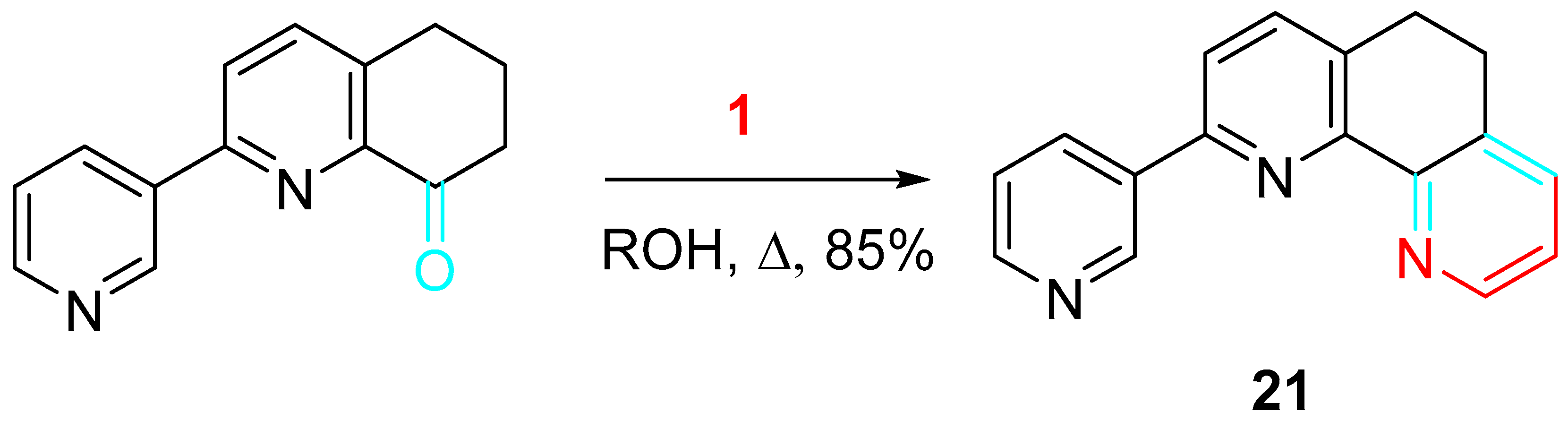
Scheme 21. Synthesis of the dihydrophenanthroline 21.
The reaction of readily available α,ß-unsaturated carbonyl compounds with propargylamine provided a high atom- and pot-economy strategy for the synthesis of polyfunctionalized pyridines under metal-free conditions with relevant functional group tolerance. The exploration of bases (CsCO3, NaHCO3, NaOAc, K2HPO4, DBU) and solvents (toluene, DCE, THF, DMSO, DMF) achieved the optimization of the reaction conditions by reacting the propargylamines with the unsaturated aldehydes in DMF in the presence of NaHCO3 at 80 °C [40]. The application to the synthesis of a variety of natural products was reported (Scheme 22) [41].
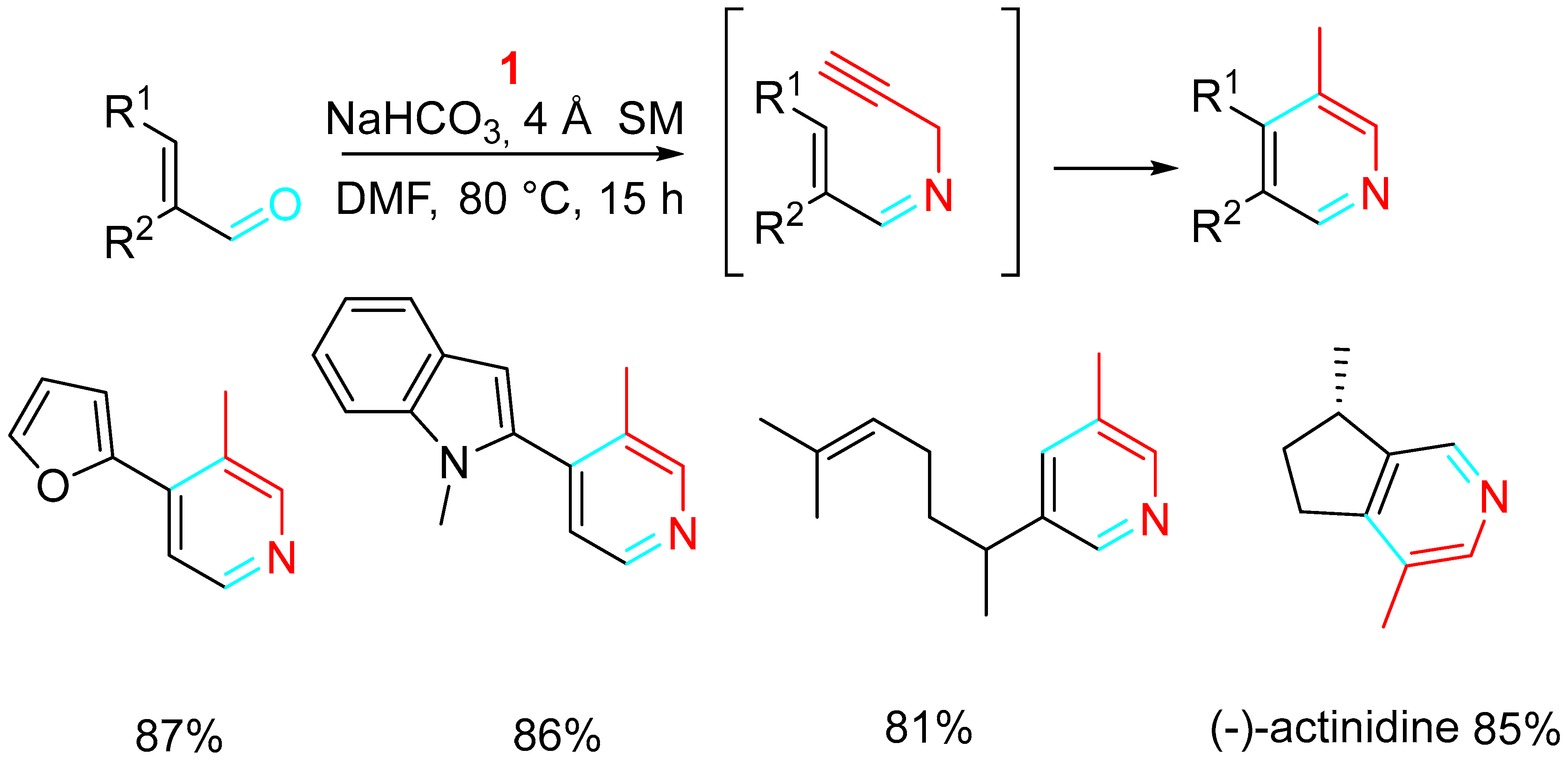
Scheme 22. Synthesis of natural products.
The method was applied to the synthesis of pyridines from the cosmetic, flavor and fragrance agent (S)-(−)-perillaldehyde and the flavoring agent found in cardamom, (1R)-myrtenal (Scheme 23).
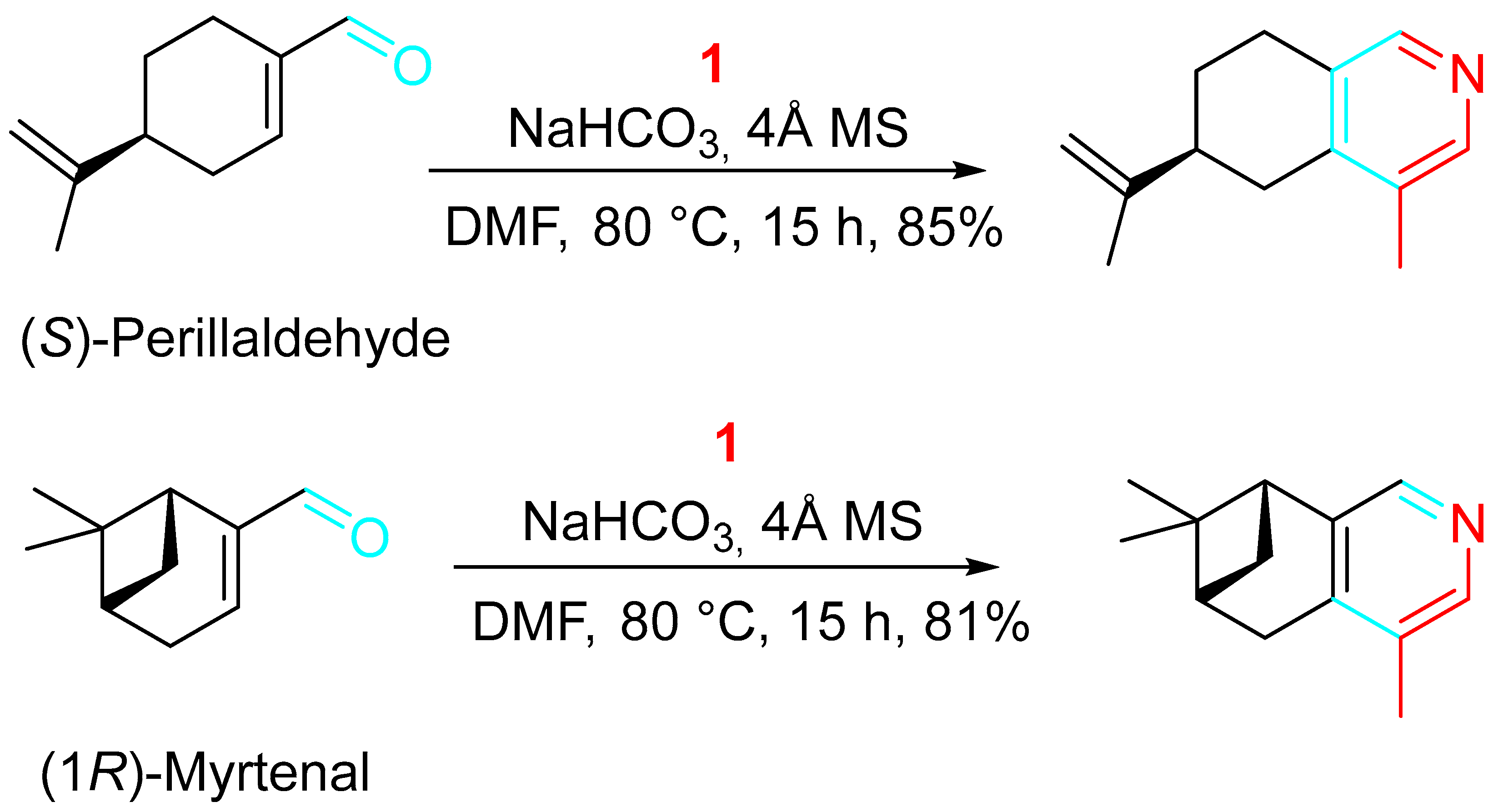
Scheme 23. Construction of pyridines starting from (S)-(−)-perillaldehyde and (1R)-myrtenal.
