Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sergiy M. Kovalenko | -- | 3945 | 2023-06-16 12:41:13 | | | |
| 2 | Lindsay Dong | + 1 word(s) | 3946 | 2023-06-19 02:52:51 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Hryhoriv, H.; Kovalenko, S.M.; Georgiyants, M.; Sidorenko, L.; Georgiyants, V. Biological Activity of the 3-Heteroaryl Fluoroquinolone Hybrids. Encyclopedia. Available online: https://encyclopedia.pub/entry/45717 (accessed on 25 June 2026).
Hryhoriv H, Kovalenko SM, Georgiyants M, Sidorenko L, Georgiyants V. Biological Activity of the 3-Heteroaryl Fluoroquinolone Hybrids. Encyclopedia. Available at: https://encyclopedia.pub/entry/45717. Accessed June 25, 2026.
Hryhoriv, Halyna, Sergiy M. Kovalenko, Marine Georgiyants, Lyudmila Sidorenko, Victoriya Georgiyants. "Biological Activity of the 3-Heteroaryl Fluoroquinolone Hybrids" Encyclopedia, https://encyclopedia.pub/entry/45717 (accessed June 25, 2026).
Hryhoriv, H., Kovalenko, S.M., Georgiyants, M., Sidorenko, L., & Georgiyants, V. (2023, June 16). Biological Activity of the 3-Heteroaryl Fluoroquinolone Hybrids. In Encyclopedia. https://encyclopedia.pub/entry/45717
Hryhoriv, Halyna, et al. "Biological Activity of the 3-Heteroaryl Fluoroquinolone Hybrids." Encyclopedia. Web. 16 June, 2023.
Copy Citation
There are promising studies in the area of 3-heteroaryl hybrids. The latter can be synthesized via different convinient methods with the formation of new derivatives with five-membered and fused heterocycles or creation of bis-fluoroquinolones with variable linking moieties. These novel compounds revealed not only good antimicrobial properties compared to the parent molecules but were also widely investigated as anticancer agents with promising activity.
fluoroquinolones
synthesis
3-heteroaryl hybrids
biological activity
1. Novel FQ Hybrids as Antimicrobials and Antiviral Medicines
Among the core fluoroquinolones (FQs) molecules that served for the development and research of new potent antimicrobials, the first place is taken by norfloxacin. The carboxylic group of the initial compound was modified mainly with five-membered heterocyles with further investigation of antibacterial, antifungal and antiviral activities.
For example, a series of 1,3,4-oxadiazoles containing FQ derivatives was synthesized and screened for antibacterial and antimycobacterial properties in ref. [1] (Figure 1). The disk diffusion method revealed potent antibacterial activities against Staphylococcus aureus, Enterococcus faecalis, Streptococcus pneumoniae, Escherichia coli and Klebsiella pneumoniae. In addition, the obtained norfloxacin derivatives showed antimycobacterial activity against Mycobacterium smegmatis H37Rv with minimal inhibitory concentrations (MICs) of 22.35, 16.20 and 20.28 μg/mL. The scholars also studied absorption, distribution, metabolism and excretion (ADME) properties and proved the promising pharmacokinetic properties and drug-likeness for the obtained compounds.
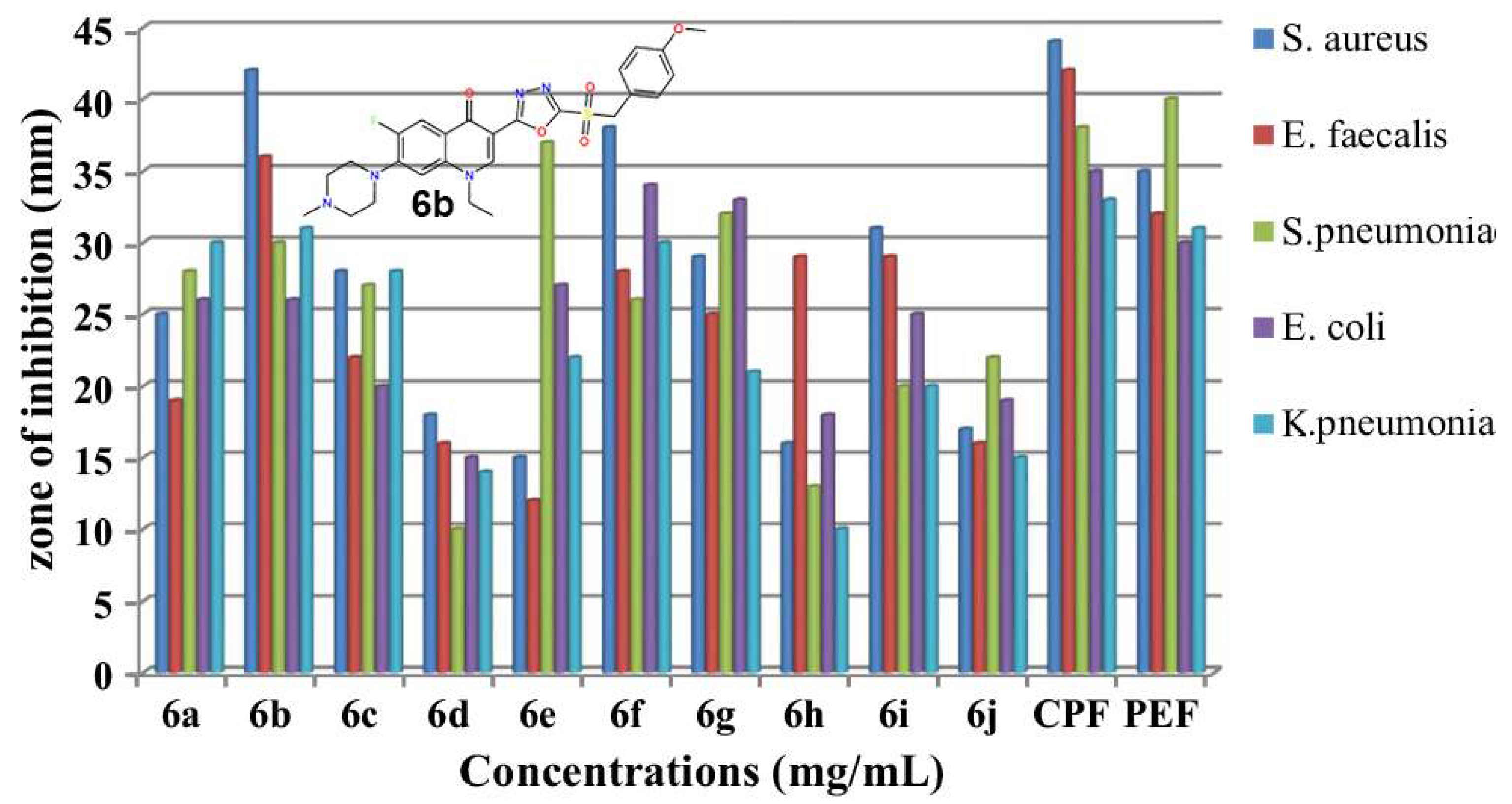
Figure 1. Antibacterial activity of new 1,3,4-oxadiazole hybrids of norfloxacin [1].
