Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Juergen Arnhold | -- | 4371 | 2023-06-20 16:59:50 | | | |
| 2 | Sirius Huang | Meta information modification | 4371 | 2023-06-21 04:06:21 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Arnhold, J. Host-Derived Cytotoxic Agents. Encyclopedia. Available online: https://encyclopedia.pub/entry/45876 (accessed on 28 July 2026).
Arnhold J. Host-Derived Cytotoxic Agents. Encyclopedia. Available at: https://encyclopedia.pub/entry/45876. Accessed July 28, 2026.
Arnhold, Jürgen. "Host-Derived Cytotoxic Agents" Encyclopedia, https://encyclopedia.pub/entry/45876 (accessed July 28, 2026).
Arnhold, J. (2023, June 20). Host-Derived Cytotoxic Agents. In Encyclopedia. https://encyclopedia.pub/entry/45876
Arnhold, Jürgen. "Host-Derived Cytotoxic Agents." Encyclopedia. Web. 20 June, 2023.
Copy Citation
At inflammatory sites, cytotoxic agents are released and generated from invading immune cells and damaged tissue cells. The further fate of the inflammation highly depends on the presence of antagonizing principles that are able to inactivate these host-derived cytotoxic agents.
cytotoxic agents
reactive species
transition metal ions
free heme
serine proteases
angiotensin II
matrix metalloproteases
chronic inflammation
1. The Balance between the Action of Cytotoxic Agents and Protective Principles
1.1. Major Classes of Host-Derived Cytotoxic Agents
The contact of cytotoxic agents with living matter worsens cell functions and can induce irreversible changes in cells and tissues including cell death. According to the source of these agents, they can be roughly divided into external and host-derived cytotoxic components. The group of external cytotoxic agents comprises pathogen-derived toxins and manifold external poisons that act on the organism by inhalation, direct contact, or uptake with food. A third group represents environmental cytotoxic agents. An overview about external cytotoxic agents is given in Figure 1. Selected examples of these agents are included.
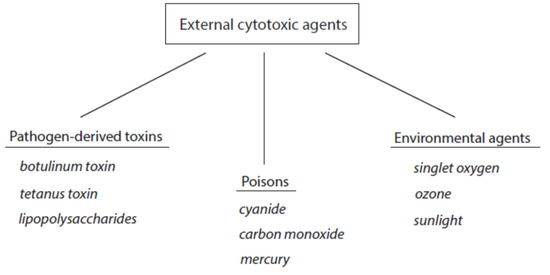
Figure 1. Classification of external cytotoxic agents. Three selected examples are indicated for each group.
Of course, these external cytotoxic agents can cause sufficient threat to the affected tissues and finally the death of the organism. However, these agents are usually not involved in long-lasting, chronic inflammation.
Host-derived cytotoxic agents result from activated immune cells such as neutrophils, eosinophils, monocytes, macrophages, and T cells, but also from affected tissue cells of non-immunological origin, for instance, muscle cells and red blood cells. Immune cells contain an arsenal of potentially cytotoxic agents that are needed to inactivate and kill pathogens and to remove and digest affected cells and destroyed tissues [1]. Usually these agents act within small, bounded compartments, e.g., within the phagosomes of neutrophils and macrophages. However, a certain amount of these cytotoxic agents is released from activated immune cells into the surrounding milieu, where they become dangerous to unperturbed cells. The following classes of immune-cell-derived cytotoxic agents are known: small reactive species, heme peroxidases, free metal ions, serine proteases, matrix metalloproteases, and small pro-inflammatory peptides. These agents are either pre-assembled or generated during cellular immune activation.
Damage to tissue cells of non-immunological origin can result in the uncontrolled release of heme proteins such as hemoglobin and myoglobin and the subsequent formation of free heme. Cellular stress is also associated with enhanced formation of reactive species and deviations in free metal ion metabolism. As a result, the metabolism of mitochondria is disturbed and numerous oxidative processes in biological constituents take place.
According to their mode of action, host-derived cytotoxic agents can be divided into oxidant- and protease-based agents (Figure 2). Products of the first group promote oxidative alterations of biological constituents, whereas members of the second group cause proteolytic cleavage in cell and tissue components.
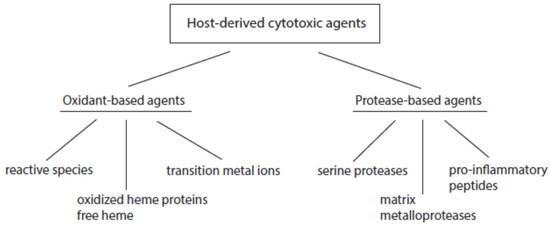
Figure 2. Major classes of host-derived cytotoxic agents.
1.2. Control of Cytotoxic Agents by Protective Principles
The destructive action of host-derived cytotoxic agents depends not only on the mass of released cytotoxic agents, but also on the current status of host-own protective principles [1][2]. In order to curtail or avoid destruction by these agents, numerous ready-to-use mechanisms exist in cells and tissues to inactivate immediately hazardous components released from activated immune and affected tissue cells. Usually, unperturbed cells and tissues are well equipped with protective principles. In this way, any threat to unperturbed tissue components is minimized.
The balance between host-derived cytotoxic agents and protective principles functions well as long as the activation of immune cells is moderate enough and neighboring tissues are well-equipped with ready-to-use protective mechanisms (Figure 3). Problems can arise with severe and long-lasting immune responses and with the decline, exhaustion, or inactivation of selected antagonizing principles despite an up-regulation of many protective proteins under stress situations. In turn, long-lasting inflammatory processes can result from the permanent release of cytotoxic agents from damaged cells in combination with insufficient inactivation of these agents. In other words, low expression of a few protective principles favors the continuous action of destructive agents and affects still-unperturbed cells. In this vicious circle of permanent cell destruction, not only novel cytotoxic elements but also novel alarmins and antigens are liberated from affected cells. In addition, pro-inflammatory peptides such as angiotensin II and bradykinin are formed by insufficient inactivation of serine proteases. Hence, the inflammation cannot be terminated sufficiently and flares up again and again. In severe cases, a very low level of protection leads to organ failure, sepsis, and septic shock.
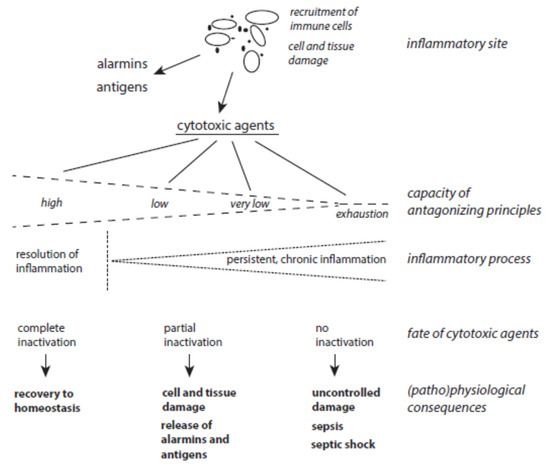
Figure 3. The interplay between host-derived cytotoxic agents and antagonizing principles. (Patho)physiological consequences of the release of cytotoxic agents at inflammatory sites highly depend on the status of protective mechanisms.
To overcome chronic inflammation, it is highly essential, besides inhibition of selected pathways in the inflammatory cascade, to improve poorly expressed protective systems to better detoxify the damaging agents.
1.3. Disturbed Balance between De Novo Synthesis and Damage of Tissue Components during Resolution of Inflammation
Termination of inflammation is characterized by the down-regulation of pro-inflammatory cells, cytokines, and signaling pathways as well as by the formation of anti-inflammatory mediators and induction of repair processes. During this phase of inflammation, cytokines of the transforming growth factor β (TGF-β) family, which are secreted from M2-type macrophages and some other cells, suppress together with interleukin 10 (IL-10)-activated immune cells [3][4]. These cytokines also promote tissue repair by stimulating fibroblasts to synthesize collagen and other components of the extracellular matrix (ECM) and by the release of tissue inhibitors of metalloproteases (TIMPs) [5][6]. The latter inhibitors down-regulate the activity of matrix metalloproteases (MMPs) and thus prevent degradation of ECM components.
1.4. Selected Environmental Cytotoxic Agents
Although not host-derived, we can also be exposed to external cytotoxic agents (see Figure 1). Of these agents, environmental cytotoxic agents act more or less intensely and permanently on our organism. As this exposure concerns nearly all persons, these agents are usually detoxified by antagonizing principles when the exposure is moderate and does not exceed a critical level.
2. Selected Cytotoxic Agents and Their Counter-Regulating Principles
2.1. Small Reactive Species and Metal Ions
2.1.1. Superoxide Anion Radicals
The stepwise reduction of dioxygen yields the species superoxide anion radical (O2•−) and hydrogen peroxide (H2O2) [7]. These species are less dangerous concerning their direct action on tissue components. However, they are involved in the formation of highly reactive and tissue-damaging agents by interaction with radicals, metal ions, and iron-containing proteins.
Activated leukocytes are able to generate large amounts of O2•− by reducing dioxygen. This reaction is catalyzed by NADPH oxidase, which is assembled from several membranous and cytoplasmic components during the activation of neutrophils, eosinophils, monocytes, and macrophages [8][9]. NADPH oxidases are also distributed in cells of the blood vessel wall, respiratory tract, gastrointestinal tract, and thyroid gland [10][11][12]. However, these enzymes are less efficient in reducing dioxygen than NADPH oxidase from immune cells. Other sources for superoxide anion radicals are reactions of xanthine oxidase [13][14], autoxidation of hemoglobin and myoglobin [15][16], cytochrome P450-driven redox recycling of some xenobiotica [17][18], and one-electron reduction of dioxygen by different mitochondrial enzymes [19][20].
Superoxide anion radicals are unstable. Two superoxide anion radicals dismutate spontaneously to hydrogen peroxide and dioxygen [21]. The rate of this dismutation highly depends on pH, with a maximal rate around pH 4.8, the pka value of O2•−, and decreasing rates with increasing pH [22]. With one unit pH increase, the dismutation rate of O2•− decreases by one order of magnitude. At pH 7.4 this rate is 2 × 105 M−1s−1 [22].