The process also achieved the total syntheses of suaveoline alkaloids (Scheme 24) [42].
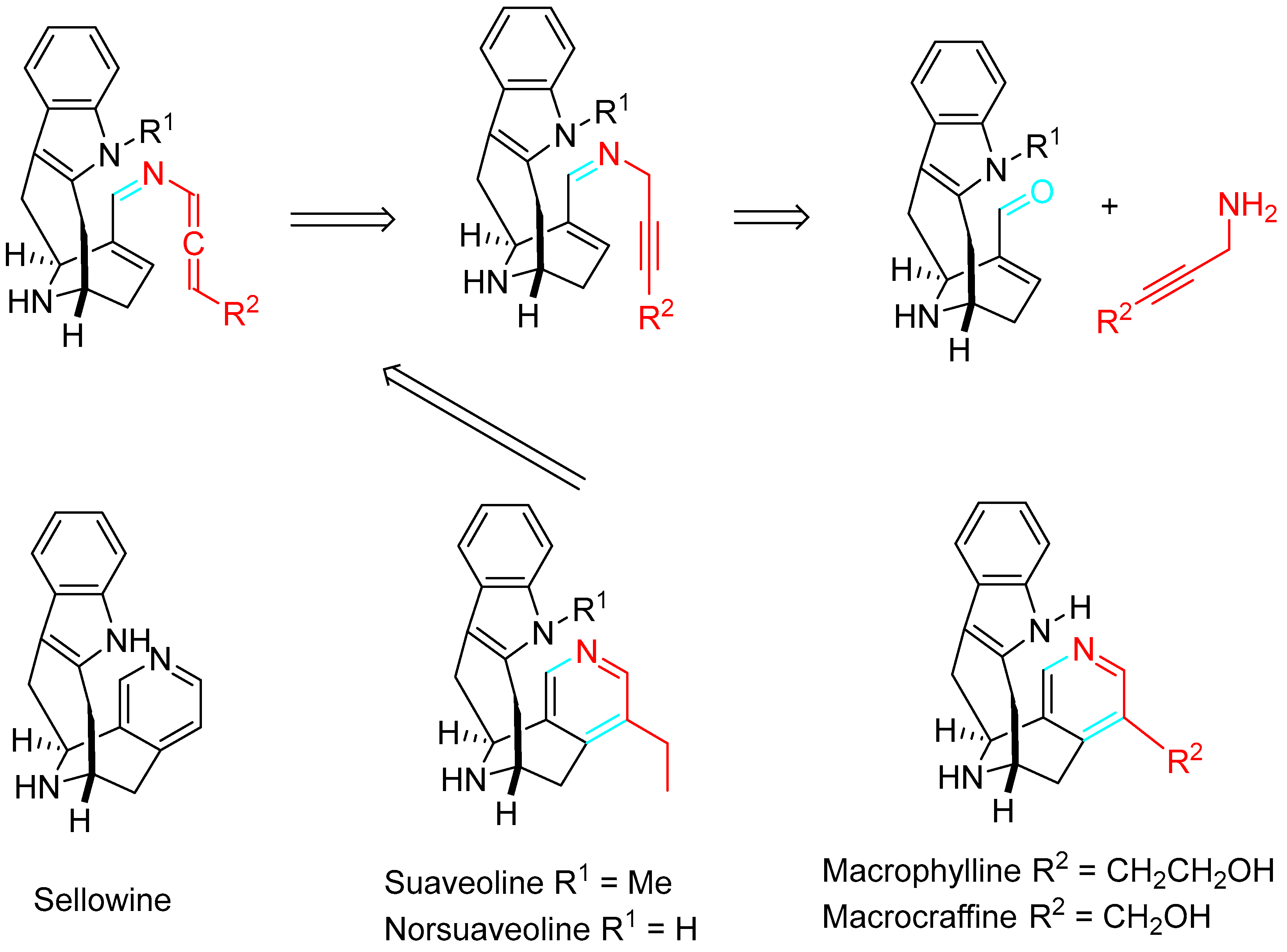
Scheme 24. Retrosynthetic pathway of suaveoline alkaloids.
The practicality of the protocol for the sustainable synthesis of these kinds of molecules through a tandem condensation–alkyne isomerization–6π-3-azatriene electrocyclization sequence was highlighted (Scheme 25) [43].
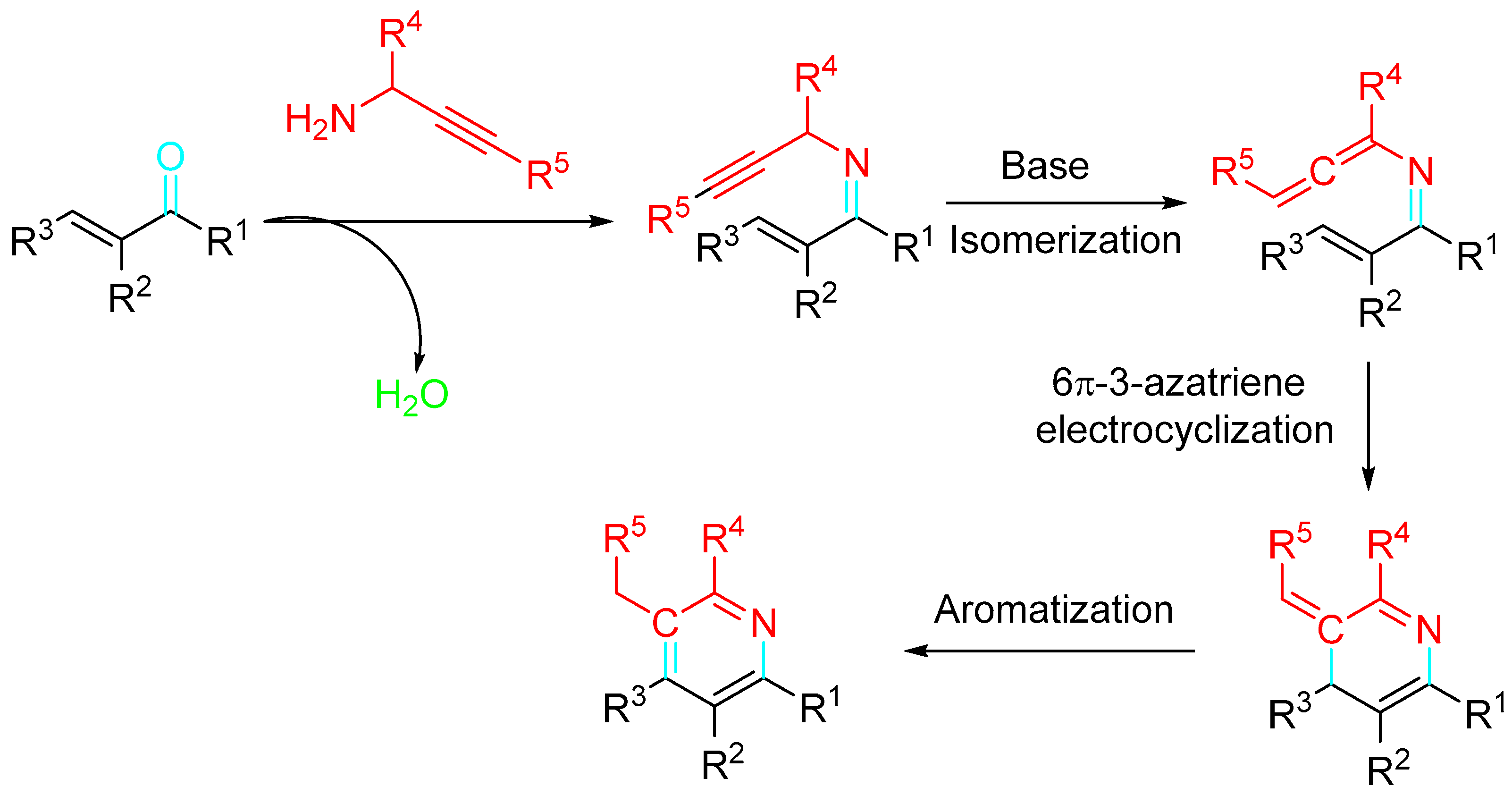
Scheme 25. Pyridine synthesis by a 6π-3-azatriene electrocyclization.
The reaction of propargylamines with (hetero)aromatic aldehydes efficiently afforded β-carbolines, γ-carbolines and other fused azaheteroaromatics under metal-free conditions (Scheme 26) [44].