It is worth noting that most of the investigations in this area are based on in silico studies of drug-likeness and binding properties. Thus, the scholars of [2] described molecular docking investigations (AutoDock Tools-1.5.6) against the receptor GlcN-6P (2VF5) that revealed good binding affinities for synthesized norfloxacin derivatives. In the work, the core molecule was modified with 1,3,4-oxadiazole, thiazolidin-4-one, 1,3,4-oxadiazoline, 1,2,4-triazole and 1,3,4-thiadiazole rings. The antimicrobial assessment via disk diffusion and serial dilution methods showed higher activity than for norfloxacin against S. aureus, Staphylococcus epidermidis, Streptococcus pyogenes, Micrococcus luteus, K. pneumoniae, Pseudomonas aeruginosa, E. coli and Proteus mirabilis.
1,2,4-Triazole and 1,3,4-oxadiazole norfloxacin hybrids with suitable druglike, antibacterial and antifungal properties were successfully obtained by researches [3]. Especially pronounced was the activity against S. pneumoniae, with minimum inhibitory concentrations of 0.89 and 0.96 mg/mL and minimal bactericidal concentrations of 2.95 and 2.80 mg/mL. Combined with simple synthetic approaches, such results are promising for further investigations.
Interestingly, similar oxadiazole norfloxacin and ciprofloxacin derivatives showed good antibacterial activity against both Gram-positive (S. aureus) and Gram-negative (E. coli) strains, combined with promising antifungal activity against fungi (Saccharomyces cerevisiae) in comparison with reference drugs ciprofloxacin and fluconazole in the study [4].
A few interesting works were devoted to an exploration of the antimicrobial potential of aminothiazolyl hybrids of norfloxacin. It was proved that the 2-aminothiazole fragment at the 3-position of the quinolone core plays an important role in exerting antibacterial activity. For instance, in this case, the antibacterial activity investigation revealed higher values in comparison with the reference drugs against methicillin-resistant Staphylococcus aureus (MRSA) and S. aureus 25923, with MIC values of 0.009 and 0.017 mM [5]. Figure 2 shows the detailed activity and concentrations of the synthesized compounds.
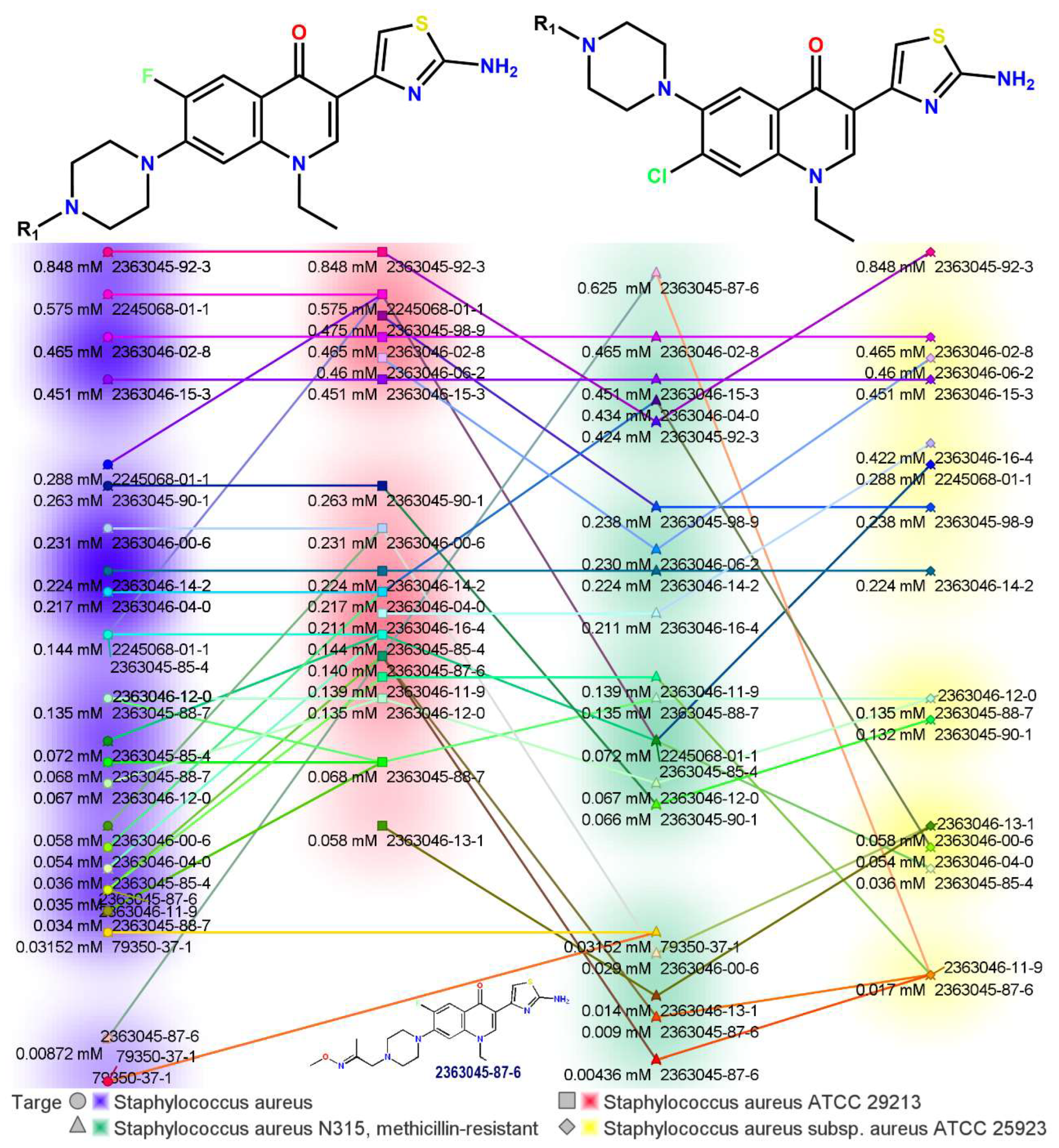
Figure 2. Antimicrobial potency of novel aminothiazolyl hybrids of norfloxacin [5].
Another series of aminothiazolyl norfloxacin analogs was synthesized by the scholars of [6] and was screened for antimicrobial properties. Most of the compounds synthesized were superior to reference drug inhibitory efficiencies against K. pneumoniae and Candida albicans, with MIC values of 0.005 and 0.010 mM. Furthermore, these compounds revealed better antibacterial activity against S. aureus ATCC 29213 and methicillin-resistant strains (Figure 3).