Superoxide anion radical reacts in a very rapid reaction with nitrogen monoxide, also a radical species, under the formation of the powerful oxidant peroxynitrite [23][24]. In mitochondria, superoxide anion radical is able to release Fe2+ from molecules containing [4Fe-4S]2+ clusters such as aconitase [25][26].
In humans, control over O2•− is realized with three isoforms of superoxide dismutase (SOD) and cytochrome c. SOD1 is distributed in the cytoplasm, intermembrane space of mitochondria, and nuclei [27][28]. In the mitochondrial matrix, SOD2 dominates [29]. SOD3 is mostly found in blood vessel walls and lungs [30]. These enzymes catalyze the dismutation of O2•− with a rate several orders higher than the spontaneous dismutation reaction of O2•−. In the intermembrane space of mitochondria, oxidized cytochrome c oxidizes O2•− to O2, thus contributing to the detoxification of O2•− [31][32].
Figure 4 depicts the major pathways for the formation of reactive species with a special focus on processes in activated neutrophils and stressed mitochondria. In both systems, the generation of small reactive species starts with the reduction of dioxygen to superoxide anion radicals.
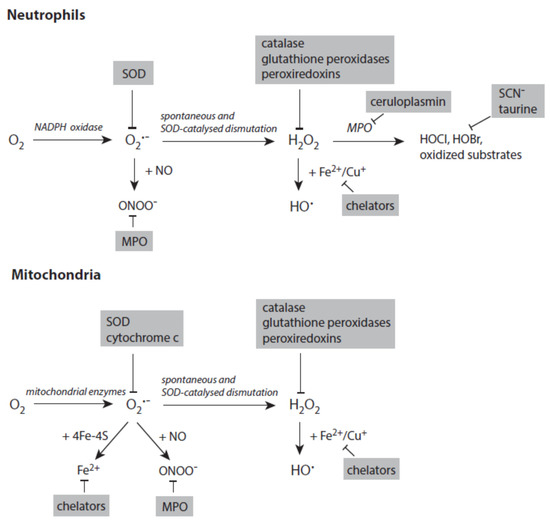
Figure 4. Major pathways in formation of reactive species in activated neutrophils (upper panel) and stressed mitochondria (lower panel). Antagonizing principles against these species are displayed on grey backgrounds. In deactivation of transition metal ions, the term chelators stands for numerous proteins that scavenge, transport, and store iron and copper ions. Further explanations are given in the text. Abbreviations: MPO—myeloperoxidase, SOD—superoxide dismutase.
2.1.2. Hydrogen Peroxide
Spontaneous and SOD-catalyzed dismutation of O2•− represent the main route of formation of H2O2. Thus, all processes generating O2•− also yield H2O2. Otherwise, different peroxisomal enzymes are able to reduce O2 directly to H2O2 [33].
Due to its electronic structure, reactions of H2O2 are restricted to transition metal ions, complexes of these ions, and some proteins with selenocysteine (or cysteine) residues at the active site [34][35]. Hydrogen peroxide is freely permeable through biological membranes, unlike O2•−. The interaction of transition metal ions such as Fe2+ and Cu+ with H2O2 yields very reactive hydroxyl radicals and metal-based reactive species that can cause manifold damaging reactions on biological material [36][37].
Heme peroxidases, different cytochromes, hemoglobin, and myoglobin are activated by H2O2 leading to reactive states of the heme in these proteins. During immune response, H2O2 activates the heme peroxidases myeloperoxidase (MPO), eosinophil peroxidase (EPO), and lactoperoxidase (LPO), which are involved in both pro- and anti-inflammatory activities [2][38][39][40][41].
Several enzymes are known to catalyze the reduction of H2O2 to H2O (Figure 4). Glutathione peroxidase (GPX) utilizes glutathione (GSH) to reduce H2O2. The highly distributed isoforms GPX1 and especially GPX4 also detoxify peroxynitrite, lipid hydroperoxides, and other organic hydroperoxides [42][43]. GSH is recovered from the resulting oxidized glutathione (GSSG) by glutathione reductase [44]. Peroxiredoxins, which are closely coupled to the thioredoxin system, also efficiently reduce H2O2 to H2O [45]. Catalase removes H2O2 by both reduction to H2O and oxidation to O2 [46].
2.1.3. Transition Metal Ions and Hydroxyl Radicals
In the reaction between H2O2 and Fe2+, which is known as the Fenton reaction, the highly reactive hydroxyl radical is formed. Alternatively, iron–oxygen complexes such as ferryl or perferryl compounds are discussed as products of this reaction [36][37]. Similarly, the reaction of H2O2 with Cu+ also yields hydroxyl radicals [47]. Organic hydroperoxides are also oxidized by Fe2+ and Cu+ under the formation of reactive radical species that are involved in subsequent destructive reactions. Beyond Fenton chemistry, further mechanisms apparently contribute to metal-ion-induced tissue damage such as the interaction of Fe2+ with biological buffer components or the formation of Fe2+–O2 and Fe2+–O2–Fe3+ complexes [48][49][50][51][52].
Hydroxyl radicals react in a nearly diffusion-controlled manner with many substrates by abstraction of an H-atom or by addition to an unsaturated system under formation of a hydroxylated product [53]. In both reaction types, substrate radicals are formed that can undergo manifold further reactions.
To avoid the disastrous formation of reactive species such as hydroxyl radicals and others, the main strategy of living matter is the tight control of transport, storage, and utilization of free metal ions (Figure 4) as both iron and copper ions are necessary constituents of many proteins [54][55]. Major components controlling iron metabolism are hepcidin (intestinal absorption), transferrin (blood transport), transferrin receptor (uptake by cells), and ferritin (intracellular storage) [56][57][58][59]. Similarly, different import and export transporters and chaperones are involved in copper metabolism [60]. Ceruloplasmin is able to oxidize both Fe2+ and Cu+ [61]. Lactoferrin released from activated neutrophils binds Fe3+ and promotes its transfer to transferrin [62].
2.1.4. Peroxynitrite
As already mentioned, peroxynitrite is formed in a very rapid reaction between O2•− and NO [23][24]. Peroxynitrite is involved in the formation of thiyl radicals and nitration of tyrosine residues, and is able to induce lipid-peroxidation processes [63][64][65][66]. In reaction with CO2, it yields nitrosoperoxycarbonate, which can decompose into radical species [67][68].
At inflammatory sites where heme peroxidases are present, peroxynitrite is decomposed in its reaction with resting MPO [69][70][71]. Other redox-active heme proteins scavenge peroxynitrite and inactivate this powerful oxidant [72][73][74].
2.1.5. Heme Peroxidases and Hypohalous Acids
At an inflammatory site, the heme protein MPO can be released from activated neutrophils (Figure 4) [38][75]. Eosinophils contain a similar peroxidase, the eosinophil peroxidase (EPO) [76]. A third immunologically relevant heme peroxidase is LPO, which is distributed in mucous surfaces [39]. All three heme peroxidases are able to oxidize SCN− to −OSCN. MPO and EPO also oxidize Br− to HOBr, whereas only MPO is able to yield HOCl from Cl− oxidation [41].
The MPO product HOCl reacts efficiently with methionine and cysteine residues of proteins. Further major protein targets for HOCl are residues of cystine, histidine, tryptophan, lysine, and α-amino groups [77][78]. HOBr , like HOCl, also oxidizes many residues in proteins, especially cysteine and methionine ones. HOBr induces ring halogenation in tyrosine residues more efficiently than HOCl [79].
Both HOCl and HOBr are inactivated at a high rate by thiocyanate (SCN−) [80][81]. HOCl is additionally inactivated by Br−. Further antagonizing principles against both hypohalous acids are ascorbate, GSH, taurine, and, additionally for HOBr, urate [82].
In blood, MPO and EPO are inactivated by ceruloplasmin through the formation of a tight inhibitory complex between heme peroxidase and ceruloplasmin [83][84][85][86].
2.2. Hemoglobin and Myoglobin Metabolites
There is always a release of intact hemoglobin from red blood cells and myoglobin from muscles at low levels. Intravascular hemolysis and rhabdomyolysis can be markedly enhanced under stress and disease situations (Figure 5). Once released from red blood cells, tetrameric ferric hemoglobin dissociates into dimers and is easily oxidized to methemoglobin. This oxidation is usually caused by nitric monoxide. Excessive intravascular hemolysis can affect the bioavailability of NO [87][88]. The serum protein haptoglobin is able to scavenge free methemoglobin. The resulting haptoglobin–methemoglobin complex is eliminated from circulating blood by spleen and liver macrophages in a CD163-dependent process [89][90]. In a similar way, haptoglobin also scavenges metmyoglobin formed after the release of myoglobin from muscle cells.
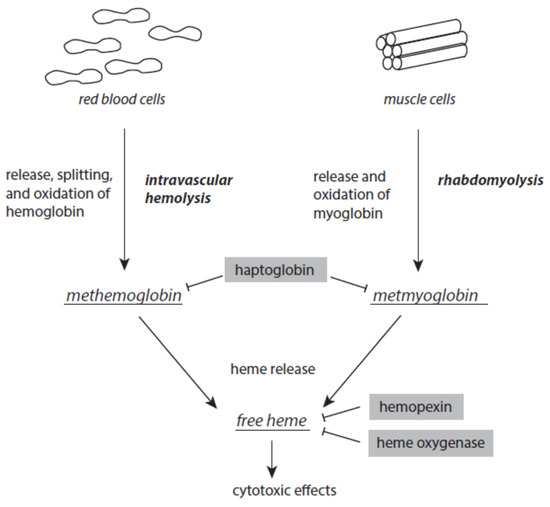
Figure 5. Formation of methemoglobin, metmyoglobin, and free heme as a result of excessive intravascular hemolysis and rhabdomyolysis. Protective mechanism are presented on grey backgrounds. Further explanations are given in the text.