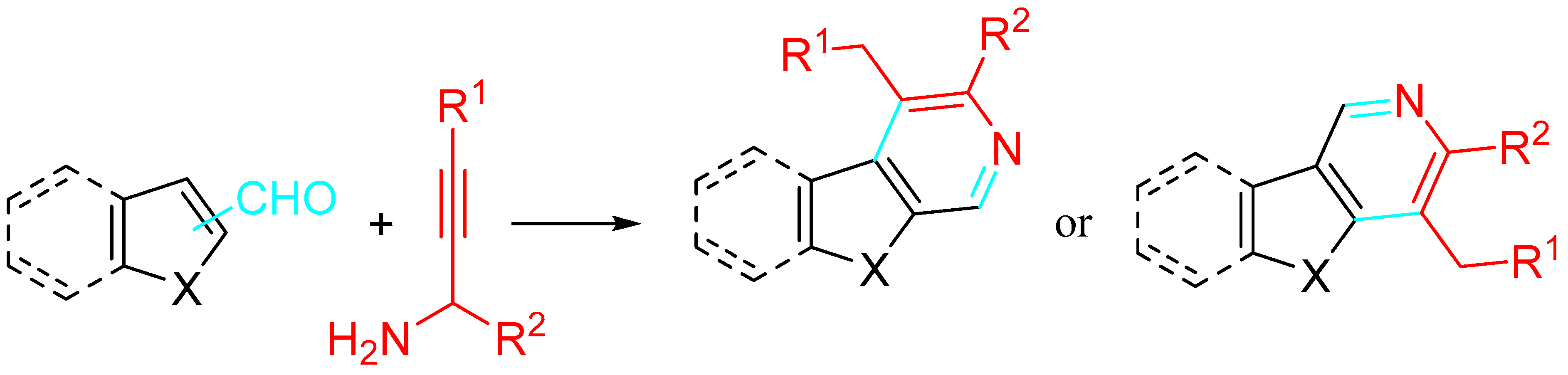
Scheme 26. Synthesis of β- and γ-carbolines from indole aldehydes and substituted propargylic amines.
A one-pot, three-component method allowed the preparation of 3-substituted pyridines and carbolines 22 via copper-free, palladium-catalyzed Sonogashira cross-coupling with aryl iodides, followed by 6π-aza cyclization. This method selectively provided the fused pyridines with good yields (67–92%) (Scheme 27) [45].
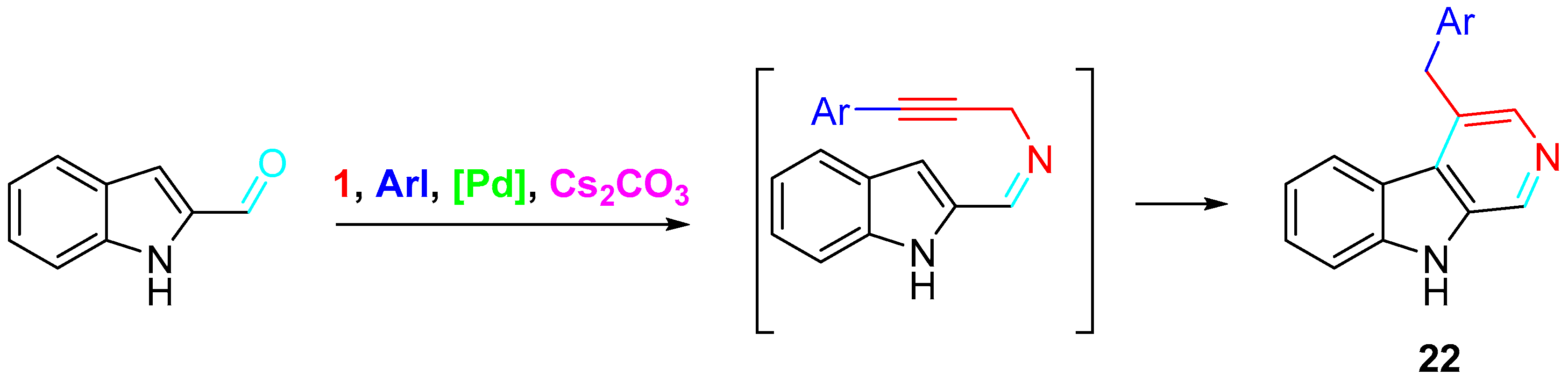
Scheme 27. Synthesis of 4-benzylated carbolines 22.
Alternatively, a further preparation method for the polysubstituted pyridine derivatives comprised the employment of an α,β-unsaturated carbonyl compound and propargylamine hydrochloride as raw materials in chlorobenzene (PhCl) with the sequential addition of 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) and magnesium sulfate (MgSO4) (Scheme 28). The advantage of this alternative procedure is a relatively strong industrial application prospect [46].
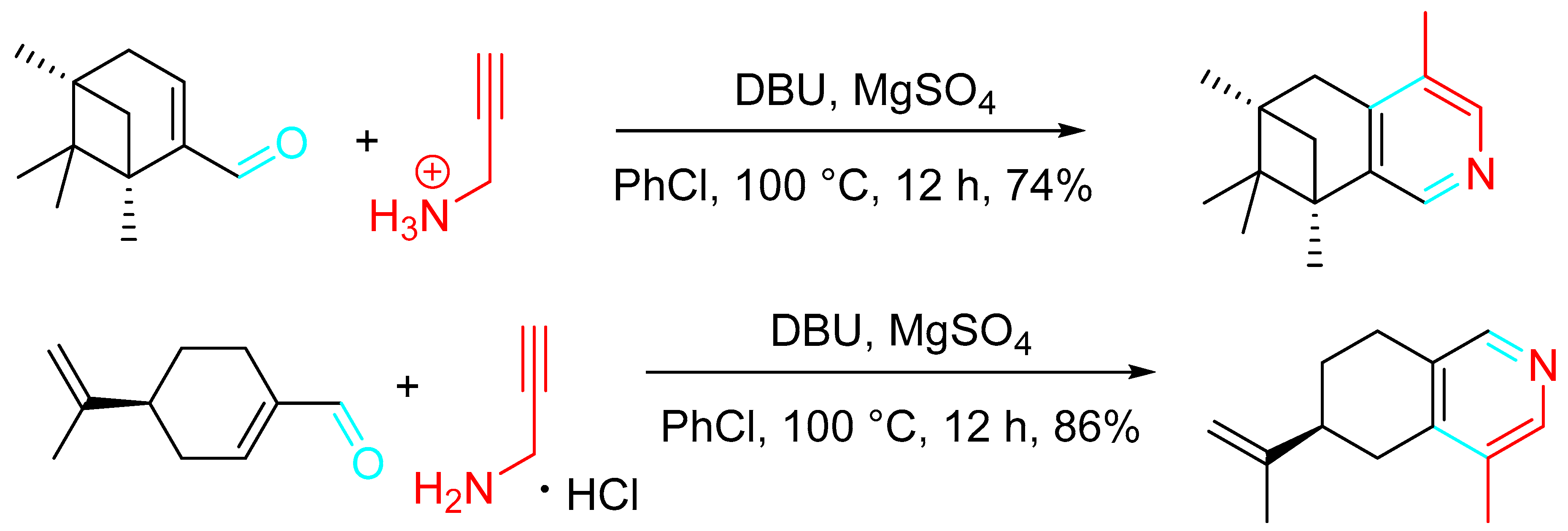
Scheme 28. Alternative synthesis of polysubstituted pyridine derivatives from α,β-unsaturated carbonyl compounds and propargylamine hydrochloride.
Accordingly, an easy synthesis of onychine 23, an azafluorenone alkaloid isolated from a plant of the Annonaceae family, was reported to occur through aza-Claisen rearrangement, tautomerization, 1,5-sigmatropic hydrogen shift, 6π-electron cyclization, and oxidation of the N-propargyl enamine, obtained in a yield of 61% by dehydration condensation of but-2-yn-1-amine with 1,3-indanedione (Scheme 29) [47].
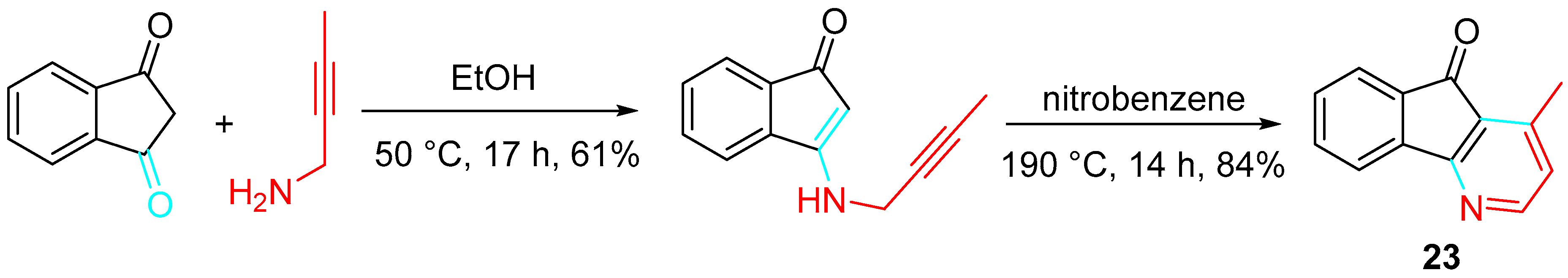
Scheme 29. Total synthesis of onychine 23.
The sequential O-propargylation of aromatic hydroxyaldehydes/condensation reaction with propargylamine allowed a simple approach to the synthesis of chromenopyridine and chromenopyridinone derivatives. The intramolecular cycloaddition reaction between the alkyne and azadiene of 24, which is formed as an intermediate, furnished the desired skeleton of chromenopyridine 25 (Scheme 30) [48].