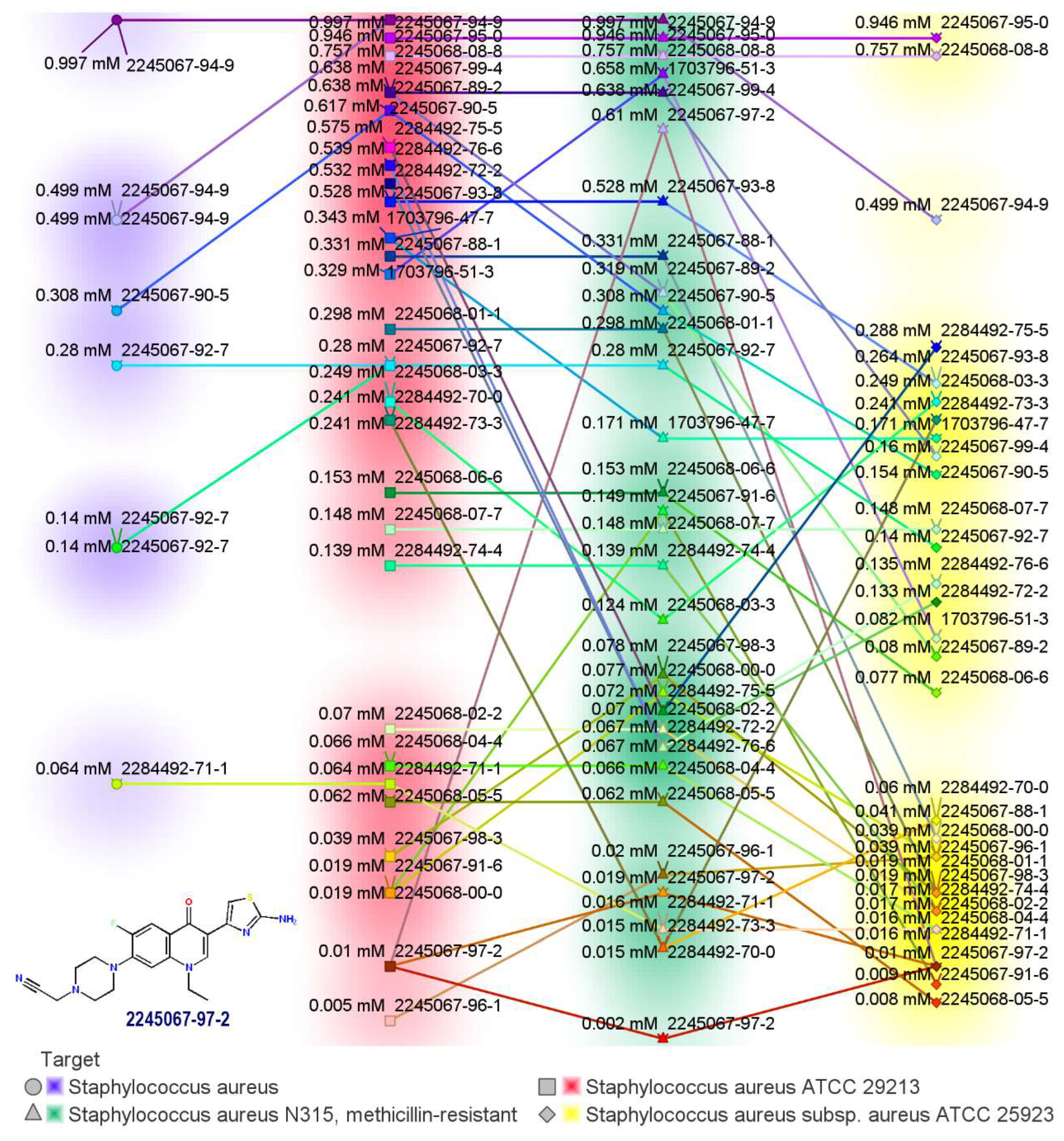
Figure 3. Antimicrobial activity of aminothiazolyl hybrids of norfloxacin [6].
In addition, investigations of the inhibitory activity against the DNA gyrase from E. coli showed that aminothiazolyl norfloxacin analogs have good inhibitory potency for DNA gyrase (IC50 ¼ 16.7 mM), which was more effective than the reference drug norfloxacin (IC50 ¼ 18.6 mM). Altogether, the scholars concluded that the replacement of the carboxyl group with the weak basic 2-aminothiazole moiety provides a similar antibacterial mechanism to norfloxacin by targeting DNA gyrase.
They additionally proved their hypothesis via docking studies with the topoisomerase IV-DNA complex and gyrase-DNA complex. Among the found interactions, the sulfur atom in the 2-aminothiazole moiety participated in the non-covalent coordination with ARG-136 residue via hydrogen bond formation that is favorable for stabilizing the compound-enzyme-DNA supramolecular complex, which further accounted for the good inhibitory efficiency against the tested strains.
The investigation in ref [7] also describes a series of novel 2-aminothiazol-4-yl norfloxacin analogs created to combat quinolone resistance. Among them, 3-(2-aminothiazol-4-yl)-7-chloro-6-(pyrrolidin-1-yl)quinolone exhibited potent antibacterial activity, strong inhibitory potency to DNA gyrase and a broad antimicrobial spectrum, including against multidrug-resistant strains. Moreover, this molecule induced bacterial resistance more slowly than initial norfloxacin. The docking evaluation gave good total scores (5.68 and 6.46) for aminothiazolquinolones against topoisomerase IV-DNA and gyrase-DNA complexes.
A tertrazolyl moiety appeared to be one more promising bioisostere that was introduced in the C-3 of norfloxacin and ciprofloxacin [8]. The scholars conducted docking studies using a Molegro Virtual Docker (MVD) to prove their idea. The tested compounds showed a very similar binding mode with DNA gyrase compared to the co-crystallized ciprofloxacin. The tetrazole formed three hydrogen bonds with Ser1084 instead of one, which is formed by ciprofloxacin. The bond lengths of the three hydrogen bonds were 1.9, 2.1 and 3.1Å. The nitrogen of the piperazine formed a hydrogen bond with base pair DNA backbone DT4. The MolDock score (kcal mol−1) and the Rerank Score were −123.54 and −74.67, respectively. Overall, the tested compound revealed a similar manner to ciprofloxacin, with additional hydrogen bonds related to the 3-tetrazole scaffold, supporting the molecular design.
In addition, this modification led to optimization of the solubility profile of the initial molecules. As for the antibacterial activity, the inhibition zones for S. aureus and MRSA were from 12.5 to 25 mM. Several derivatives revealed activity at 12.5 and 25 mM, respectively, against Salmonella typhi, while reference drugs were active at 100 mM. Moreover, high activity against Vibrio cholerae and E. coli was observed.
A few more investigations described modifications of ciprofloxacin. Thus, ref. [9] evidenced the synthesis and evaluation of antibacterial activity of ciprofloxacin C3 hybrids with isatins, phthalimides and oxadiazoles. In vitro antibacterial evaluation was made using disk diffusion and serial dilution methods, and antitubercular activity was measured using the Lowenstein–Jensen (LJ) method. All the obtained compounds were highly effective against E. coli, K. pneumonia (Gram-negative) and S. aureus (Gram-positive) at concentrations of 75 and 100 μg/mL. In addition, they possessed antitubercular activity against normal, multidrug-resistant and extensively drug-resistant strains of Mycobacterium tuberculosis. Therefore, they are more potent antimicrobial agents than ciprofloxacin.
Similar research was conducted by scientists [10] who successfully obtained 1,3,4-oxadiazole hybrids of ciprofloxacin and tested them against the standard group of Gram-positive and Gram-negative microorganisms. Here, again, promising activity was observed for both groups that exceeded the reference drug ciprofloxacin.