Although it is an acute-phase protein, the capacity of haptoglobin is limited when severe intravascular hemolysis or rhabdomyolysis occur. Both methemoglobin and metmyoglobin spontaneously liberate ferric protoporphyrin IX, briefly known as free heme, a very dangerous molecule [91]. Free heme easily intercalates into the lipid phases of membranes and lipoproteins and the hydrophobic areas of proteins. At these loci, it catalyzes oxidative processes [92][93]. In intact red blood cells, free heme induces hemolytic processes, thus enhancing existing intravascular hemolysis [94][95]. Free heme is highly cytotoxic to kidney and liver [96][97]. It is also a ligand to toll-like receptor 4 and thus contributes to the intensification of inflammatory processes [98][99]. In the nucleus, free heme interacts with parallel guanine-rich quadruplex DNA and RNA structural elements, known as G4 structures [100][101].
In order to avoid the disastrous activities of free heme, different serum proteins are able to complex and inactivate free heme. Hemopexin binds free heme with high affinity. This free-heme–hemopexin complex is liberated from circulating blood via CD91-mediated internalization by hepatocytes [102]. In humans, unlike mice, hemopexin is not an acute-phase protein [103].
2.3. Oxidation of Cell and Tissue Components
In addition to proteolytic cleavage, lipids, proteins, nucleic acids, and carbohydrates are subjected under stress conditions to numerous chemical processes, whereby oxidative modifications predominate [106]. The major initiating agents of these oxidative processes are highly reactive species, free transition metal ions, free heme, and aldehydes. Besides the open chain form of glucose [107], aldehydes result mostly from oxidative modifications of lipids [108][109].
Oxidative alterations of biological substrates are counterbalanced by lipid- and water-based antioxidant mechanisms. In lipid phases, major natural antioxidants are α-tocopherol, β-carotene, ubiquinol, and dehydrolipoic acid. They are mainly involved in the scavenging of lipid peroxyl radicals [110][111][112]. Inactivation of lipid hydroperoxides is a further strategy to prevent oxidative processes. This is achieved most of all by the action of glutathione peroxidase 4 (GPX4). A high intracellular level of GSH is essential for the proper action of GPX4 and other glutathione peroxidases [42][43]. In addition, perturbed acyl residues in phospholipids are cleaved by phospholipases [113]. A thorough control over transition free metal ions also contributes to the prevention of oxidative processes in membranes and lipoproteins.
Urate and ascorbate are the main water-soluble antioxidants in our organism [114][115]. Different polyphenols are important dietary antioxidants [116]. They exert their protective action by radical scavenging, sequestration of free metal ions, and interaction with activated complexes of heme proteins [117][118][119].
2.4. Serine Proteases
2.4.1. Release of Serine Proteases from Immune Cells
At inflammatory sites, activated neutrophils can release the serine proteases elastase, cathepsin G, proteinase 3, and neutrophil serine protease 4. These proteases are primarily involved in the deactivation, killing, and digestion of phagocytosed microorganisms in neutrophils. Their pH optimum is around 8–9, a condition that predominates in early phagosomes of neutrophils [120][121]. Elastase exhibits a killing activity against Gram-negative bacteria [122][123] and a variety of cancer cells [124]. In cancer cells, unlike non-cancer cells, elastase cleaves CD95 to liberate a death domain fragment that acts cytotoxically together with histone H1 [124].
Serine proteases participate in the recruitment of neutrophils to a destination site by digestion of the surrounding tissue components and the induction and regulation of immune signaling. Elastase and proteinase 3 are able to cleave a broad range of chemokines and cytokines [125]. The substrate specificity of cathepsin G is also relatively broad but more restricted than that observed for elastase and proteinase 3 [126]. In these experiments, only a few cytokines and chemokines, such as TNF-α, interleukin 5 (IL-5), interleukin 8 (IL-8), macrophage colony-stimulating factor (M-CSF), monocyte chemoattractant protein 1 (MCP-1), IL-1α, and Rantes, were resistant to neutrophil serine proteases.
Elastase and other serine proteases are attached together with other neutrophil proteins to a DNA network in neutrophil extracellular traps. These traps can kill external microbes independent of phagocytosis [127][128].
Activated mast cells release the serine proteases chymase, tryptase, and cathepsin G [129]. These proteases are involved in matrix destruction, tissue remodeling, and regulation of inflammation. Mast cell tryptase and chymase are more restrictive than neutrophil serine proteases in the cleavage of chemokines and cytokines [125][126].
2.4.2. Activities of Neutrophil Serine Proteases
Although all serine proteases contribute to damaging reactions, the focus is mostly directed on elastase. An overview about multiple activities of neutrophil elastase during immune response is given in Figure 6. Once released from activated neutrophils, elastase can affect healthy tissues. Elastase is involved in the destruction of extracellular matrix components such as elastin, collagens, proteoglycans, and laminin [130].
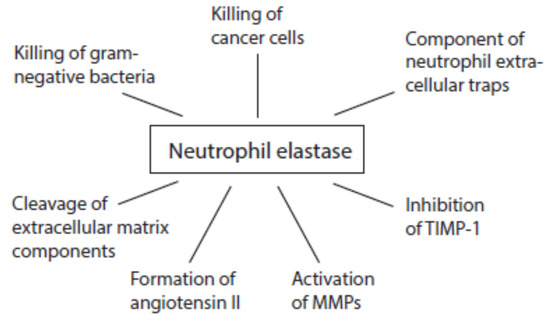
Figure 6. Activities of neutrophil elastase at inflammatory sites.
Like cathepsin G, proteinase 3, and cathepsin B, neutrophil elastase is able to convert angiotensinogen and angiotensin I into angiotensin II [131][132][133]. This pro-inflammatory peptide can further foment inflammatory processes.
At inflammatory sites, neutrophil elastase activates MMP2, MMP3, and MMP9 from inactive precursors by cleaving an inhibitory protein residue [134][135][136][137]. Cathepsin G is also able to activate MMP3 [134]. Cathepsin G and proteinase 3 are involved in MMP2 activation [135]. Elastase might additionally degrade TIMP-1 [136][138].
2.4.3. Mast Cell Serine Proteases
Human mast cells contain several types of tryptases and two members of chymase-like proteins, namely α-chymase and cathepsin G, which are secreted in response to allergens and pathogens [129]. Mast cell proteases are known to stimulate the production of pro-inflammatory mediators such as IL-6 and IL-8 from bronchial epithelial cells and promote procollagen cleavage. With these activities they contribute to the recruitment of neutrophils and eosinophils at inflamed epithelium [139][140][141][142].
Other inflammation-promoting activities of chymase are the cleavage of angiotensin I into angiotensin II, activation of MMPs, and release of selected extracellular matrix elements [143]. Tryptase is involved in the degradation of fibronectin and chemokines [143]. Both tryptases and chymases contribute to the activation of different MMPs [144][145][146]. MMPs are implicated in the pathogenesis of atherosclerosis and abdominal aortic aneurysms [147][148][149][150][151]. Mast cell proteases are implied in airway epithelial remodeling and alterations in epithelium functions [152]. They also contribute to angiogenesis induction during tumor growth [153]. Chymase promotes the formation of active TGF-β from its precursor [154].
Tetrameric tryptase is stabilized by heparin and some other glycosoaminoglycans [155]. In this complex, tryptase is not accessible to anti-proteases such as A1AT, SPLI, and α2-macroglobulin [156][157]. Lactoferrin, myeloperoxidase, and antithrombin III, which are known to have heparin-binding domains, can inhibit tryptase activity [158][159][160][161]. Spontaneous dissociation of the tryptase tetramer is a further mechanism to control tryptase activity [155][162].
2.4.4. Antiproteases
Several antagonizing proteins against elastase and other serine proteases exist in blood and tissues (Figure 7). The most abundant anti-protease is the serpin α1-antitrypsin (A1AT). This serum protein is synthesized in the liver and represents an acute-phase protein. A1AT inhibits elastase and cathepsin G but not in the presence of heparin [163][164]. The activity of proteinase 3 is affected by A1AT to a lesser degree. Heparin, however, enhanced the inactivation of proteinase 3 [165].
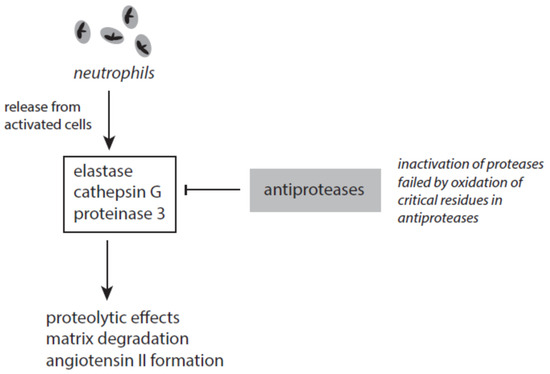
Figure 7. The interplay between neutrophil-derived serine proteases and antiproteases.
Several factors contribute to the failure of A1AT to inhibit elastase. The inactivation of elastase requires two unperturbed methionine residues (Met-351 and Met-358) at the active site of A1AT. By oxidation of these residues A1AT loses its ability to inhibit elastase [166]. Under stress conditions methionine oxidation in A1AT can be initiated by highly reactive species such as hydroxyl radicals, peroxynitrite, HOCl, HOBr, and others [167]. Tobacco smoke and activated phagocytes are under discussion to contribute to methionine oxidation in A1AT and thus cause an acquired A1AT deficiency [167]. Furthermore, neutrophil elastase can bind to negatively charged surfaces and polymers. Surface-bound elastase cannot be inhibited by endogenous antiproteases [168].
Serpin A3, also known as α1-antichymotrypsin, is, like A1AT, an acute-phase protein. This antiprotease efficiently inactivates cathepsin G and mast cell chymase [169][170][171].