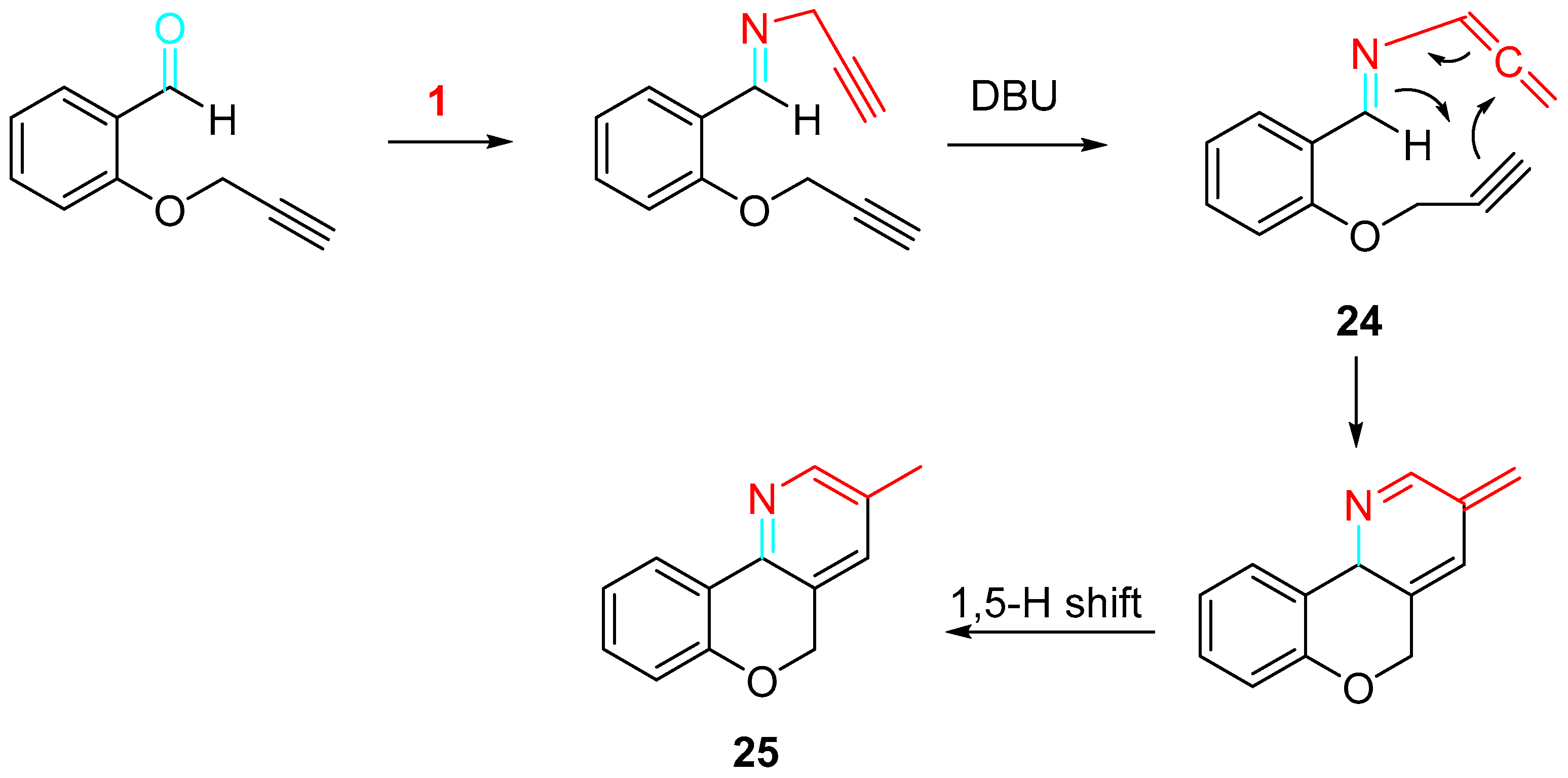
Scheme 30. Mechanism for the formation of chromenopyridines 25.
Moreover, the N-propargylation of aromatic aminobenzaldehydes, followed by reaction with propargylamine in the presence of DBU, gave the corresponding benzo[h][1,6]-naphthyridines 30 (Scheme 31) [49]. The lack of reactivity of the 2-(prop-2-yn-1-ylamino)benzaldehyde was surmounted by double propargylation of the aniline derivative leading to the intermediate 27, which cyclized in refluxing ethanol to afford the N-propargyl derivative 28 with 80% yield. The 3-methylbenzo[h][1,6]-naphthyridine 30 was isolated by increasing the reaction time to 48 h. Oxidation of 28 with CrO3 in pyridine in dichloromethane at room temperature gave the desired product 29 with 95% yield.
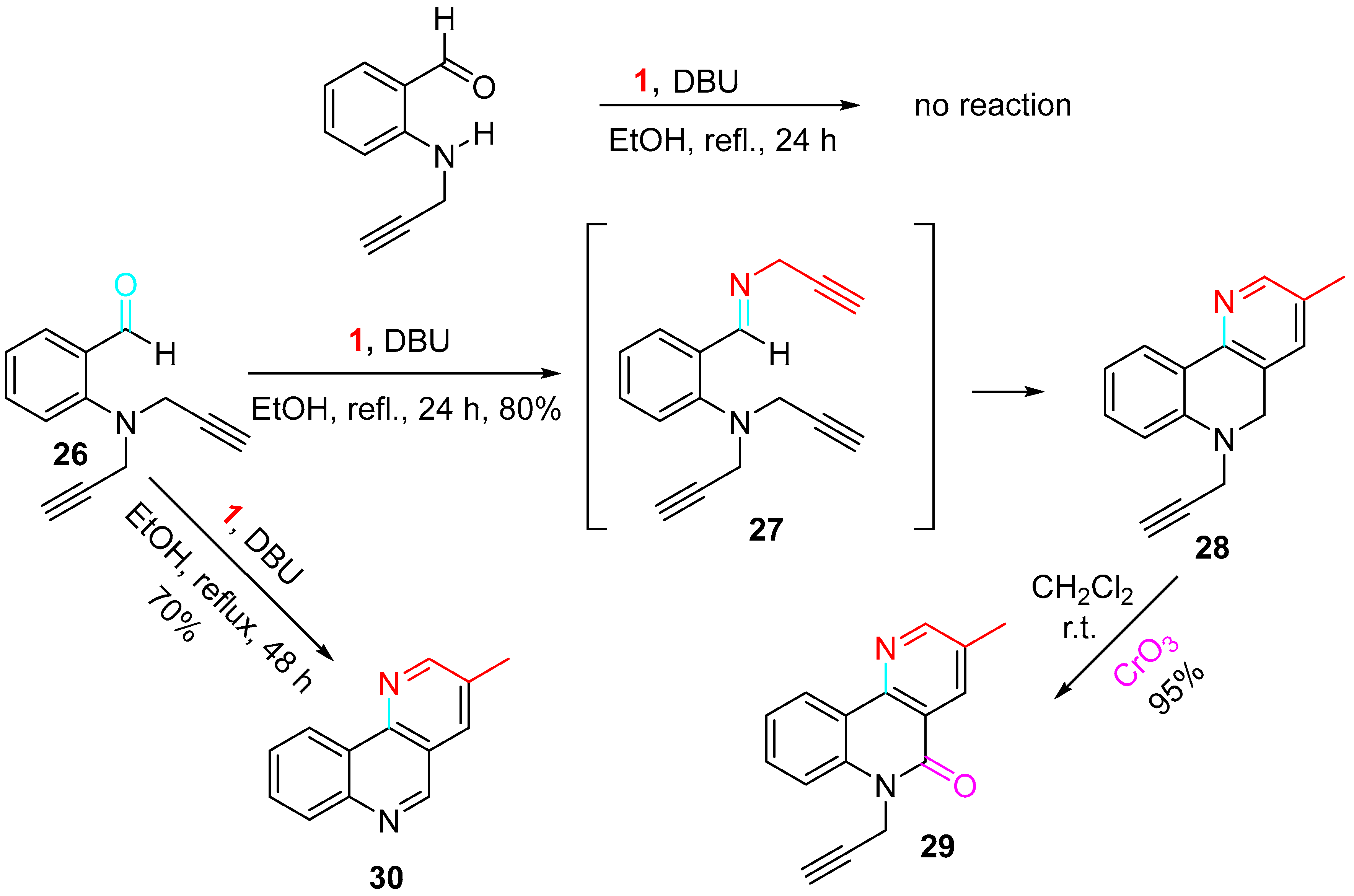
Scheme 31. Sequential reaction of 2-(bis(prop-2-yn-1-ylamino)methyl benzaldehyde 26 with propargylamine.
Moreover, a variety of starting materials 31 synthesized by Sonogashira coupling reactions afforded the corresponding naphthyridine derivatives 32 by reacting with propargylamine in refluxing EtOH in the presence of DBU (Scheme 32).
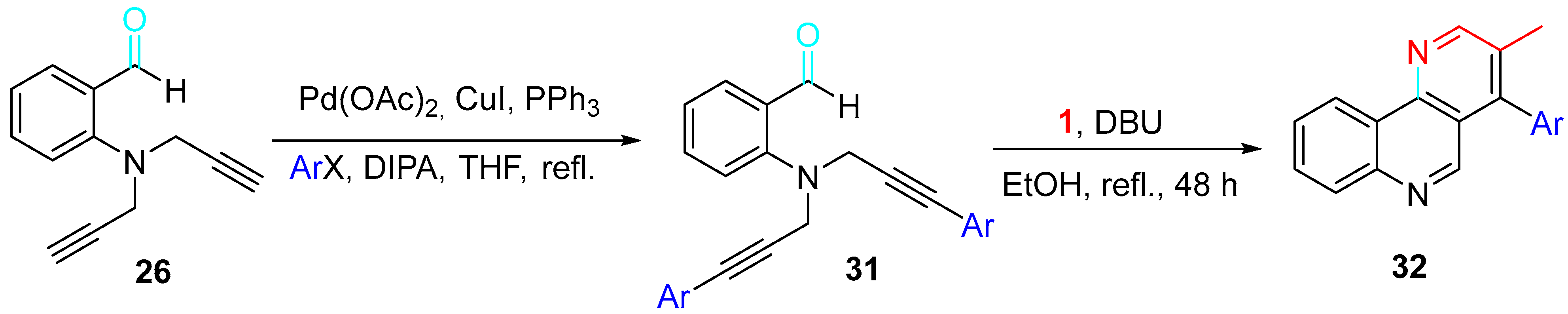
Scheme 32. Synthesis of naphthyridines 32.