More pronounced activity against Gram-negative strains is described in ref. [11] for new C3 triazole ciprofloxacin derivatives. Antibacterial activity against Gram-positive strains, in this case, remained at the ciprofloxacin level. In addition, molecular docking studies using topoisomerase (3ILW) protein revealed correlation between the antibacterial activity and binding free energy of the molecules. The tested compound showed high affinity with low energy of −6.2 kcal/mol with the employed protein (for ciprofloxacin, it was −6.7 kcal/mol).
Furthermore, a series of new ofloxacin analogs was synthesized by modifying it by triazoles [12]. In the first stage of the research, in silico docking studies using Autodock vina 4.0 program were performed. Almost all the compounds used for docking showed a best-fit Root Mean Square Difference (RMSD) value of 0.000 with topoisomerase II (3ILW), and good inhibition, with an affinity range between −7.4 and −6.4 kcal/mol. The obtained data were verified via in vitro antimicrobial screening, where the obtained compounds showed promising activity against S. aureus, S. epidermidis and Bacillus subtilis (MIC 0.125 μg/mL).
Several patents on FQs hybridized via five-membered heterocyles at C3 were obtained by Chinese scientists. They claim antibacterial (against Gram-positive and Gram-negative strains), antifungal and DNA intercalating properties in the obtained thiazole [13] and aminothiazole derivatives [14][15]. In addition, they describe a simple and affordable preparation technique based on available raw materials. Another patent [16][17] describes the preparation of novel hybrids with rhodamine as promising antibacterial agents.
Among fused hybrids, the scholars of [18] describe novel benzimidazole–quinolinone derivatives of ciprofloxacin and levofloxacin. All the synthesized compounds revealed promising antifungal activity when compared with the reference drug griseofulvin.
A series of new triazolothiadiazole derivatives of ciprofloxacin was obtained and tested against S. aureus and E. coli [19]. They showed stronger inhibitory activity against E. coli than that of S. aureus compared to ciprofloxacin. Therefore, the fused heterocycle-based substituted FQs are valuable for further investigations.
Lastly, a novel class of FQ oxadiazole derivatives that inhibit NS5B polymerase, a key enzyme of the Hepacivirus viral life cycle, is described in ref. [20] that may be the first step toward exploration of this dimension of FQs.
Altogether, the published studies definitely prove the logic and efficiency of these investigations. Even if they are now underestimated, their continuation gives a perfect chance to obtain fruitful results, namely, new potent antimicrobials to combat the problem of resistance to antibiotics.
2. Novel FQ Hybrids as Promising Antitumor Agents
Surprisingly, literature data revealed that many research groups searched for antitumor agents among new FQ hybrids. This approach is based on the concept of bioisosters that gained popularity in medicinal chemistry in recent years. Namely, the bioisosteric replacement of the carboxylic group with different heterocyclic moieties and synthesis of bis-fluoroquinolones linked via a heterocycle at C3 are two main strategies that are widely presented.
Another valuable point is the variety of cancer types against which the compounds synthesized were tested in the above-mentioned investigations.
Thus, new 1,3,4-thiadiazole derivatives of ciprofloxacin were synthesized and investigated via thorough in silico and in vitro studies (Figure 4). Theoretical and experimental DNA binding research revealed good correlation with human hepatocellular carcinoma (Huh-7) cell line activity. IC50 values from Huh-7 cell line studies (25.75 μM) revealed synthesized compounds as potent anticancer agents, promising for further investigations [21].
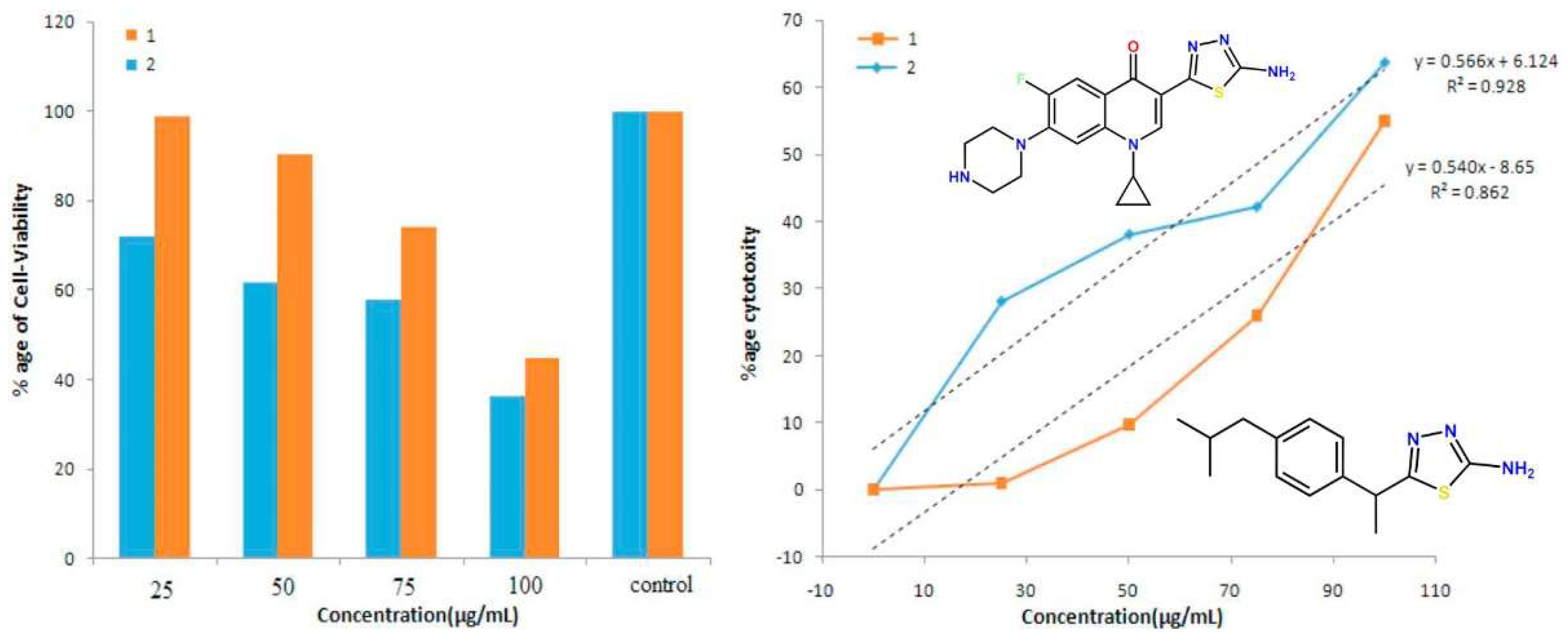
Figure 4. New 1,3,4-thiadiazole ciprofloxacin derivatives and their antitumor activity [21].