Secretory leukocyte protease inhibitor (SLPI) is able to inactivate several serine proteases such as neutrophil elastase, cathepsin G, tryptase, and chymase [172]. SLPI is constitutively expressed in mucous secretions [173][174] and also secreted from activated immune cells. It is assumed that SLPI exhibits an anti-apoptotic effect on immune cells and thus contributes to a better removal of dying cells and microbes at inflammatory sites [175].
Elafin, which is also known as proteinase inhibitor 3, is able to inactivate neutrophil elastase and proteinase 3 [176][177]. It exerts anti-inflammatory, anti-microbial, and wound-healing effects [176][177]. Contradictory results were reported about the action of elafin on tumorigenesis. These results range from promotion of cell proliferation and induction of resistance against chemotherapy to tumor-suppressive effects [178][179]. In early-stage hepatocellular carcinoma, elafin promotes metastasis formation via activation of EGFR/AKT signaling [180].
The antiprotease serpin B1 efficiently inactivates elastase, cathepsin G, and proteinase 3 [181]. Under oxidative stress, the cysteine residue at the active site in serpin B1 is oxidized with the loss of the antiprotease activity.
In contrast to the aforementioned antiproteases, which directly interact with the active site of proteases, α2-macroglobulin forms a tetrameric cage around active proteases, thus inhibiting the direct contact between protease and substrate molecules. In this way, large substrate molecules such as collagen are excluded from direct contact, whereas small peptide substrates can be digested [182][183]. Although α2-macroglobulin inhibits the activities of elastase, cathepsin G, proteinase 3, and MMP9 released from neutrophils [184][185][186], the complex between elastase and α2-macroglobulin is still active against small substrates [185]. Moreover, neutrophil-derived reactive species such as HOCl can hinder α2-macroglobulin to form tetramers and promote stabilization of dimers with the loss of the antiprotease activity [187][188].
High-affinity complexes are also known between ceruloplasmin and serine proteases of neutrophils [62]. In this way, a destructive action of serine proteases on tissue components is minimized.
2.5. Small Pro-Inflammatory Peptides
2.5.1. Angiotensin II
The peptide hormone angiotensin II is an essential part of the renin–angiotensin–aldosteron system. It is involved in the regulation of blood pressure and water metabolism. During this activity, angiotensin II is formed from angiotensin I by the angiotensin-converting enzyme (ACE).
At inflammatory sites, angiotensin II can also be produced from cleavage of both angiotensinogen and angiotensin I by serine proteases released from immune cells such as elastase, cathepsin G, proteinase 3, and mast cell chymase (Figure 8) [131][132][133][189]. Increased angiotensin II contributes via docking to AT1 and AT2 receptors to proteolysis, actin cleavage, apoptosis induction, and activation of the ubiquitin-mediated protein degradation [190][191][192]. It also promotes superoxide anion radical production via activation of NADH/NADPH oxidases [193]. Generally, these pro-inflammatory activities of angiotensin II mediate the prolonged existence of inflammatory states [194].
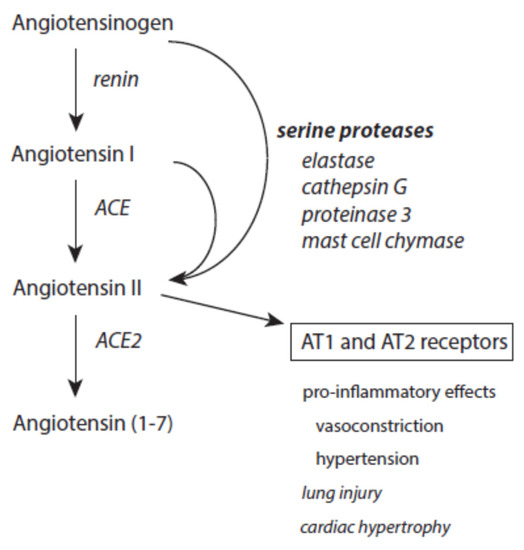
Figure 8. Effects of serine proteases on the renin–angiotensin–aldosteron system. Further explanations are given in the text.
Angiotensin II is under the control of ACE2, which converts this octapeptide to angiotensin 1–7 [195]. This limits the devastating activity of angiotensin II. Moreover, angiotensin 1–7 exerts an anti-inflammatory activity [196].
2.5.2. Bradykinin
As an essential member of the contact system, the nonapeptide bradykinin is responsible for increased vascular permeability, vasodilation, hypotension, and other effects via interaction with its constitutively expressed B2 receptor [197][198]. A further pro-inflammatory metabolite is des-Arg9-bradykinin, which is formed from bradykinin by carboxypeptidase N. At inflammatory sites, des-Arg9-bradykinin acts selectively via bradykinin B1 receptors, which are only expressed in inflamed and injured tissue [199][200][201].
Bradykinin is a short-lived mediator of inflammation. It is inactivated by aminopeptidase P and the angiotensin-converting enzyme (ACE). Inhibition of ACE enhances bradykinin’s effects [200].
2.6. Inhibition of Matrix Metalloproteases
In human tissues, 23 MMPs and four TIMPs are found. Most MMPs are normally not expressed in healthy tissue. The activity of MMPs is essential in tissue remodeling, such as angiogenesis, bone growth, wound healing, and repair processes during the resolution of inflammation [202][203].
MMPs are secreted as inactive enzymes bearing an inhibitory prodomain that must be cleaved. In addition to neutrophil serine proteases, plasmin, chymases, and other MMPs are involved in MMP activation [204]. At low concentrations, highly reactive species such as HOCl, •OH, and ONOO− can activate MMPs. However, higher concentrations of these species inactivate active MMPs [205].
During the exudation and infiltration phase of inflammation, MMP2 and MMP9 are mainly secreted from invading immune cells, smooth muscle cells, and fibroblasts [206][207][208]. These and other MMPs contribute to cleaving the matrix components collagen and elastin.
The activity of MMPs is tightly controlled by TIMPs and α2-macroglobulin. The latter inhibitor, which has a very broad activity range against proteases, acts in blood and other biological fluids [209]. Generally, TIMPs have a broad spectrum of inhibition of MMPs. The constitutively expressed TIMP-2, like TIMP-3 and TIMP-4, is able to inhibit nearly all MMPs. TIMP-1 has a low activity against membrane-bound MMPs [203]. TIMP-3 additionally inhibits members of disintegrin metalloproteinases. Moreover, it is the only TIMP that binds to the ECM [210].
References
- Arnhold, J. Immune response and tissue damage. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 155–204.
- Arnhold, J. The dual role of myeloperoxidase in immune response. Int. J. Mol. Sci. 2020, 21, 8057.
- Li, M.O.; Flavell, R.A. Contextual regulation of inflammation: A duet by transforming growth factor-beta and interleukin-10. Immunity 2008, 28, 468–476.
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146.
- Frangogiannis, N.G. Transforming growth factor-β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103.
- Budi, E.H.; Schaub, J.R.; Decaris, M.; Turner, S.; Derynck, R. TGF-β as a driver of fibrosis: Physiological roles and therapeutic opportunities. J. Pathol. 2021, 254, 358–373.
- Arnhold, J. Role of reactive species in destructions. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 23–54.
- Segal, A.W.; Jones, O.T.G. Novel cytochrome b system in phagocytic vacuoles from human granulocytes. Nature 1976, 276, 515–517.
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47.
- Cheng, G.; Cao, Z.; Xu, X.; van Meir, E.G.; Lambeth, J.D. Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001, 269, 131–140.
- Geiszt, M.; Witta, J.; Baffi, J.; Lekstrom, K.; Leto, T.L. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal host defense. FASEB J. 2003, 17, 1502–1504.
- Harper, R.W.; Xu, C.; Eiserich, J.P.; Chen, Y.; Kao, C.Y.; Thai, P.; Setiadi, H.; Wu, R. Differential regulation of dual NADPH/peroxidases. Duox1 and Duox2, by Th1 and Th2 cytokines in the respiratory tract epithelium. FEBS Lett. 2005, 579, 4911–4917.
- Olson, J.S.; Ballou, D.P.; Palmer, G.; Massey, V. The reaction of xanthine oxidase with molecular oxygen. J. Biol. Chem. 1974, 249, 4350–4362.
- Anderson, R.F.; Hille, R.; Massey, V. The radical chemistry of milk xanthine oxidase as studies by radiation chemistry technique. J. Biol. Chem. 1986, 261, 15870–15876.
- Carrell, R.W.; Winterbourn, C.C.; Rachmilewitz, E.A. Activated oxygen and hemolysis. Br. J. Hematol. 1975, 30, 259–264.
- Harel, S.; Kanner, J. Hemoglobin and myoglobin as inhibitors of hydroxyl radical generation in a model system of “iron redox” cycle. Free Radic. Res. Commun. 1989, 6, 1–10.
- Land, E.J.; Mukherfee, T.; Swallow, A.J.; Bruce, J.M. One-electron reduction of adriamycin: Properties of the semiquinone. Arch. Biochem. Biophys. 1983, 225, 116–121.
- Camhi, S.L.; Lee, P.; Choi, A.M.K. The oxidative stress response. New Horizons 1995, 3, 170–182.
- Goncalves, R.L.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Site of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 2015, 290, 209–227.
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31.
- Koppenol, W.H. Thermodynamics of reactions involving oxyradicals and hydrogen peroxide. Bioelectrochem. Bioenerg. 1987, 18, 3–11.
- Bielski, B.H.J.; Cabelli, D.E.; Arudi, R.L. Reactivity of HO2/O2− radicals in aqueous solution. J. Phys. Chem. Ref. Data 1985, 14, 1041–1100.
- Huie, R.E.; Padmaja, S. Reactions of NO and O2−. Free Radic. Res. Commun. 1993, 18, 195–199.
- Kissner, R.; Nauser, T.; Bugnon, P.; Lye, P.G.; Koppenol, W.H. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem. Res. Toxicol. 1997, 10, 1285–1292.
- Gardner, P.R. Superoxide-driven aconitase Fe-S cycling. Biosci. Rep. 1997, 17, 33–42.