The approach was extended to the synthesis of the chromenopyrazinone 33 (Scheme 33).
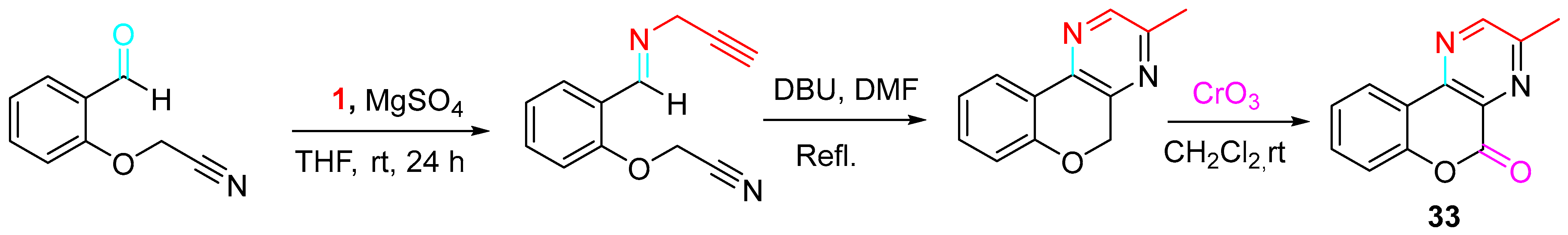
Scheme 33. Synthesis of the chromenopyrazinone 33.
Pyrazines 34 were also synthesized through the gold-catalyzed coupling reaction of aldehydes with propargylamine by means of a different sequential process; 1,2-dichloroethane (DCE) was the best choice as solvent. The addition of five equivalents of H2O under otherwise identical conditions was advantageous for the reaction outcome. The [(Ph3P)AuNTf2] catalyst (5 mol%) was identified as the most effective. The feature of the phosphine showed little effect. Any significant difference was observed by the substitution of [(Ph3P)AuNTf2] with [P(tBu)2(o-biphenyl)AuNTf2]. AuCl3 resulted in a less effective catalyst, while different Lewis acid catalysts, such as PtCl2, InCl3, Bi(OTf)3, ZnCl2, and AgNTf2, failed to afford the product. The reaction of aromatic and α,β-unsaturated aldehydes with two equivalents of propargylamine gave the corresponding pyrazine derivatives with high yields. The catalyst loading could be reduced from 5 mol% to 1 mol% without significant loss of yield of the product when the reaction was carried on a 1 gram scale (Scheme 34) [50].
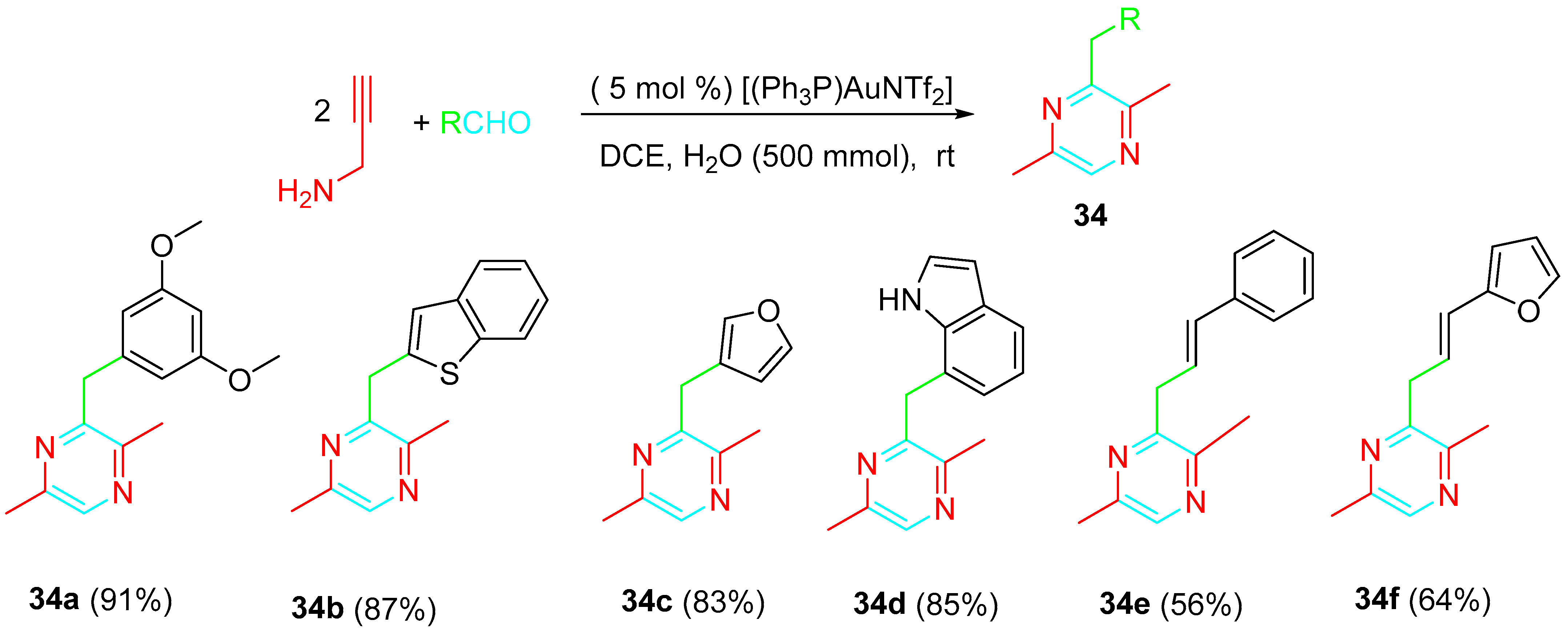
Scheme 34. Gold-catalyzed synthesis of pyrazines 34 from the reaction of propargylamine with aldehydes.
The following reaction mechanism was suggested on the base of labeling experiments and density functional theory (DFT) (Scheme 35). In situ generated gold(I)-imine complex A undergoes a chemo- and regioselective hydroamination reaction with propargylamine to produce the intermediate B. The following protonolysis of the Au-C bond generates the intermediate C, which cyclizes to afford the intermediate D. This new cationic species readily releases benzaldehyde by hydrolysis, regenerating the gold catalyst and producing the 2,5-dimethylenepiperazine E, which readily isomerizes to its more stable 2,5-dimethyl-1,4-dihydropyrazine isomer F. Then, an intermolecular enamine addition from F towards the gold-activated benzaldehyde occurs to produce the intermediate G. Finally, the subsequent isomerization–aromatization sequence gives the reaction product 34.
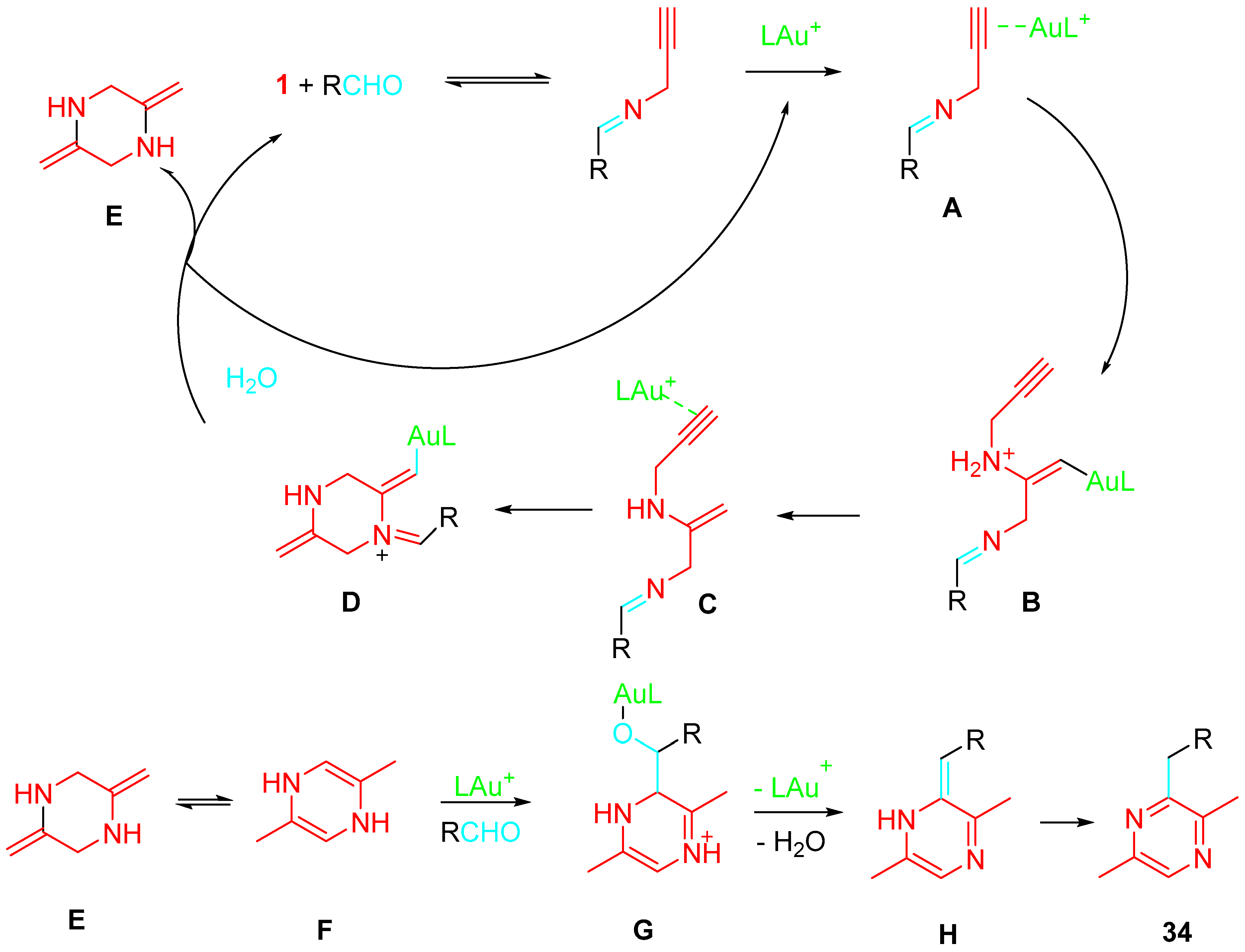
Scheme 35. Proposed mechanism for the Au-catalyzed formation of pyrazines 34.