Other isosteres of the C-3 carboxylic group for the pefloxacin 1,3,4-oxadiazole-thione ring and oxadiazole thione Mannich bases were suggested by the scholars of [1]. The obtained compounds were tested in vitro against a liver cancer (Hep-3B) cell line and, according to the results, all the title compounds showed more significant potency than parent compounds. In addition, derivatives of aliphatic amines appeared to be more active than the derivatives of aromatic amines. Furthermore, similar derivatives were reported in patent [22], and it was shown that molecules with an electron-withdrawing group attached on the aryl ring had more potency than compounds with an electron-donating group.
The scholars of [23] patented similar oxadiazole norfloxacin derivatives that were screened in vitro against the same liver cancer (Hep-3B) cell line. Evaluation was performed via an MTT assay. The results revealed higher cytotoxicity compared to norfloxacin for fifteen title compounds. Correspondent quaternary ammonium salts exhibited promising anticancer activity with IC50 values below 25.0 μmol/L.
Another search for agents against the human hepatoma (Hep-3B) cancer cell line and human pancreatic (Capan-1) cell line was made based on comparative molecular field analysis techniques [24]. Three-dimensional quantitative structure–activity relationship (3D-QSAR) investigations on the antitumor activity of s-triazole sulfide-ketone derivatives of ciprofloxacin and levofloxacin gave the possibility to design four novel molecules with promising anti-tumor activity and to plan further in vitro research.
A series of ciprofloxacin and norfloxacin oxadiazole derivatives was evaluated for their antiproliferative activities against human lung tumor (A549) cell lines. Among them, the most active compound, 1-cyclopropyl-6-fluoro-3-[5-(4-nitrophenyl)-1,3,4-oxadiazol-2-yl]-7-piperazinyl-1,4-dihydro-quinolin-4-one, was found, with a half-maximal inhibitory concentration (IC50) of 9.0 μg/mL [25].
Furthermore, the scope of cancer cell lines expands via other organs. For instance, the scholars of [12] patented novel ofloxacin 1,3,4-triazole derivatives as antitumor agents for treating bladder, stomach or pancreatic cancer (Figure 5).
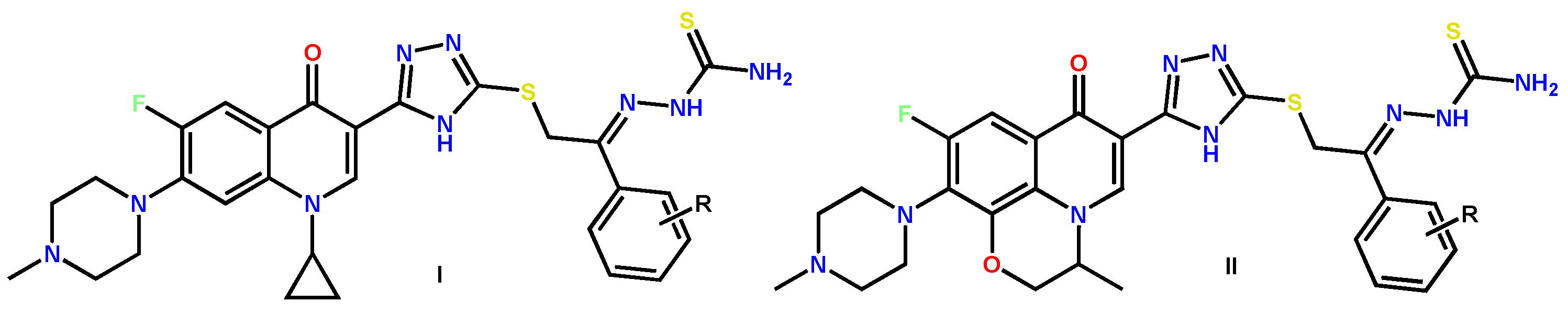
Figure 5. Novel ofloxacin 1,3,4-triazole derivatives as antitumor agents [12].
Research related to bladder tumors is also patented [26] and describes similar triazole derivatives for which an IC50 value of 0.6μM against a bladder tumor was detected.
The series of C-3 s-triazole thioether ketone semicarbazone ciprofloxacin hybrids was synthesized via multi-step synthesis and patented as promising antitumor agents for treating stomach, pancreatic or bladder cancer [27]. Inhibitory activity with an IC50 of 6.4, 10.5, 9.7, 3,8, 3.6 and 48.6 μM against bladder cancer (T24), gastric cancer (HGC823, HGC27), pancreatic cancer (Panc-1, Capan-1) and VERO cancer cell lines, respectively, was revealed.
A simple synthetic approach led to the development of novel citrate-triazole-oxadiazole norfloxacin hybrids that exhibited remarkable anticancer activity against cervical cancer (HeLa) cell lines with an IC50 value 11.3 ± 0.41, comparable to the standard drug [3]. The compounds also revealed suitable druglike properties and are expected to present a good bioavailability profile.
Bis-lomefloxacin derivatives linked via an oxadiazole-carbazide bridge showed high activity against the human non-small-cell lung cancer (A549) cell line, human pancreatic cancer (Capan-1) cell line and human skin melanoma (A375) cell line, exceeding the parent compound, as well as isomerase inhibitor hydroxycamptothecin (HC) and tyrosine kinase inhibitors Ragofini (RRF) and cabozantinib (CZT) [28][29].
Similar bis-fluoroquinolone oxadiazole carbazide N-methyl-ciprofloxacin derivatives were synthesized, tested against human A549, Capan-I and A375 cell lines; they showed promising activity and were patented [30][31]. In addition, they reduced toxic side effects on normal cells.
As anti-lung cancer and antihepatoma drugs, bis-fluoroquinolone oxadiazole urea N-acetyl norfloxacin derivatives were also patented [32]. In addition, rufloxacin bis-fluoroquinolone oxadiazole urea derivatives revealed inhibiting activities on the human non-small-cell lung cancer (A549) cell line, human liver cancer (SMCC-7722) cell line, human gastric cancer (HGC27) cell line, human pancreatic cancer (Capan-I) cell line, human skin melanoma (A375) cell line and human leukemia (HL60) cell line [33].
Bis-acetylciprofloxacin linked via thiadiazole and urea pharmacophore showed higher antitumor activity and selectivity as well as a reduction in toxic side effects on normal cells [34]. The compounds synthesized were checked on lung cancer (A549), human papillomavirus-related endocervical adenocarcinoma (SMCC-7721), human gastric cancer (HGC27), human pancreatic cancer Capan-1, melanoma (A375), human leukemia (HL60) and myelogenous leukemia (K562G) cell lines, with promising results. N-acetylnorfloxacin thiadiazole norfloxacin derivatives exhibited IC50 values in a range of 0.36 μM to 4.66 μM toward cancer cell lines [35].
Many published papers are devoted to the search for potential antileukemic agents among 3-heteroaryl FQ hybrids. For instance, a series of [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine and pyrazolo[5,1-c][1,2,4]triazole derivatives of norfloxacin, ciprofloxacin and levofloxacin was successfully obtained [36]. Their in vitro antitumor activity was tested against murine leukemia (L1210) and Chinese hamster ovary (CHO) cell lines via the standard MTT assay. The results showed poor inhibitory activity for parent fluoroquinolones (IC50 > 150 mmol/L), while isolated fused compounds had a potential activity with an IC50 value within 10.0 mmol/L.