- Gardner, P.R. Aconitase: Sensitive target and measure of superoxide. Meth. Enzymol. 2002, 349, 9–23.
- McCord, J.; Fridovich, I. Superoxide dismutase: An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1960, 224, 6049–6055.
- Chang, L.Y.; Slot, J.W.; Geuze, H.J.; Crapo, J.D. Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J. Cell Biol. 1988, 107, 2169–2179.
- Weisiger, R.A.; Fridovich, I. Mitochondrial superoxide dismutase. Site of synthesis and intramolecular localization. J. Biol. Chem. 1973, 248, 4793–4796.
- Antonyuk, S.V.; Strange, R.W.; Marklund, S.L.; Hasnain, S.S. The structure of human extracellular copper-zinc superoxide dismutase at 1.7 Å resolution: Insights into heparin and collagen binding. J. Mol. Biol. 2009, 388, 310–326.
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52.
- Pasdois, P.; Parker, J.E.; Griffiths, E.J.; Halestrap, A.P. The role of oxidized cytochrome c in regulating mitochondrial reactive species production and its perturbation in ischemia. Biochem. J. 2011, 436, 493–505.
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta 2006, 1763, 1755–1766.
- Koppenol, W.H.; Butler, J. Mechanisms of reactions involving singlet oxygen and the superoxide anion. FEBS Lett. 1977, 83, 1–6.
- Reich, H.J.; Hondal, R.J. Why nature chose selenium. ACS Chem. Biol. 2016, 11, 821–841.
- Halliwell, B.; Gutteridge, J.M.C. Iron as a biological pro-oxidant. ISI Atlas Sci. Biochem. 1988, 1, 48–52.
- Koppenol, W.H. The centennial of the Fenton reaction. Free Radic. Biol. Chem. 1993, 15, 645–651.
- Arnhold, J.; Flemmig, J. Human myeloperoxidase in innate and acquired immunity. Arch. Biochem. Biophys. 2010, 500, 92–106.
- Flemmig, J.; Gau, J.; Schlorke, D.; Arnhold, J. Lactoperoxidase as potential drug target. Expert Opin. Ther. Targets 2016, 20, 447–461.
- Arnhold, J. Heme peroxidases at unperturbed and inflamed mucous surfaces. Antioxidants 2021, 10, 1805.
- Arnhold, J.; Malle, E. Halogenation activity of mammalian heme peroxidases. Antioxidants 2022, 11, 890.
- Ursini, F.; Maiorino, M.; Roveri, A. Phospholipid hydroperoxide glutathione peroxidase (PHGPx): More than an antioxidant enzyme? Biomed. Environm. Sci. 1997, 10, 327–332.
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997.
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208.
- Low, F.M.; Hampton, M.P.; Winterbourn, C.C. Prx2 and peroxide metabolism in the erythrocyte. Antioxid. Redox Signal. 2008, 10, 1621–1630.
- Goyal, M.M.; Basak, A. Human catalase: Looking for complete identity. Prot. Cell 2010, 1, 888–897.
- Gunther, M.R.; Hanna, P.M.; Mason, R.P.; Cohen, M.S. Hydroxyl radical formation from cuprous ion and hydrogen peroxide: A spin-trapping study. Arch. Biochem. Biophys. 1995, 316, 515–522.
- Ryan, T.P.; Aust, S.D. The role of iron in oxygen-mediated toxicities. Crit. Rev. Toxicol. 1992, 22, 119–141.
- Reinke, L.A.; Rau, J.M.; McCay, P.B. Characteristics of an oxidant formed during iron(II) autoxidation. Free Radic. Biol. Med. 1994, 16, 485–492.
- Qian, S.Y.; Buettner, G.R. Iron and dioxygen chemistry is an important route to initiation of biological free radical oxidations: An electron paramagnetic resonance spin trapping study. Free Radic. Biol. Med. 1999, 26, 1447–1456.
- Urbanski, N.K.; Beresewicz, A. Generation of •OH initiated by interaction of Fe2+ and Cu+ with dioxygen; comparison with the Fenton chemistry. Acta Biochim. Pol. 2000, 47, 951–962.
- Flemmig, J.; Arnhold, J. Ferrous ion-induced strand breaks in the DNA plasmid pBR322 are not mediated by hydrogen peroxide. Eur. Biophys. J. 2007, 36, 377–384.
- Bors, W.; Erben-Russ, M.; Saran, M. Fatty acid peroxyl radicals: Their generation and reactivities. Bioelectrochem. Bioenerg. 1987, 18, 37–49.
- Crichton, R.R.; Ward, R.J. Iron homeostasis. Metal Ions Biol. Syst. 1998, 35, 633–665.
- Ponka, P. Cellular iron metabolism. Kidney Int. 1999, 55, S2–S11.
- Zhao, N.; Zhang, A.-S.; Enns, C.A. Iron regulation by hepcidin. J. Clin. Investig. 2013, 123, 2337–2343.
- Gkouvatsos, K.; Papanikolaou, G.; Pantopoulos, K. Regulation of iron transport and the role of transferrin. Biochim. Biophys. Acta 2011, 1820, 188–202.
- Gamella, E.; Buratti, P.; Cairo, G.; Recalcati, S. The transferrin receptor: The cellular iron gate. Metallomics 2017, 9, 1367–1375.
- Massover, W.H. Ultrastructure of ferritin and apoferritin: A review. Micron 1993, 24, 389–437.
- Prohaska, J.R. Role of copper transporters in copper homeostasis. Am. J. Clin. Nutr. 2008, 88, 826S–829S.
- Stoj, C.; Kosman, D.J. Cuprous oxidase activity of yeast Fet3p and human ceruloplasmin: Implication for function. FEBS Lett. 2003, 554, 422–426.
- Sokolov, A.V.; Pulina, M.O.; Ageeva, K.V.; Runova, O.I.; Zakharova, E.T.; Vasilyev, V.B. Identification of leukocyte cationic proteins that interact with ceruloplasmin. Biochemistry 2007, 72, 872–877.
- Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. Peroxynitrite-induced membrane lipid peroxidation: The cytotoxic potential of superoxide and nitric oxide. Arch. Biochem. Biophys. 1991, 288, 481–487.
- Darley-Usmar, V.M.; Hogg, N.; O’Leary, V.J.; Wilson, M.T.; Moncada, S. The simultaneous generation of superoxide and nitric oxide can initiate lipid peroxidation in human low density lipoprotein. Free Radic. Res. Commun. 1992, 17, 9–20.
- Goldstein, S.; Czapski, G. Mechanism of the nitrosation of thiols and amines by oxygenated ∙NO solutions: The nature of the nitrosating intermediates. J. Am. Chem. Soc. 1996, 118, 3419–3425.
- Schopfer, F.J.; Baker, P.R.S.; Freeman, B.A. NO-dependent protein nitration: A cell signaling event or an oxidative inflammatory response? Trends Biochem. Sci. 2003, 28, 646–654.
- Augusto, O.; Bonini, M.G.; Amanso, A.M.; Linares, E.; Santos, C.C.; de Menezes, S.L. Nitrogen dioxide and carbonate radical anion: Two emerging radicals in biology. Free Radic. Biol. Med. 2002, 32, 841–859.
- Ferrer-Sueta, G.; Radi, R. Chemical biology of ONOO-: Kinetics, diffusion, and radicals. ACS Chem. Biol. 2009, 4, 161–177.
- Furtmüller, P.G.; Jantschko, W.; Zederbauer, M.; Schwanninger, M.; Jakoptisch, C.; Herold, S.; Koppenol, W.H.; Obinger, C. Peroxynitrite efficiently mediates the interconverion of redox intermediates of myeloperoxidase. Biochem. Biophys. Res. Commun. 2005, 337, 944–954.
- Galijasevic, S.; Maitra, D.; Lu, T.; Sliskovic, I.; Abdulhamid, I.; Abu-Soud, H.M. Myeloperoxidase interaction with peroxynitrite: Chloride deficiency and heme depletion. Free Radic. Biol. Med. 2009, 47, 431–439.
- Koyani, C.N.; Flemmig, J.; Malle, E.; Arnhold, J. Myeloperoxidase scavenges peroxynitrite: A novel anti-inflammatory action of the heme enzyme. Arch. Biochem. Biophys. 2015, 571, 1–19.
- Su, J.; Groves, J.T. Mechanisms of peroxynitrite interactions with heme proteins. Inorg. Chem. 2010, 49, 6317–6329.
- Keng, T.; Privalle, C.T.; Gilkeson, G.S.; Weinberg, J.B. Peroxynitrite formation and decreased catalase activity in autoimmune MRL-lpr/lpr mice. Mol. Med. 2000, 6, 779–792.
- Gebicka, L.; Gebicki, J.L. Reaction of heme peroxidases with peroxynitrite. IUBMB Life 2000, 49, 11–15.
- Klebanoff, S.J. Myeloperoxidase: Friend and foe. J. Leukoc. Biol. 2005, 77, 598–625.
- Rothenberg, M.E.; Hogan, S.P. The eosinophil. Annu. Rev. Immunol. 2006, 24, 147–174.
- Pattison, D.I.; Davies, M.J. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem. Res. Toxicol. 2001, 14, 453–464.
- Hawkins, C.L.; Pattison, D.I.; Davies, M.J. Hypochlorite-induced oxidation of amino acids, peptides, and proteins. Amino Acids 2003, 25, 259–274.
- Pattison, D.I.; Davies, M.J. Kinetic analysis of the reaction of hypobromous acid with protein components: Implication for cellular damage and the use of 3-bromotyrosine as a marker of oxidative stress. Biochemistry 2004, 43, 4799–4809.
- Ashby, M.T.; Carlson, A.C.; Scott, M.J. Redox buffering of hypochlorous acid by thiocyanate in physiologic fluids. J. Am. Chem. Soc. 2004, 126, 15976–15977.
- Nagy, P.; Beal, J.L.; Ashby, M.T. Thiocyanate is an efficient endogenous scavenger of the phagocytic killing agent hypobromous acid. Chem. Res. Toxicol. 2006, 19, 587–593.