The heterogeneous gold(I)-catalyzed version of the cascade reaction of aldehydes with propargylamine occurred in 1,2-dichloroethane (DCE) at 40 °C under the presence of the readily available mesoporous MCM-41-immobilized phosphine gold(I) complex (MCM-41-PPh3-AuNTf2). The easy-to-prepare heterogeneous gold(I) catalyst could be recovered by filtration and recycled (Scheme 36) [51].
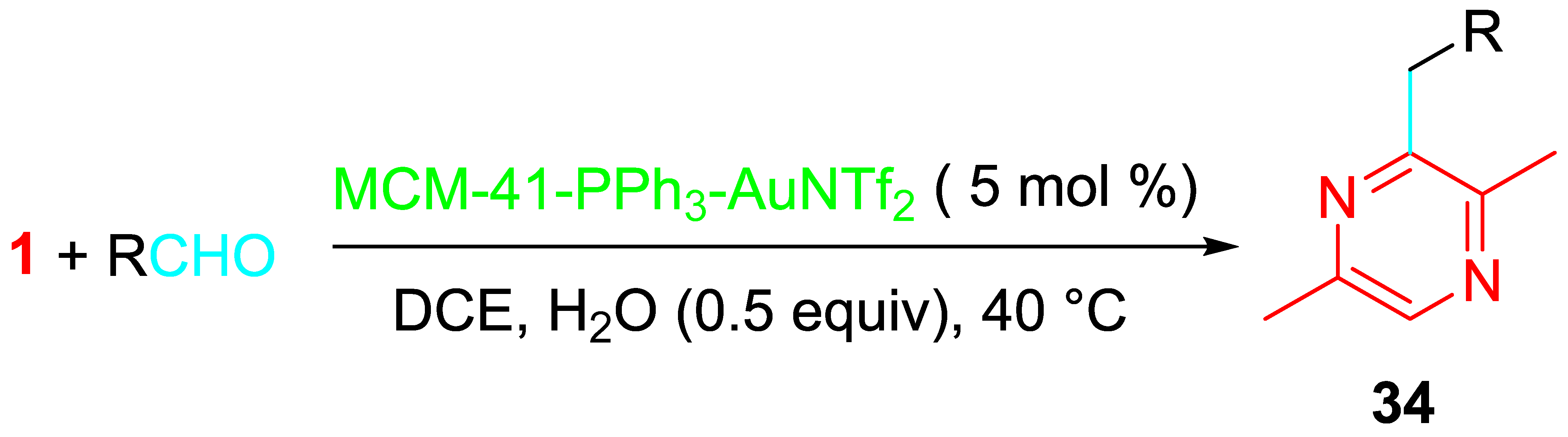
Scheme 36. Sequential reaction of propargylamine with aldehydes catalyzed by MCM-41-immobilized phosphine gold(I) complex [MCM-41-PPh3-AuNTf2].
A variant of a sequential multicomponent assembly process (MCAPs)–cyclization approach in accord with the plan outlined in Scheme 37 was explored for preparing a variety of 1,2,3-triazolo-1,4-benzodiazepines 35 of possible medical relevance by a sequential reductive amination of 2-azidobenzaldeyde derivatives with propargylamine/intramolecular Huisgen cycloaddition [52].
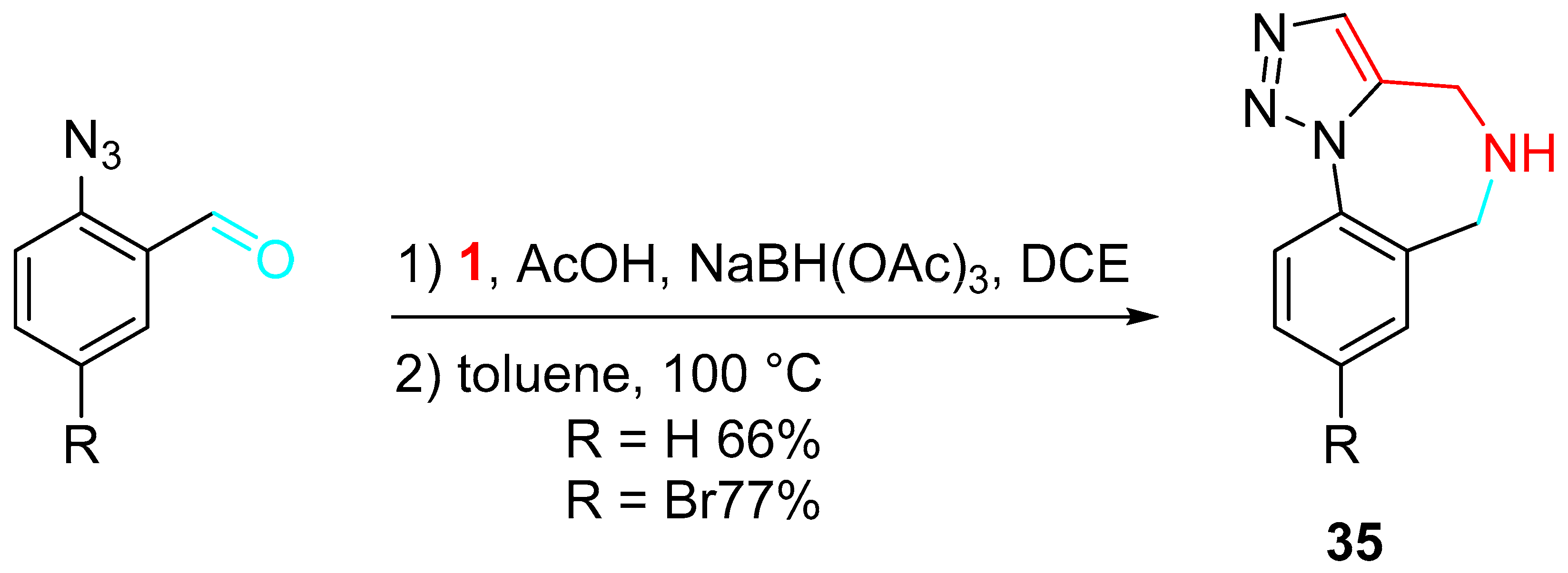
Scheme 37. Synthesis of 1,2,3-triazolo-1,4-benzodiazepines 35.
A wide library was obtained through N-functionalizations, palladium-catalyzed cross-coupling reactions, and applications of α-aminonitrile chemistry (Scheme 38). [53]
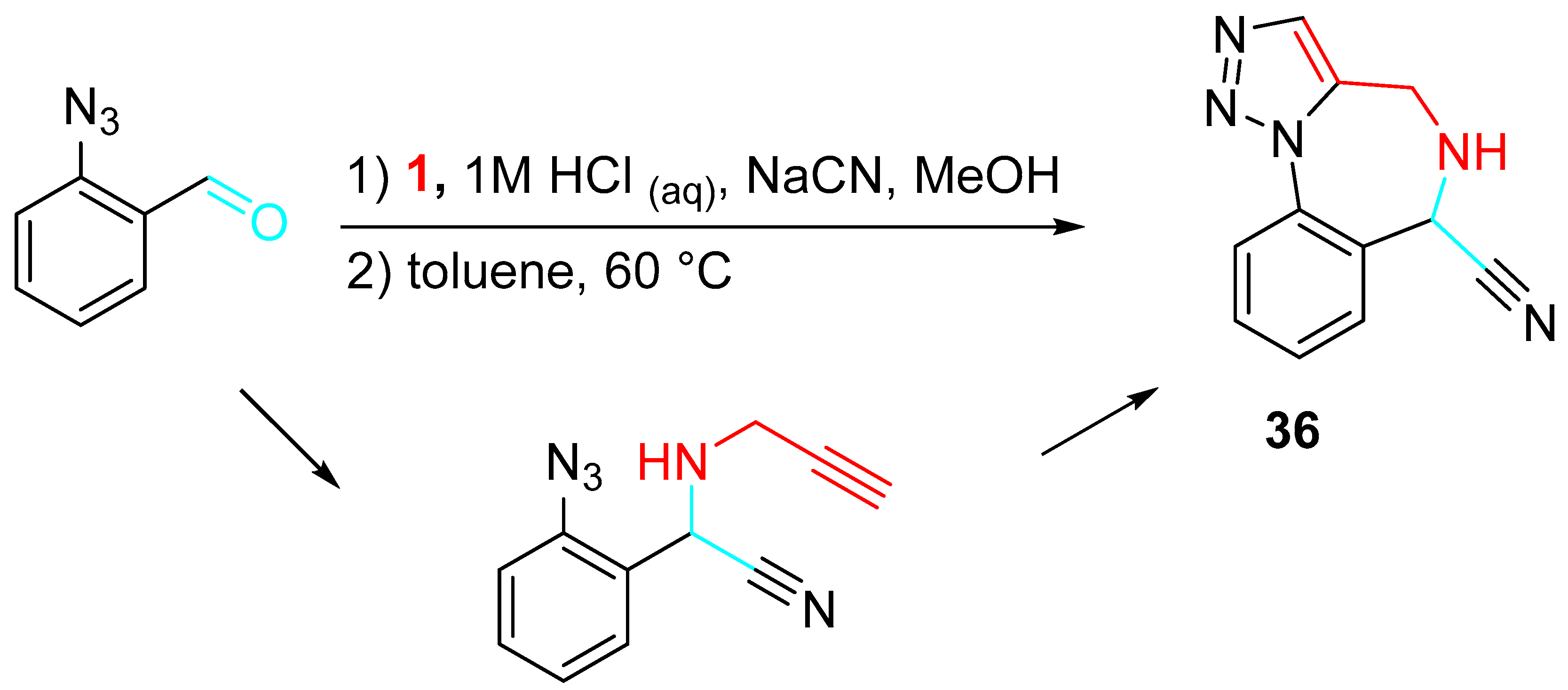
Scheme 38. Diversely substituted 1,2,3-triazolo-1,4-benzodiazepine 36.