Several in silico studies prove this idea. Thus, for the therapy of T-cell lymphoma, a molecular electronegativity distance vector of levofloxacin-thiadiazole histone deacetylase (HDAC) inhibitor was measured [37] and the compounds were described as promising.
Quantitative structure–activity relationship (QSAR) and molecular docking study of levofloxacin and thiadiazole as antitumor agents with HDAC1, HDAC2 and HDAC6 also revealed that the main factors affecting their biological activity are hydrogen bonding and hydrophobic interactions [38]. From the docking results, it can be seen that the active part of the molecule formed a hydrogen bond with the active part of the macromolecule, while the hydrophobic part of the small molecule had a hydrophobic interaction with non-polar amino acid residues in the active part of the macromolecule.
Novel C-3 s-triazole-oxadiazole sulfide Mannich base pefloxacin derivatives were also screened against SMMC-7721, L1210 and HL60 cell lines and were evaluated by an MTT assay. The investigation revealed that the sulfides and their corresponding Mannich base compounds are more potent inhibitors than the starting compounds, especially against SMMC-7721 [39][40].
New s-triazole ofloxacine derivatives with functionalized side chains of Schiff bases and Schiff–Mannich bases are described in ref. [41]. Their in vitro antitumor activity against L1210, Chinese hamster ovary (CHO) and HL60 cell lines was evaluated in the MTT assay. Higher inhibitory activity was detected for compounds possessing a free phenol group. Other novel s-triazole-thiosemicarbazone ofloxacine derivatives were also evaluated by the MTT assay and revealed more significant antiproliferative activity than parent ofloxacin. Thiosemicarbazones, especially those containing a nitro group or fluorine atom, showed activity comparable to doxorubicin. Therefore, an azole ring modified with a functional side chain is a favorable bioisosteric replacement of the C-3 carboxylic group for an improvement in antitumor activity [42].
Aminothiadiazole ciprofloxacin derivatives and their Schiff bases were screened against SMMC-7721, HL60 and L1210 cell lines via MTT assay [43]. They showed potential cytotoxicity with IC50 values that reached micro-molar concentration and were patented.
Another patent was obtained for the invention of floxacin diazole derivatives that exhibited IC50 values of approximately 2, 5 and 2.5 μM against CHO, HL60 and L1210 cell lines, respectively [44].
Several papers describe screening of anticancer activities against CHO, HL60 and L1210 cancer cell lines of fused FQ hybrids. Thus, s-triazolothiadiazole ciprofloxacin derivatives revealed significant antitumor activity against HL60, with IC50 values from 50.0 to 8.0 μmol/L [19][45], s-triazolothiadiazine ciprofloxacin derivatives showed more significant inhibitory activity (IC50 < 25.0 μmol/L) than parent ciprofloxacin (IC50 > 150.0 μmol/L), s-triazolothiadiazinone enrofloxacin derivatives exhibited significant antitumor activity, with a range of micromole concentrations for IC50 value [46], and C-3 thiazolo[3,2-b][1,2,4]triazole ofloxacin derivatives exhibited more significant antiproliferative activity than parent ofloxacin [47]. Novel C-3 thiazolotriazole levofloxacin derivatives showed more significant activity than levofloxacin [48]. The compounds with fluorophenyl or o-methoxyphenyl displayed comparable activity to doxorubicin. Antitumor agents, prepared by cyclization of norfloxacin with o-phenylenediamine, 2-aminophenol or 2-aminobenzenethiol in the presence of polyphosphoric acid (PPA), showed strong antitumor activity in cows [49]. Therefore, a fused heterocyclic moiety as an isostere of the C-3 carboxylic acid group appears to be an alternative approach for further design of active antitumor FQs.
Moreover, four different bis-fluoroquinolones investigations in this direction were also made. For example, a series of C3/C3 bis-fluoroquinolones tethered with an 1,3,4-oxadiazole ring was screened against L1210, CHO and HL60 cell lines, showed promising inhibitory activity and was patented [50]. 1,3,4-Oxadiazole-linked norfloxacin showed antitumor activity with IC50 values of 15.6, 20.5 and 7.6 μM against CHO, HL-60 and L1210 cell lines, respectively [51]. Bis-oxadiazole methylsulfide derivatives derived from ciprofloxacin [52] and levofloxacin [53] were tested against CHO, HL60 and L1210 cancer cells and evaluated by MTT assay. The preliminary results showed that piperazinium compounds possess more potent activity than that of corresponding free bases.
Furthermore, ciprofloxacin cross-linked with a [1,2,4]-triazolo[3,4-b][1,3,4]-thiadiazole core as a common bioisostere of two carboxylic acid groups appeared to be highly potent against the HL60 cell line [54]. In vitro antitumor activity of norfloxacin dimers linked with a s-triazolo[2,1-b][1,3,4]thiadiazole moiety against L1210 and CHO cell lines was evaluated and appeared to be promising [55]. 1,2,4-Triazolo[3,4-b][1,3,4]thiadiazole-linked ciprofloxacin and levofloxacin dimers were prepared in a multi-step synthesis and revealed inhibitory activities with IC50 values of 1.1, 0.25 and 0.15 μM against CHO, HL60 and mouse lymphocytic leukemia (L1210) cell lines, respectively [56].
In addition, there are papers devoted to the hybridization of ofloxacin at C-3 with an s-triazole ring [5], oxadiazole-5-sulfanylacetylhydrazone moiety [57], triazole-oxadiazole methylsulfide [58] and oxadiazole ring [10]. All of them state higher antitumor activity than for the parent ofloxacin in the MTT assay. Similar derivatives patented by the scholars of [6] gave a CHO IC50 value of 1.3 μM. A patent for bis-oxadiazolyl methylsulfides derived from ofloxacin describes in vitro antitumor activity evaluation against three cancer cell lines by the MTT method. The compounds synthesized showed potential anticancer activity (IC50 < 25μmol/L). The activity of the quaternary ammonium salts was higher than that of the corresponding free bases [24].
Further, there are many inventions on bis-fluoroquinolones as antitumor agents that were patented. Namely, levofloxacin-containing bis-fluoroquinolone oxadiazole carbamide derivative is described as useful in the treatment of cancer [59]. A novel bis-fluoroquinolone thiadiazole urea-series fleroxacin derivative was developed by the scholars of [60] to increase the antitumor activity and selectivity of fluoroquinolones and reduce the toxic side effects on normal cells. Novel thiadiazole urea rufloxacin derivatives exhibited IC50 values in a range of 0.46 μM to 2.36 μM [61]. A series of bis-fluoroquinolone thiadiazole urea N-Me lomefloxacin derivatives is described in patent [62], similar bis-fluoroquinolone thiadiazole urea-based N-Me moxifloxacin derivatives designed as promising antitumor agents were patented [63] and the scholars continued this project with thiadiazole urea-based pefloxacin [64], ofloxacin [65], levofloxacin [66] and gatifloxacin [67] derivatives. Furthermore, N-Me moxifloxacin-containing bis-fluoroquinolone oxadiazole urea derivatives for cancer treatment are proposed in the patent [68], N-Me gatifloxacin-containing bis-fluoroquinolone oxadiazole urea derivative in [69], oxadiazole urea fleroxacin derivatives with IC50 values from 0.14 μM to 1.36 μM in [70] and oxadiazole urea pefloxacin bis-derivatives in [71].