- Davies, M.J.; Hawkins, C.L. The role of myeloperoxidase in biomolecule modification, chronic inflammation, and disease. Antioxid. Redox Signal. 2020, 32, 957–981.
- Sokolov, A.V.; Ageeva, K.V.; Pulina, M.O.; Cherkalina, O.S.; Samygina, V.R.; Vlasova, I.I.; Panasenko, O.M.; Zakharova, E.T.; Vasilyev, V.B. Ceruloplasmin and myeloperoxidase in complex affect the enzymatic properties of each other. Free Radic. Res. 2008, 42, 989–998.
- Chapman, A.L.P.; Mocatta, T.J.; Shiva, S.; Seidel, A.; Chen, B.; Khalilova, I.; Paumann-Page, M.E.; Jameson, G.N.L.; Winterbourn, C.C.; Kettle, A.J. Ceruloplasmin is an endogenous inhibitor of myeloperoxidase. J. Biol. Chem. 2013, 288, 6464–6477.
- Samygina, V.R.; Sokolov, A.V.; Bourenkov, G.; Petoukhov, M.V.; Pulina, M.O.; Zakharova, E.T.; Vasilyev, V.B.; Bartunik, H.; Svergun, D.I. Ceruloplasmin: Macromolecular assemblies with iron-containing acute phase proteins. PLoS ONE 2013, 8, e67145.
- Sokolov, A.V.; Kostevich, V.A.; Zakharova, E.T.; Samygina, V.R.; Panasenko, O.M.; Vasilyev, V.B. Interaction of ceruloplasmin with eosinophil peroxidase as compared to its interplay with myeloperoxidase: Reciprocal effect on enzymatic properties. Free Radic. Res. 2015, 49, 800–811.
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon III, R.O.; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389.
- Rother, R.P.; Bell, L.; Hillman, P.; Gladwin, M.T. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin. J. Am. Med. Assoc. 2005, 293, 1653–1662.
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffmann, H.J.; Law, S.K.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201.
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Tolosano, E. Haptoglobin and hemopexin in heme detoxification and iron recycling. In Acute Phase Proteins- Regulation and Functions of Acute Phase Proteins; Veas, F., Ed.; Intech: Rijeka, Croatia, 2011; pp. 261–288.
- Bunn, H.F.; Jandl, J.H. Exchange of heme among hemoglobins and hemoglobin and albumin. J. Biol. Chem. 1968, 243, 465–475.
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercelotti, G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. 1991, 11, 1700–1711.
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercelotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887.
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188.
- Flemmig, J.; Schlorke, D.; Kühne, F.-W.; Arnhold, J. Inhibition of the heme-induced hemolysis of red blood cells by the chlorite-based drug WF10. Free Radic. Res. 2016, 50, 1386–1395.
- Lin, T.; Sammy, F.; Yang, H.; Thundivalappil, S.; Hellman, J.; Tracey, K.C.; Warren, H.S. Identification of hemopexin as an anti-inflammatory factor that inhibits synergy of hemoglobin with HMGB1 in sterile and infectious inflammation. J. Immunol. 2012, 189, 2017–2022.
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercelotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284.
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliviera, M.F.; Oliviera, P.L.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229.
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercelotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390.
- Poon, L.C.; Methot, S.P.; Morabi-Pazocki, W.; Pio, F.; Bennet, A.J.; Sen, D. Guanine-rich RNAs and DNAs that bind heme robustly catalyze oxygen transfer reactions. J. Am. Chem. Soc. 2011, 133, 1877–1884.
- Gray, L.T.; Lombardi, E.P.; Verga, D.; Nicolas, A.; Teulade-Fichou, M.-P.; Londoño-Vallejo, A.; Maizels, N. G-Quadruplexes sequester free heme in living cells. Cell Chem. Biol. 2019, 26, 1681–1689.
- Hvidberg, V.; Maniecki, M.B.; Jacobson, C.; Hojrup, P.; Moller, H.J.; Moestrup, S.K. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579.
- Lin, T.; Maita, D.; Thundivalappil, S.R.; Riley, F.E.; Hambsch, J.; van Marter, L.J.; Christou, H.A.; Berra, L.; Fagan, S.; Christiani, D.C.; et al. Hemopexin in severe inflammation and infection: Mouse models and human diseases. Crit. Care 2015, 19, 166.
- Santoro, A.M.; Lo Giudice, M.C.; D’Urso, A.; Lauceri, R.; Purello, R.; Milardi, D. Cationic porphyrins are reversible proteasome inhibitors. J. Am. Chem. Soc. 2012, 134, 10451–10457.
- Vallelian, F.; Deuel, J.W.; Opitz, L.; Schaer, C.A.; Puglia, M.; Lönn, M.; Engelsberger, W.; Schauer, S.; Karnaukhova, E.; Spahn, D.R.; et al. Proteasome inhibition and oxidative reactions disrupt cellular homeostasis during heme stress. Cell Death Differ. 2015, 22, 597–611.
- Arnhold, J. Oxidation and reduction of biological material. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 55–97.
- Hayward, L.D.; Angyal, S.J. A symmetry rule for the circular dichroism of reducing sugar, and the proportion of carbonyl forms in aqueous solutions thereof. Carbohydr. Res. 1977, 53, 13–20.
- Pamplona, R. Advanced lipoxidation end-products. Chem. Biol. Interact. 2011, 192, 14–20.
- Vistoli, G.; de Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs amd ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47 (Suppl. 1), 3–27.
- Burton, G.W.; Joyce, A.; Ingold, K.U. Is vitamin E the only lipid-soluble, chain-breaking antioxidant in human blood plasma and erythrocyte membrane? Arch. Biochem. Biophys. 1983, 221, 281–290.
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, α-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543.
- Niki, E. Antioxidants in relation to lipid peroxidation. Chem. Phys. Lipids 1987, 44, 227–253.
- Van Kujik, F.J.G.M.; Sevanian, A.; Handelman, G.J.; Dratz, E.A. A new role for phospholipase A2: Protection of membranes from lipid peroxidation damage. Trends Biochem. Sci. 1987, 12, 31–34.
- Hochstein, P.; Hatch, L.; Sevanian, A. Uric acid: Functions and determinations. Methods Enzymol. 1984, 105, 162–166.
- Nyyssönen, K.; Porkkala-Sarataho, E.; Kaikkonen, J.; Salonen, J.T. Ascorbate and urate are the strongest determinants of plasma antioxidative capacity and serum lipid resistance to oxidation in Finnish men. Atherosclerosis 1997, 130, 223–233.
- Spencer, J.P.E.; Abd El Mohsen, M.M.; Minihane, A.-M.; Mathers, J.C. Biomarkers of the intake of dietary polyphenols: Strengths, limitations and application in nutrition research. Br. J. Nutr. 2008, 99, 12–22.
- Steenken, S.; Neta, P. One-electron redox potentials of phenols. Hydroxy- and aminophenols and related compounds of biological interest. J. Phys. Chem. 1982, 86, 3661–3667.
- Perron, N.R.; Brumaghim, J.L. A review of the antioxidant mechanisms of polyphenol compounds related to iron binding. Cell Biochem. Biophys. 2009, 53, 75–100.
- Gau, J.; Furtmüller, P.G.; Obinger, C.; Prévost, M.; van Antwerpen, P.; Arnhold, J.; Flemmig, J. Flavonoids as promoters of the (pseudo-)halogenating activity of lactoperoxidase and myeloperoxidase. Free Radic. Biol. Med. 2016, 97, 307–319.
- Wardlaw, A.C. The complement-dependent bacteriolytic activity of normal human serum. I. The effect of pH and ionic strength and the role of lysozyme. J. Exp. Med. 1962, 115, 1231–1249.
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759.
- Weinrauch, Y.; Drujan, D.; Shapiro, S.D.; Weiss, J.; Zychlinsky, A. Neutrophil elastase targets virulence factors of enterobacteria. Nature 2002, 417, 91–94.
- Belaaouaj, A. Neutrophil elastase-mediated killing of bacteria: Lessons from targeted mutagenesis. Microb. Infect. 2002, 4, 1259–1264.
- Cui, C.; Chakraborty, K.; Tang, X.A.; Zhou, G.; Schoenfelt, K.Q.; Becker, K.M.; Hoffmann, A.; Chang, Y.-F.; Blank, A.; Reardon, C.A.; et al. Neutrophil elastase selectively kills cancer cells and attenuates tumorigenesis. Cell 2021, 184, 3163–3177.
- Fu, Z.; Akula, S.; Thorpe, M.; Hellman, L. Potent and broad but not unselectively cleavage of cytokines and chemokines by human neutrophil elastase and proteinase 3. Int. J. Mol. Sci. 2020, 21, 651.
- Fu, Z.; Thorpe, M.; Alemayehu, R.; Roy, A.; Kervinen, J.; de Garavilla, L.; Abrink, M.; Hellman, L. Highly selective cleavage of cytokines and chemokines by the human mast cell chymase and neutrophil cathepsin G. J. Immunol. 2017, 198, 1474–1483.
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535.
- Papayannopoulos, V.; Zychlinsky, A. NETs: A new strategy for using old weapons. Trends Immunol. 2009, 30, 513–521.
- Caughey, G.H. Mast cell tryptases and chymases in inflammation and host defense. Immunol. Rev. 2007, 217, 141–151.
- Korkmaz, B.; Moreau, T.; Gauthier, F. Neutrophil elastase, proteinase 3 and cathepsin G: Physicochemical properties, activity and physiopathological functions. Biochimie 2008, 90, 227–242.
- Ramaha, A.; Patston, P.A. Release and degradation of angiotensin I and II from angiotensinogen by neutrophil serine proteases. Arch. Biochem. Biophys. 2002, 397, 77–83.