An atom-economical multicomponent sequential InCl3-catalyzed cyclocondensation/azide-alkyne 1,3-dipolar cycloaddition of 2-azidobenzaldehydes with propargylamines under the presence of α-diketone and ammonium acetate efficiently afforded the corresponding 9H-benzo[f]imidazo [1,2-d][1,2,3]triazolo [1,5-a][1,4]diazepines 37 (Scheme 39) [54].
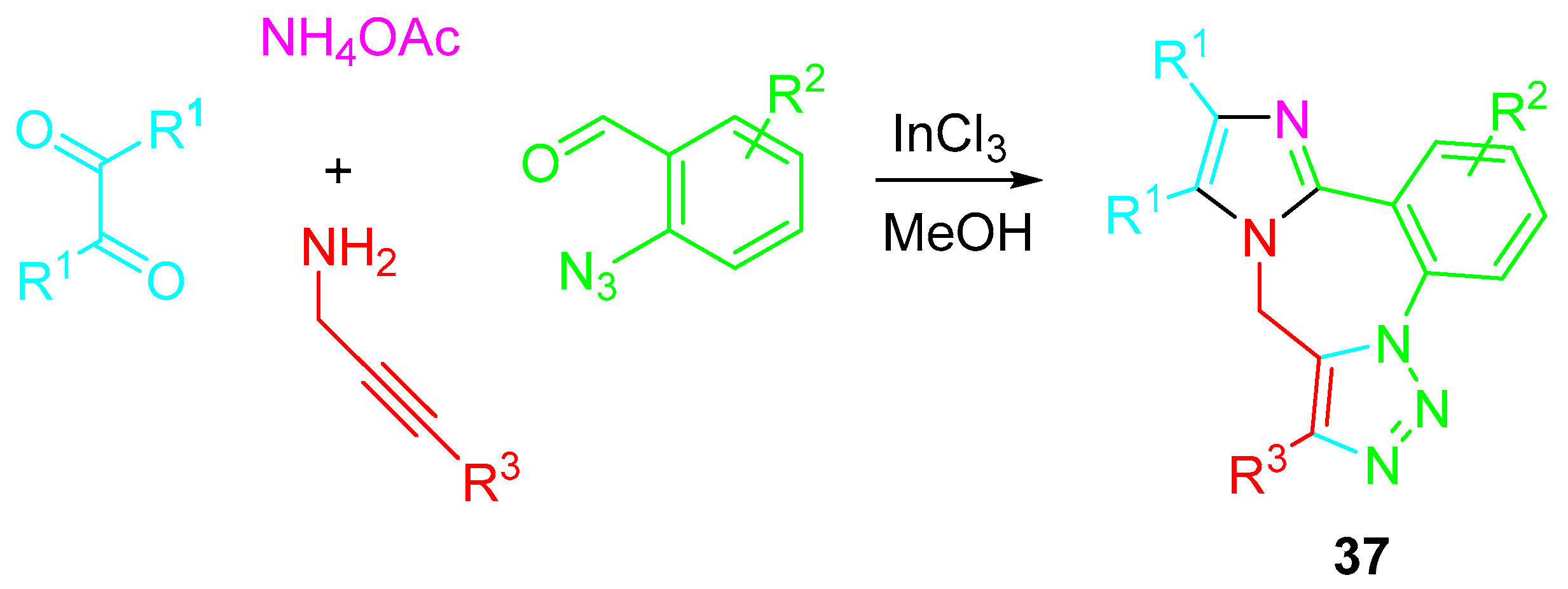
Scheme 39. Indium-catalyzed multicomponent synthesis of 9H-benzo[f]imidazo [1,2-d][1,2,3]triazolo [1,5-a][1,4]diazepines 37.
References
- Hwang, E.T.; Lee, S. Multienzymatic cascade reactions via enzyme complex by immobilization. ACS Catal. 2019, 9, 4402–4425.
- Nicolaou, K.C.; Edmonds, D.J.; Bulger, P.G. Cascade reactions in total synthesis. Angew. Chem. Int. Ed. 2006, 45, 7134–7186.
- Nicolaou, K.C.; Chen, J.S. The art of total synthesis through cascade reactions. Chem. Soc. Rev. 2009, 38, 2993–3009.
- Oishi, M.; Nakanishi, Y.; Suzuki, H. Selective incorporation of primary amines into a trizirconium imido system and catalytic cyclization of aminoalkynes. Inorg. Chem. 2017, 56, 9802–9813.
- Arcadi, A. Gold-catalyzed synthesis of nitrogen heterocyclic compounds via hydroamination reactions. In Au-Catalyzed Synthesis and Functionalization of Heterocycles; Topics in Heterocyclic Chemistry Book Series; Bandini, M., Ed.; Springer: Cham, Switzerland, 2016; Volume 46, pp. 53–86.
- Arcadi, A.; Abbiati, G.; Rossi, E. Tandem imination/annulation of γ- and δ-ketoalkynes in the presence of ammonia/amines. J. Organomet. Chem. 2011, 696, 87–98.
- Chang, S.; Lee, M.; Jung, D.Y.; Yoo, E.J.; Cho, S.H.; Han, S.K. Catalytic one-pot synthesis of cyclic amidines by virtue of tandem reactions involving intramolecular hydroamination under mild conditions. J. Am. Chem. Soc. 2006, 128, 12366–12367.
- Liu, X.-Y.; Che, C.-M. A highly efficient and selective AuI-catalyzed tandem synthesis of diversely substituted pyrroloquinolines in aqueous media. Angew. Chem. Int. Ed. 2008, 47, 3805–3810.
- Zhou, Y.; Feng, E.; Liu, G.; Ye, D.; Li, J.; Jiang, H.; Liu, H. Gold-catalyzed one-pot cascade construction of highly functionalized pyrroloquinolin-1(2H)-ones. J. Org. Chem. 2009, 74, 7344–7348.
- Ma, C.-L.; Zhao, J.-H.; Yang, Y.; Zhang, M.-K.; Shen, C.; Sheng, R.; Dong, X.-W.; Hu, Y.-Z. A copper-catalyzed tandem cyclization reaction of aminoalkynes with alkynes for the construction of tetrahydropyrroloquinolines scaffold. Sci. Rep. 2017, 7, 16640.
- Ma, C.-L.; Li, X.-H.; Yu, X.-L.; Zhu, X.-L.; Hu, Y.-Z.; Dong, X.-W.; Tan, B.; Liu, X.-Y. Gold-catalyzed tandem synthesis of bioactive spiro-dipyrroloquinolines and its application in the one-step synthesis of incargranine B aglycone and seneciobipyrrolidine (I). Org. Chem. Front. 2016, 3, 324–329.
- Galvàn, A.; Calleja, J.; Faňanás, F.J.; Rodríguez, F. Synthesis of pyrrolidine derivatives by a platinum/Brønsted acid relay catalytic cascade reaction. Chem. Eur. J. 2015, 21, 3409–3414.
- Huple, D.B.; Liu, R.-S. One-pot stereocontrolled synthesis of bicyclic pyrrolidine derivatives by a platinum-Brønsted acid relay cascade reaction. ChemCatChem 2015, 7, 2824–2825.
- Fujiwara, S.-I.; Shikano, Y.; Shin-ike, T.; Kambe, N.; Sonoda, N. Stereoselective synthesis of new selenium-containing heterocycles by cyclocarbonylation of aminoalkynes with carbon monoxide and selenium. J. Org. Chem. 2002, 67, 6275–6278.
- Hu, Y.; Huang, H. Highly selective construction of medium-sized lactams by palladium-catalyzed intramolecular hydroaminocarbonylation of aminoalkynes. Org. Lett. 2017, 19, 5070–5073.
- Li, X.; Wang, S.; Wang, H.; Wang, W.; Liu, L.; Chang, W.; Li, J. Synthesis of eight-membered nitrogen heterocycles via a heterogeneous PtI2-catalyzed cascade cycloaddition reaction of δ-aminoalkynes with electron-deficient alkynes. Adv. Synth. Catal. 2020, 362, 1525–1531.
- Li, X.; Jiang, C.; Wang, X.; Ren, J.; Zeng, T.; Xu, X.; Li, J.; Liu, L. Platinum iodide-catalyzed formal three-component cascade cycloaddition reactions between γ-aminoalkynes and electron-deficient alkynes. J. Org. Chem. 2021, 86, 16614–16624.
- Abbiati, G.; Arcadi, A.; Bianchi, G.; Di Giuseppe, S.; Marinelli, F.; Rossi, E. Sequential amination/annulation/aromatization reaction of carbonyl compounds and propargylamine: A new one-pot approach to functionalized pyridines. J. Org. Chem. 2003, 68, 6959–6966.
- Yang, D.; Liu, H.; Wang, D.-L.; Lu, Y.; Zhao, X.-L.; Liu, Y. Au complex containing phosphino and imidazolyl moieties as a bifunctional catalyst for one-pot synthesis of pyridine derivatives. J. Mol. Catal. A Chem. 2016, 424, 323–330.