At last, there are two unusual papers that are unique according to their research strategy. Thus, a novel series of topoisomerase I (Top I) inhibitors was designed via condensation of FQs with o-phenylenediamine, o-aminophenol or o-aminobenzenethiol in polyphosphoric acid (PPA). The most potent compound 1-ethyl-3-(6-nitrobenzoxazol-2-yl)-6,8-difluoro-7-(3-methylpiperazin-1-yl)-4(1H)-quinolone revealed a significant inhibitory effect on Top I, leading to Top I-mediated cleavage and influencing Top I expression at the cellular level. Moreover, it induced cell death via apoptosis and accelerated DNA strand breaks without significant alteration in cell cycle populations. The in vivo evaluation on the growth of HT-29 tumor xenografts in nude mice showed its therapeutic potential for further development [72].
The scholars of [73] searched for P-glycoprotein ABCB1 inhibitors among fused FQ derivatives. The ABCB1 is involved in multidrug resistance of tumor cells by preventing intracellular accumulation of cytotoxic drugs. In addition, its overexpression limits drug oral bioavailability. To find new potent ABCB1 inhibitors, a 3D pharmacophore model was created based on known inhibitors. The inhibitory activities of the best hits were evaluated by several biological assays, such as rhodamine 123 accumulation assay, chemosensitization assay and multidrug resistance 1-Madin-Darby canine kidney cell/Madin-Darby canine kidney cell permeability assay. The most promising compounds were identified and taken for further development.
3. Other Types of Biological Activity
Apart from antimicrobial and anticancer potency of FQs, it should emphasize the possibility to broaden the horizons of their utilization as biologically active molecules.
First of all, in line with the problem of combating infectious diseases, the scholars of [74] searched for potent molecules to cure protozoal infections. It was already known that N-benzylamide derivative of norfloxacin is promising so they synthesized thiosemicarbazide of 1-butyl-6-fluoro-7-morpholino-4-oxo-1,4-dihydroquinoline-3-carboxylic acid and its heterocyclic derivatives—1,3,4-thiadiazole and 1,2,4-triazole. Furthermore, modification of the 1,2,4-triazole ring with maleimides was also described. For the obtained compounds, antitrypanosomal activity was also typical but lower than for N-benzylamide derivative. Still, the C3 substitution plays a key role in this type of activity.
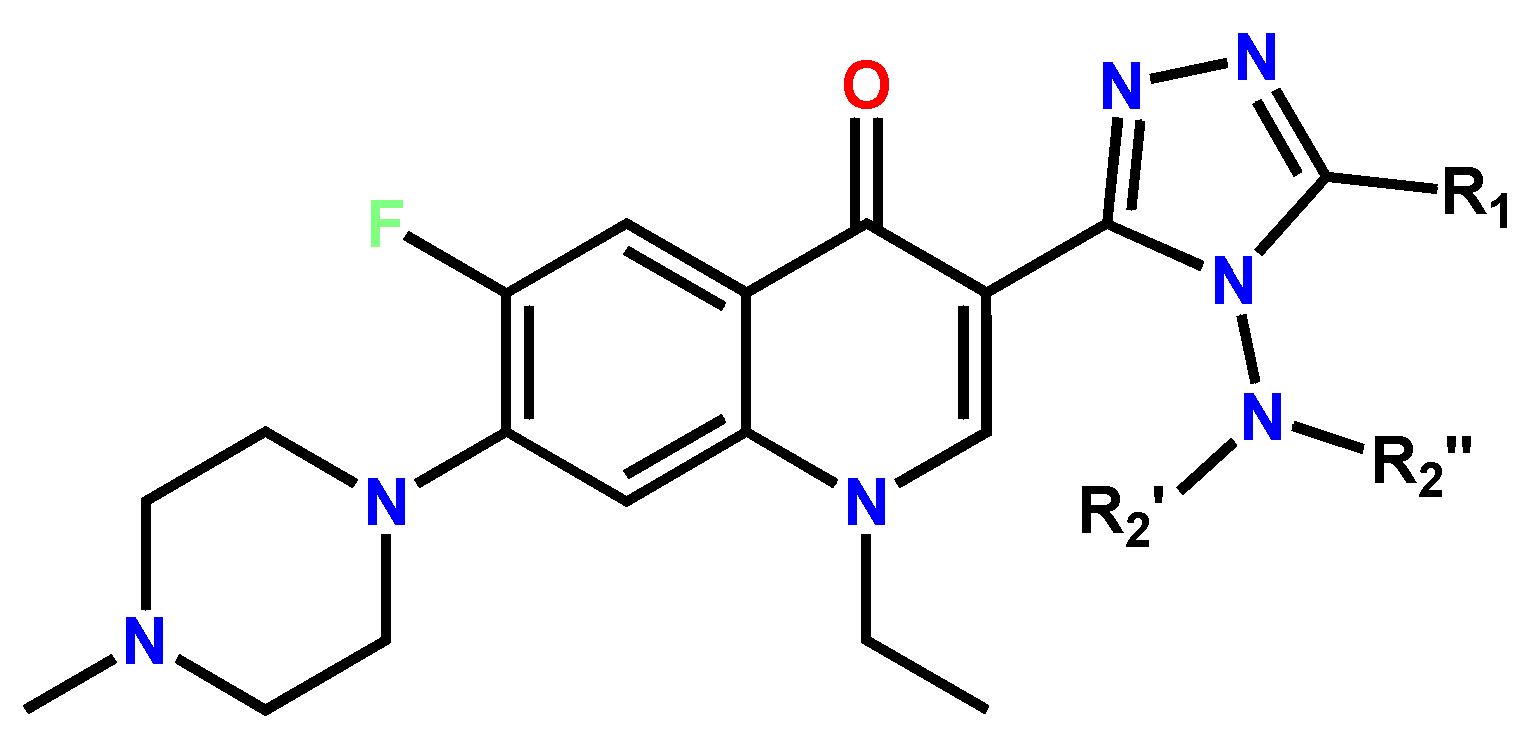
Triazole derivatives of norfloxacin and their Schiff bases were successfully obtained and described in the paper [35]. The Schiff bases (Figure 6) that were screened for analgesic and anti-inflammatory activity on the carrageenan-induced rat paw edema model revealed encouraging results and exceeded the reference drug ibuprofen. At the same time, unsubstituted triazoles revealed antibacterial and antifungal activity. Therefore, further investigations in this area may result in compounds with a versatile pharmacological profile.
Figure 6. Triazole derivatives of norfloxacin as analgesic and anti-inflammatory agents [35].
The synthesized levofloxacin triazole-3-thiol, oxazole, oxadiazole and thiadiazol derivatives revealed antioxidant activity equivalent to ascorbic acid (IC50 = 31.95 g/mL) in the investigation [75].