- Vidotti, D.B.; Casarini, D.E.; Christovam, P.C.; Leite, C.A.; Schor, N.; Boim, M.A. High glucose concentration stimulates intracellular renin activity and angiotensin II generation in rat mesangial cells. Am. J. Physiol. Renal Physiol. 2004, 286, F1039–F1045.
- Penafuerte, C.A.; Gagnon, B.; Sirois, J.; Murphy, J.; MacDonald, N.; Tremblay, M.L. Identification of neutrophil-derived proteases and angiotensin II as biomarkers of cancer cachexia. Br. J. Cancer 2016, 114, 680–687.
- Okada, Y.; Nakanishi, I. Activation of matrix metalloproteinase 3 (stromelysin) and matrix metalloproteinase 2 (‘gelatinase’) by human neutrophil elastase and cathepsin G. FEBS Lett. 1989, 249, 353–356.
- Shamanian, P.; Schwartz, J.D.; Pocock, B.J.Z.; Monea, S.; Whiting, D.; Marcus, S.G.; Mignatti, P. Activation of progelatinase (MMP-2) by neutrophil elastase, cathepsin G, and proteinase-3: A role for inflammatory cells in tumor invasion and angiogenesis. J. Cell. Physiol. 2001, 189, 197–206.
- Gaggar, A.; Li, Y.; Weathington, N.; Winkler, M.; Kong, M.; Jackson, P.; Blalock, J.E.; Clancy, J.P. Matrix metalloprotease-9 dysregulation in lower airway secretions of cystic fibrosis patients. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L96–L104.
- Garratt, L.W.; Sutanto, E.N.; Ling, K.-M.; Looi, K.; Iosifidis, T.; Martinovich, K.M.; Shaw, N.C.; Kicic-Starcevich, E.; Knight, D.A.; Ranganathan, S.; et al. on behalf of the Australian respiratory early surveillance team for cystic fibrosis. Matrix metalloproteinase activation by free neutrophil elastase contributes to bronchiectasis progression in early cystic fibrosis. Eur. Respir. J. 2015, 46, 384–394.
- Jackson, P.I.; Xu, X.; Wilson, L.; Weathington, N.M.; Clancy, J.P.; Blalock, J.E.; Gaggar, A. Human neutrophil elastase-mediated cleavage sites of MMP-9 and TIMP-1: Implications to cystic fibrosis proteolytic dysfunction. Mol. Med. 2010, 16, 159–166.
- Cairns, J.A.; Walls, A.F. Mast cell tryptase is a mitogen for epithelial cells. Stimulation of IL-8 production and intercellular adhesion molecule-1 expression. J. Immunol. 1996, 156, 275–283.
- Dougherty, R.H.; Sidhu, S.S.; Raman, K.; Solon, M.; Solberg, O.D.; Caughey, G.H.; Woodruff, P.G.; Fahy, J.V. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J. Allergy Clin. Immunol. 2010, 125, 1046–1053.
- Ramu, S.; Akbarshshi, H.; Mogren, S.; Berlin, F.; Cerps, S.; Menzel, M.; Hvidtfeldt, M.; Porsbjerg, C.; Uller, L.; Andersson, C.K. Direct effects of mast cell proteases, tryptase and chymase, on bronchial epithelial integrity proteins and anti-viral responses. BMC Immunol. 2021, 22, 35.
- Karimi, N.; Morovati, S.; Chan, L.; Napoleoni, C.; Mehrani, Y.; Bridle, B.W.; Karimi, K. Mast cell tryptase and implications for SARS-CoV-2 pathogenesis. Bio. Med. 2021, 1, 136–149.
- He, A.; Shi, G.-P. Mast cell chymase and tryptase as targets for cardiovascular and metabolic diseases. Curr. Pharm. Des. 2013, 19, 1114–1125.
- Gruber, B.L.; Marchese, M.J.; Suzuki, K.; Schwartz, L.B.; Okada, Y.; Nagae, H.; Ramamurthy, N.S. Synovial procollagenase activation by human mast cell tryptase dependence upon matrix metalloproteinase 3 activation. J. Clin. Investig. 1989, 84, 1657–1662.
- Lees, M.; Taylor, D.J.; Woolley, D.E. Mast cell proteinases activate precursor forms of collagenase and stromelysin, but not of gelatinases A and B. Eur. J. Biochem. 1994, 223, 171–177.
- Yamamoto, K.; Kumagai, N.; Fukuda, K.; Fujitsu, Y.; Nishida, T. Activation of corneal fibroblast-derived matrix metalloproteinase-2 by tryptase. Curr. Eye Res. 2006, 31, 313–317.
- Pyo, R.; Lee, J.K.; Shipley, J.M.; Curci, J.A.; Mao, D.; Ziporin, S.J.; Ennis, T.L.; Shapiro, S.D.; Senior, R.M.; Thompson, R.W. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J. Clin. Investig. 2000, 105, 1641–1649.
- Galis, Z.S.M.; Johnson, C.; Godin, D.; Magid, R.; Shipley, J.M.; Senior, R.M.; Ivan, E. Targeted disruption of the matrix metalloproteinase-9 gene impairs smooth muscle cell migration and geometrical arterial remodeling. Circ. Res. 2002, 91, 852–859.
- Longo, G.M.; Xiong, W.; Greiner, T.C.; Zhao, Y.; Fiotti, N.; Baxter, B.T. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J. Clin. Investig. 2002, 110, 625–632.
- Kuzuya, M.; Nakamura, K.; Sasaki, T.; Cheng, X.W.; Itohara, S.; Iguchi, A. Effect of MMP-2 deficiency on atherosclerotic lesion formation in apoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1120–1125.
- Wågsäter, D.; Zhu, C.; Björkegren, J.; Skogsberg, J.; Eriksson, P. MMP-2 and MMP-9 are prominent matrix metalloproteinases during atherosclerosis development in the Ldlr(−/−)Apob(100/100) mouse. Int. J. Mol. Med. 2011, 28, 247–253.
- Berlin, F.; Mogren, S.; Tutzauer, J.; Andersson, C.K. Mast cell proteases tryptase and chymase induce migratory and morphological alterations in bronchial epithelial cells. Int. J. Mol. Sci. 2021, 22, 5250.
- De Souza Junior, D.A.; Santana, A.C.; da Silva, E.Z.M.; Oliver, C.; Jamur, M.C. The role of mast cell specific chymases and tryptases in tumor angiogenesis. BioMed Res. Int. 2015, 2015, 142359.
- Arooj, M.; Kim, S.; Sakkiah, S.; Cao, G.P.; Lee, Y.; Lee, K.W. Molecular modeling study for inhibition mechanism of human chymase and its application in inhibitor design. PLoS ONE 2013, 8, e62740.
- Schwartz, L.B.; Bradford, T.R. Regulation of tryptase from human lung mast cells by heparin. Stabilization of the active tetramer. J. Biol. Chem. 1986, 261, 7372–7379.
- Schwartz, L.B.; Lewis, R.A.; Austen, K.F. Tryptase from human pulmonary mast cells. Purification and characterization. J. Biol. Chem. 1981, 256, 11939–11943.
- Alter, S.C.; Yates, P.; Margolius, H.S.; Schwartz, L.B. Tryptase and kinin generation: Tryptase from human mast cells does not activate human urinary prokallikrein. Int. Arch. Allergy Appl. Immunol. 1987, 83, 321–324.
- Cregar, L.; Elrod, K.C.; Putnam, D.; Moore, W.R. Neutrophil myeloperoxidase is a potent and selective inhibitor of mast cell tryptase. Arch. Biochem. Biophys. 1999, 366, 125–130.
- Elrod, K.C.; Moore, W.R.; Abraham, W.M.; Tanaka, R.D. Lactoferrin, a potent tryptase inhibitor, abolishes late-phase airway responses in allergic sheep. Am. J. Respir. Crit. Care Med. 1997, 156, 375–381.
- Alter, S.C.; Kramps, J.A.; Janoff, A.; Schwartz, L.B. Interactions of human mast cell tryptase with biological protease inhibitors. Arch. Biochem. Biophys. 1990, 276, 26–31.
- Samoszuk, M.; Corwin, M.; Hazen, S.L. Effects of human mast cell tryptase and eosinophil granule proteins on the kinetics of blood clotting. Am. J. Hematol. 2003, 73, 18–25.
- Schechter, N.M.; Eng, G.Y.; McCaslin, D.R. Human skin tryptase: Kinetic characterization of its spontaneous inactivation. Biochemistry 1993, 32, 2617–2625.
- Frommherz, K.J.; Faller, B.; Bieth, J.G. Heparin strongly decreases the rate of inhibition of neutrophil elastase by α1-proteinase inhibitor. J. Biol. Chem. 1991, 266, 15356–15362.
- Ermolieff, J.; Boudier, C.; Laine, A.; Meyer, B.; Bieth, J.G. Heparin protects cathepsin G against inhibition by protein proteinase inhibitors. J. Biol. Chem. 1994, 269, 29502–29508.
- Fleddermann, J.; Pichert, A.; Arnhold, J. Interaction of serine proteases from polymorphonuclear leucocytes with the cell surface and heparin. Inflammation 2012, 35, 81–88.
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J. Biol. Chem. 2000, 275, 27258–27265.
- Evans, M.D.; Pryor, W.A. Cigarette smoking. emphysema and damage to alpha 1-proteinase inhibitor. Am. J. Physiol. Lung Cell. Mol. Physiol. 1994, 266, L593–L611.
- Dittrich, A.S.; Kühbandner, I.; Gehrig, S.; Rickert-Zacharias, V.; Twigg, M.; Wege, S.; Taggart, C.C.; Herth, F.; Schultz, C.; Mall, M.A. Elastase activity on sputum neutrophils correlates with severity of lung disease in cystic fibrosis. Eur. Respir. J. 2018, 51, 1701910.
- Duranton, J.; Adam, C.; Blieth, J.G. Kinetic mechanism of the inhibition of cathepsin G by α1-antichymotrypsin and α1-proteinase inhibitor. Biochemistry 1997, 37, 11239–11245.