- Sotnik, S.O.; Subota, A.I.; Kliuchynskyi, A.Y.; Yehorov, D.V.; Lytvynenko, A.S.; Rozhenko, A.B.; Kolotilov, S.V.; Ryabukhin, S.V.; Volochnyuk, D.M. Cu-catalyzed pyridine synthesis via oxidative annulation of cyclic ketones with propargylamine. J. Org. Chem. 2021, 86, 7315–7325.
- Savić, M.P.; Ajduković, J.J.; Plavša, J.J.; Bekić, S.S.; Ćelić, A.S.; Klisurić, O.R.; Jakimov, D.S.; Petri, E.T.; Djurendić, E.A. Evaluation of A-ring fused pyridine D-modified androstane derivatives for antiproliferative and aldo-keto reductase 1C3 inhibitory activity. Med. Chem. Commun. 2018, 9, 969–981.
- Yan, J.-Z.; Li, J.; Rao, G.-W. One-pot synthesis of new A-ring fused steroidal pyridines. Steroids 2007, 7, 736–739.
- Laavola, M.; Haavikko, R.; Hämäläinen, M.; Leppänen, T.; Nieminen, R.; Alakurtti, S.; Moreira, V.M.; Yli-Kauhaluoma, J.; Moilanen, E. Betulin derivatives effectively suppress inflammation in vitro and in vivo. J. Nat. Prod. 2016, 79, 274–280.
- Haavikko, R.; Nasereddin, A.; Sacerdoti-Sierra, N.; Kopelyanskiy, D.; Alakurtti, S.; Tikka, M.; Jaffe, C.L.; Yli-Kauhaluoma, J. Heterocycle-fused lupane triterpenoids inhibit Leishmania donovani amastigotes. Med. Chem. Commun. 2014, 5, 445–451.
- Hodoň, J.; Frydrych, I.; Trhlíková, Z.; Pokorný, J.; Borková, L.; Benická, S.; Vlk, M.; Lišková, B.; Kubíčková, A.; Medvedíková, M.; et al. Triterpenoid pyrazines and pyridines—Synthesis, cytotoxicity, mechanism of action, preparation of prodrugs. Eur. J. Med. Chem. 2022, 243, 114777.
- Rabe, S.; Moschner, J.; Bantzi, M.; Heretsch, P.; Giannis, A. C-H-Functionalization logic guides the synthesis of a carbacyclopamine analog. Beilstein J. Org. Chem. 2014, 10, 1564–1569.
- Simcere Pharmaceutical Group. Compound as Potassium Channel Modulating Agents. CN108250128 A, 6 July 2018.
- Suzhou Yunxuan Pharmaceutical Co Ltd. Heterocyclic Compound with Wnt Signal Path Inhibitory Activity and Application Thereof. CN105254613 A, 20 January 2016.
- Zhang, X.; Zheng, J.; Ma, H. Heteroaryl Compounds as cxcr4 Inhibitors, Composition and Method Using the Same. Patent WO2019/60860 A1, 28 March 2019.
- Wortmann, L.; Lindenthal, B.; Muhn, P.; Walter, A.; Nubbemeyer, R.; Heldmann, D.; Sobek, L.; Morandi, F.; Schrey, A.K.; Moosmayer, D.; et al. Discovery of BAY-298 and BAY-899: Tetrahydro-1,6-naphthyridine-based, potent, and selective antagonists of the luteinizing hormone receptor which reduce sex hormone levels in vivo. J. Med. Chem. 2019, 62, 10321–10341.
- Luo, G.; Chen, L.; Conway, C.M.; Denton, R.; Keavy, D.; Gulianello, M.; Huang, Y.; Kostich, W.; Lentz, K.A.; Mercer, S.E.; et al. Discovery of BMS-846372, a potent and orally active human CGRP receptor antagonist for the treatment of migraine. ACS Med. Chem. Lett. 2012, 3, 337–341.
- C.H. Boehringer Sohn AG & Co. KG. Aryl- and Heroarylcarbonyl Derivatives of Benzomorphanes and Related Scaffolds, Medicaments Containing Such Compounds and Their Use. EP2220048 B1, 25 January 2017.
- Lowe, R.A.; Taylor, D.; Chibale, K.; Nelson, A.; Marsden, S.P. Synthesis and evaluation of the performance of a small molecule library based on diverse tropane-related scaffolds. Biorg. Med. Chem. 2020, 28, 115442.
- Eckhardt, M.; Peters, S.; Nar, H.; Himmelsbach, F.; Zhuang, L. Boehringer Ingheleim Int, Aryl-and Heteroarylcarbonyl Derivatives of Hexahydroindenopyridine and Octahydrobenzoquinoline. U.S. Patent 2011/0136800 A1, 9 June 2011.
- Park, N.Y.; Lee, H.S.; Piao, L.H.; Park, S.Y.; Jeong, W. Transition Metal Compound for Olefin Polymerization Catalyst, and Olefin Polymerization Catalyst Including Same. U.S. Patent 11254759 B2, 22 February 2022.
- Faňanás, F.J.; Arto, T.; Mendoza, A.; Rodríguez, F. Synthesis of 2,5-dihydropyridine derivatives by gold-catalyzed reactions of β-ketoesters and propargylamines. Org. Lett. 2011, 13, 4184–4187.
- Lee, S.; Yoo, H.; Park, S.; Yoon, R.; Kim, S. Facile one-pot synthesis of polysubstituted pyridinium salts by annulation of enamines with alkynes. Chem. Eur. J. 2023, e202300059.
- Changchun Haipurunsi Tech Co Ltd. A Kind of Miscellaneous Anthracene Derivant and Preparation Method Thereof and Organic Luminescent Device. CN108218860 A, 29 June 2018.
- University Zhejlang Technology. Method for Synthesizing Substituted Dihydrophenanthroline Compound. CN112480112 A, 15 October 2021.
- Watkins, E.B.; Uredi, D.; Motati, D.R. Novel Methods for Preparation of Substituted Pyridines and Related Novel Compounds. U.S. Patent 2020/0095245 A1, 10 August 2020.
- Uredi, D.; Motati, D.R.; Watkins, E.B. A simple, tandem approach to the construction of pyridine derivatives under metal-free conditions: A one-step synthesis of the monoterpene natural product, (-)-actinidine. Chem. Commun. 2019, 55, 3270–3273.
- Zhao, Z.; Wei, H.; Xiao, K.; Cheng, B.; Zhai, H.; Li, Y. Facile synthesis of pyridines from propargyl amines: Concise total synthesis of suaveoline alkaloids. Angew. Chem. Int. Ed. 2019, 58, 1148–1152.
- Wei, H.; Li, Y. Quick access to pyridines through 6π-3-azatriene electrocyclization: Concise total synthesis of suaveoline alkaloids. Synlett 2019, 30, 1615–1620.
- Uredi, D.; Motati, D.R.; Watkins, E.B. A unified strategy for the synthesis of β-carbolines, γ-carbolines, and other fused azaheteroaromatics under mild, metal-free conditions. Org. Lett. 2018, 20, 6336–6339.
- Uredi, D.; Burra, A.G.; Watkins, E.B. Rapid access to 3-substituted pyridines and carbolines via a domino, copper-free, palladium-catalyzed Sonogashira cross-coupling/6π-aza cyclization sequence. J. Org. Chem. 2021, 86, 17748–17761.
- Lanzhou University. A Kind of Polysubstituted Pyridine Derivative and Preparation Method Thereof. CN108358834 A, 3 August 2018.
- Chikayuki, Y.; Miyashige, T.; Yonekawa, S.; Kirita, A.; Matsuo, N.; Teramoto, H.; Sasaki, S.; Higashiyama, K.; Yamauchi, T. Transition-metal-free synthesis of pyridine derivatives by thermal cyclization of N-propargyl enamines. Synthesis 2020, 52, 1113–1121.
- Keskin, S.; Balci, M. Intramolecular heterocyclization of O-propargylated aromatic hydroxyaldehydes as an expedient route to substituted chromenopyridines under metal-free conditions. Org. Lett. 2015, 17, 964–967.
- Hoplamaz, E.; Keskin, S.; Balci, M. Regioselective synthesis of benzo-naphthyridines and chromenopyrazinones through alkyne cyclization. Eur. J. Org. Chem. 2017, 1489–1497.
- Alcaide, B.; Almendros, P.; Alonso, J.M.; Fernández, I.; Gómez-Campillosa, G.; Torres, M.R. A gold-catalysed imine-propargylamine cascade sequence: Synthesis of 3-substituted-2,5-dimethylpyrazines and the reaction mechanism. Chem. Commun. 2014, 50, 4567–4570.
- Nie, Q.; Yao, F.; Yi, F.; Cai, M. A heterogeneous gold(I)-catalyzed cascade annulation of aldehydes with propargylamine leading to 3-substituted 2,5-dimethylpyrazines. J. Organomet. Chem. 2017, 846, 343–350.
- Donald, J.R.; Martin, S.F. Synthesis and diversification of 1,2,3-triazole-fused 1,4-benzodiazepine scaffolds. Org. Lett. 2011, 13, 852–855.
- Donald, J.R.; Wood, R.R.; Martin, S.F. Application of a sequential multicomponent assembly process/Huisgen cycloaddition strategy to the preparation of libraries of 1,2,3-triazole-fused 1,4-benzodiazepines. ACS Comb. Sci. 2012, 14, 135–143.
- Nguyen, H.H.; Palazzo, T.A.; Kurth, M.-J. Facile one-pot assembly of imidazotriazolobenzodiazepines via indium(III)-catalyzed multicomponent reactions. Org. Lett. 2013, 15, 4492–4495.
More
Information
Subjects:
Chemistry, Organic
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
918
Revisions:
4 times
(View History)
Update Date:
26 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No