- Travis, J.; Bowen, J.; Baugh, R. Human α1-antichymotrypsin: Interaction with chymotrypsin-like proteinases. Biochemistry 1978, 26, 5651–5656.
- Kalsheker, N.A. α1-Antichymotrypsin. Int. J. Biochem. Cell Biol. 1996, 28, 961–964.
- Thompson, R.C.; Ohlsson, K. Isolation, properties, and complete amino acid sequence of human secretory leukocyte protease inhibitor, a potent inhibitor of leukocyte elastase. Proc. Natl. Acad. Sci. USA 1986, 83, 6692–6696.
- Franken, C.; Meijer, C.J.L.M.; Dijkman, J.H. Tissue distribution of antileukoprotease and lysozyme in humans. J. Histochem. Cytochem 1989, 37, 493–498.
- Lee, C.H.; Igarashi, Y.; Hohman, R.J.; Kaulbach, H.; White, M.V.; Kaliner, M.A. Distribution of secretory leukoprotease inhibitor in the human nasal airway. Am. Rev. Respir Dis 1993, 147, 710–716.
- McGarry, N.; Greene, C.M.; McElvaney, N.G.; Weldon, S.; Taggart, C.C. The ability of secretory leukocyte protease inhibitor to inhibit apoptosis in monocytes is independent of its antiprotease activity. J. Immunol. Res. 2015, 2015, 507315.
- Williams, S.E.; Brown, T.I.; Roghanian, A.; Sallenave, J.M. SLPI and elafin: One glove, many fingers. Clin. Sci. 2006, 110, 21–35.
- Verrier, T.; Solhonne, B.; Sallenave, J.M.; Garcia-Verdugo, I. The WAP protein Trappin-2/Elafin: A handyman in the regulation of inflammatory and immune responses. Int. J. Biochem. Cell Biol. 2012, 44, 1377–1380.
- Labidi-Galy, S.I.; Clauss, A.; Ng, V.; Duraisamy, S.; Elias, K.M.; Piao, H.Y.; Bilal, E.; Davidowitz, R.A.; Lu, Y.; Badalian-Very, G.; et al. Elafin drives poor outcome in high-grade serous ovarian cancers and basal-like breast tumors. Oncogene 2015, 34, 373–383.
- Hunt, K.K.; Wingate, H.; Yokota, T.; Liu, Y.; Mills, G.B.; Zhang, F.; Fang, B.; Su, C.H.; Zhang, M.; Yi, M.; et al. Elafin, an inhibitor of elastase, is a prognostic indicator in breast cancer. Breast Cancer Res. 2013, 15, R3.
- Wang, C.; Liao, Y.; He, W.; Zhang, H.; Zuo, D.; Liu, W.; Yang, Z.; Qiu, J.; Yuan, Y.; Li, K.; et al. Elafin promotes tumour metastasis and attenuates the anti-metastatic effects of erlotinib via binding to EGFR in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 113.
- Cooley, J.; Takayama, T.K.; Shapiro, S.D.; Schechter, N.M.; Remold-O’Donnell, E. The serpin MNEI inhibits elastase-like and chymotrypsin-like serine proteases through efficient reactions at two active sites. Biochemistry 2001, 40, 15762–15770.
- Marrero, A.; Duquerroy, S.; Trapani, S.; Goulas, T.; Guevara, T.; Andersen, G.R.; Navaza, J.; Sottrup-Jensen, L.; Gomis-Rüth, F.X. The crystal structure of human alpha2-macroglobulin reveals a unique molecular cage. Angew. Chem. Int. Ed. Engl. 2012, 51, 3340–3344.
- Vandooren, J.; Itoh, Y. Alpha-2-macroglobulin in inflammation, immunity and infections. Front. Immunol. 2021, 12, 803244.
- Salvesen, G.; Virca, G.D.; Travis, J. Interaction of alpha 2-macroglobulin with neutrophil and plasma proteinases. Ann. N. Y. Acad. Sci. 1983, 421, 316–326.
- Wewers, M.D.; Herzyk, D.J.; Gadek, J.E. Alveolar fluid neutrophil elastase activity in the adult respiratory distress syndrome is complexed to alpha-2-macroglobulin. J. Clin. Investig. 1988, 82, 1260–1267.
- Rao, N.V.; Wehner, N.G.; Marshall, B.C.; Gray, W.R.; Gray, B.H.; Hoidal, J.R. Characterization of proteinase-3 (PR-3), a neutrophil serine proteinase. Structural and functional properties. J. Biol. Chem. 1991, 266, 9540–9548.
- Siddiqui, T.; Zia, M.K.; Ali, S.S.; Ahsan, H.; Khan, F.H. Insight into the interactions of proteinase inhibitor- alpha-2-macroglobulin with hypochlorite. Int. J. Biol. Macromol. 2018, 117, 401–406.
- Reddy, V.Y.; Desorchers, P.E.; Pizzo, S.V.; Gonias, S.L.; Sahakian, J.A.; Levine, R.L.; Weiss, S.J. Oxidative dissociation of human alpha 2-macroglobulin tetramers into dysfunctional dimers. J. Biol. Chem. 1994, 269, 4683–4691.
- Ahmad, S.; Simmons, T.; Varagic, J.; Moniwa, N.; Chappell, M.C.; Ferrario, C.M. Chymase-dependent generation of angiotensin II from angiotensin-(1-12) in human atrial tissue. PLoS ONE 2011, 6, e28501.
- Song, Y.H.; Li, Y.; Du, J.; Mitch, W.E.; Rosenthal, N.; Delafontaine, P. Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J. Clin. Investig. 2005, 115, 451–458.
- Sanders, P.M.; Russell, S.T.; Tisdale, M.J. Angiotensin II directly induces muscle protein catabolism through the ubiquitin-proteasome proteolytic pathway and may play a role in cancer cachexia. Br. J. Cancer 2005, 93, 425–434.
- Trobec, K.; von Haehling, S.; Anker, S.D.; Lainsack, M. Growth hormone, insulin-like growth factor 1, and insulin signaling—A pharmacological target in body wasting and cachexia. J. Cachexia Sarcopenia Muscle 2011, 2, 191–200.
- Semprun-Prieto, L.C.; Sukhanov, S.; Yoshida, T.; Rezk, B.M.; Gonzalez-Villalobos, R.A.; Vaughn, C.; Tabony, A.M.; Delafontaine, P. Angiotensin II induced catabolic effect and muscle atrophy are redox dependent. Biochem. Biophys. Res. Commun. 2011, 409, 217–221.
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology, and aging. EMBO Mol. Med. 2010, 2, 247–257.
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pey, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828.
- Ferrario, C.M.; Chappell, M.C. Novel angiotensin peptides. Cell. Mol. Life Sci. 2004, 61, 2720–2727.
- Muller, F.; Renné, T. Novel roles for factor XII-driven plasma contact activation system. Curr. Opin. Hematol. 2008, 15, 516–521.
- Oehmcke-Hecht, S.; Köhler, J. Interaction of the human contact system with pathogens—An update. Front. Immunol. 2018, 9, 312.
- Marceau, F.; Hess, H.J.; Rachvarov, D.R. The B1 receptors for kinins. Pharmacol. Rev. 1998, 50, 357–386.
- Cyr, M.; Lepage, Y.; Blais Jr., C.; Gervais, N.; Cugno, M.; Rouleau, J.-L.; Adam, A. Bradykinin and des-Arg9-bradykinin metabolic pathways and kinetics of activation of human plasma. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H275–H283.
- Hamza, M.; Wang, X.M.; Adam, A.; Brahim, J.S.; Rowan, J.S.; Carmona, G.N.; Dionne, R.A. Kinin B1 receptors contributes to acute pain following minor surgery in humans. Mol. Pain 2010, 6, 12.
- Murphy, G.; Nagase, H. Progress in matrix metalloproteinase research. Mol. Aspects Med. 2008, 29, 290–308.
- Löffek, S.; Schilling, O.; Franzke, C.W. Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208.
- Ra, H.-J.; Parks, W.C. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007, 26, 587–596.
- Fu, X.; Kao, J.L.; Bergt, C.; Kassim, S.Y.; Huq, N.P.; d’Avignon, A.; Parks, W.C.; Mecham, R.P.; Heinecke, J.W. Oxidative cross-linking of tryptophan to glycine restrains matrix metalloproteinase activity: Specific structural motifs control protein oxidation. J. Biol. Chem. 2004, 279, 6209–6912.
- Thompson, R.W.; Holmes, D.R.; Mertens, R.A.; Liao, S.; Botney, M.D.; Mecham, R.P.; Welgus, H.G.; Parks, W.C. Production and localization of 92-kilodalton gelatinase in abdominal aortic aneurysms. An elastolytic metalloproteinase expressed by aneurysm-infiltrating macrophages. J. Clin. Investig. 1995, 96, 318–326.
- Davis, V.; Persidskaia, R.; Baca-Regen, L.; Itoh, Y.; Nagase, H.; Persidsky, Y.; Ghorpade, A.; Baxter, B.T. Matrix metalloproteinase-2 production and its binding to the matrix are increased in abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1625–1633.
- Zhang, X.; Shen, Y.H.; LeMaire, S.A. Thoracic aortic dissection: Are matrix metalloproteinases involved? Vascular 2009, 17, 147–157.
- Strickland, D.K.; Ashcom, J.D.; Williams, S.; Burgess, W.H.; Migliorini, M.; Argraves, W.S. Sequence identity between the alpha 2-macroglobulin receptor and low density lipoprotein receptor-related protein suggests that this molecule is a multifunctional receptor. J. Biol. Chem. 1990, 265, 17401–17404.
- Lee, M.H.; Atkinson, S.; Murphy, G. Identification of the extracellular matrix (ECM) binding motifs of tissue inhibitor of metalloproteinases (TIMP)-3 and effective transfer to TIMP-1. J. Biol. Chem. 2007, 282, 6887–6898.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
585
Revisions:
2 times
(View History)
Update Date:
21 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No