A series of novel benzimidazole-quinolinone derivatives of ciprofloxacin and levofloxacin was screened for in vitro antidiabetic activity by α-glucosidase inhibitory action and appeared to be promising at a 200 μg/mL concentration compared to acarbose [18].
Novel 5-amino-1,3,4-thiazidazole hybrids of norfloxacin and levofloxacin were synthesized, characterized and assessed for their acetyl cholinesterase enzyme (AChE) inhibitory activity [76]. The obtained derivatives showed promising results, especially levofloxacin derivative (IC50 18.1 ± 0.9 nM), which substantially exceeded the reference drug neostigmine (IC50 2186.5 ± 98.0 nM). In addition, the scholars evaluated the ADMET parameters, and values of the hybrids showed appropriate correlation with the binding energy values (Kcal/mol). Combined with high drug-likeness scores and the results of molecular docking studies, this research can be a promising background for finding a treatment for Alzheimer’s disease.
Altogether, it can assume that the scope of the probable biological activity of FQ hybrids is wide. That makes this area of investigation even more versatile and attractive for medicinal chemists.
References
- Allaka, T.R.; Kummari, B.; Polkam, N.; Kuntala, N.; Chepuri, K.; Anireddy, J.S. Novel heterocyclic 1,3,4-oxadiazole derivatives of fluoroquinolones as a potent antibacterial agent: Synthesis and computational molecular modeling. Mol. Divers. 2021, 26, 1581–1596.
- Arshad, M.; Khan, M.S.; Nami, S.A.A. Norfloxacin Analogues: Drug Likeness, Synthesis, Biological, and Molecular Docking Assessment. Russ. J. Bioorganic Chem. 2021, 47, 483–495.
- Allaka, T.R.; Katari, N.K.; Jonnalagadda, S.B.; Malkhed, V.; Anireddy, J.S. Design, Synthesis and Biological Evaluation of Novel Heterocyclic Fluoroquinolone Citrate Conjugates as Potential Inhibitors of Topoisomerase IV: A Computational Molecular Modeling Study. Curr. Cancer Drug Targets 2021, 18, 11–30.
- Khan, N.M.; Kumar, P.; Hemanth, S.K.K.; Bharath, R.K.P. Synthesis, molecular bioinformatics modelling, and antimicrobial evaluation of some novel oxadiazole fluoroquinolone derivatives. Asian J. Pharm. Clin. Res. 2022, 15, 40–46.
- Gao, L.-z.; Li, T.; Xie, Y.-s.; Hu, G.-q.; Huang, W.-l. Synthesis and antitumor activity of fluoroquinolon-3-yl s-triazole sulfanylacetylhydrazones and s-triazole hydrazone derivatives (V). Zhongguo Yaoxue Zazhi 2015, 50, 545–549.
- Al-Mathkuri, T.S.F.; Al-Jubori, H.M.S.; Saleh, A.T. Synthetic and Study the Chelating Activity of Some Polymers Containing Heterocyclic Rings Which Derivative from 1,2,4- Trizol Levofloxacin Acid. Orient. J. Chem. 2018, 34, 2031.
- Cui, S.-F.; Addla, D.; Zhou, C.-H. Novel 3-aminothiazolylquinolones: Design, synthesis, bioactive evaluation, SARs, and preliminary antibacterial mechanism. J. Med. Chem. 2016, 59, 4488–4510.
- Azad, C.S.; Narula, A.K. An operational transformation of 3-carboxy-4-quinolones into 3-nitro-4-quinolones via ipso-nitration using polysaccharide supported copper nanoparticles: Synthesis of 3-tetrazolyl bioisosteres of 3-carboxy-4-quinolones as antibacterial agents. RSC Adv. 2016, 6, 19052–19059.
- Niveditha, N.; Begum, M.; Prathibha, D.; Sirisha, K.; Mahender, P.; Chitra, C.; Rao, V.R.; Reddy, V.M.; Achaiah, G. Design, Synthesis and Pharmacological Evaluation of Some C3 Heterocyclic-Substituted Ciprofloxacin Derivatives as Chimeric Antitubercular Agents. Chem. Pharm. Bull. 2020, 68, 1170–1177.
- Hou, L.-l.; Yin, J.; Wang, W.; Xie, S.-q.; Huang, W.-l.; Hu, G.-q. Synthesis and antitumor activity of fluoroquinolone C-3 heterocycles-oxadiazole derivatives (II). Zhongguo Yaoxue Zazhi 2013, 48, 1194–1196.
- Jubie, S.; Kalirajan, R.; Yadav, P. Design, Synthesis and Docking Studies of a Novel Ciprofloxacin Analogue as an Antimicrobial AGENT. E-J. Chem. 2012, 9, 980–987.
- Jubie, S.; Prabitha, P.; Kumar, R.R.; Kalirajan, R.; Gayathri, R.; Sankar, S.; Elango, K. Design, synthesis, and docking studies of novel ofloxacin analogues as antimicrobial agents. Med. Chem. Res. 2011, 21, 1403–1410.
- Zhou, C.; Cui, S.; Dinesh, A. Quinolone Thiazole Compound and the Preparation Method and Application Thereof. CN Patent CN104530034A, 22 April 2015.
- Zhou, C.; Wang, L.; Battini, N.; Chen, J.; Xie, Y. Aminothiazole Quinolone Oxime Compound, Its Preparation Method and Application in Preparing Antibacterial Drug, Antifungal Drug and DNA Intercalator. CN Patent CN109651353A, 19 April 2019.
- Zhou, C.; Wang, L.; Battini, N. Preparation of the 3-(2-Aminothiazol-4-yl)-7-(4-substituent-piperazin-1-yl)quinolin-4(1H)-one Compound and Their Application as the Antifungal Drugs. CN Patent CN108440518A, 24 August 2018.
- Yang, Y.; Chen, D.; Qian, X.; Luo, X.; Shao, L.; Dong, X.; Qian, L. Preparation of Novel Rhodamine Dyes Useful as Antibacterial Agents. WO Patent WO2020019289A1, 30 January 2020.
- Yang, Y.; Chen, D.; Qian, X.; Luo, X.; Shao, L.; Dong, X.; Qian, L. Preparation of Novel Rhodamine Dyes Useful as Antibacterial Agents. CN Patent CN108570032A, 25 September 2018.
- Muluk, R.; Kothawade, P.; Kulkarni, G.; Ingale, P. Synthesis and evaluation of some novel benzimidazole-quinolinone derivatives for their antifungal and antidiabetic activity. World J. Pharm. Pharm. Sci. 2018, 7, 1263–1278.
- Hu, G.-Q.; Wu, X.-K.; Xie, S.-Q.; Du, G.-J.; Huang, W.-L.; Zhang, H.-B. Synthesis and bioactivity of water-soluble fused s-triazolothiadiazole systems (II): Fluoroquinolone piperazine derivatives. Huaxue Xuebao 2008, 66, 2157–2162.
- Kumar, D.V.; Rai, R.; Brameld, K.A.; Riggs, J.; Somoza, J.R.; Rajagopalan, R.; Janc, J.W.; Xia, Y.M.; Ton, T.L.; Hu, H.; et al. 3-Heterocyclyl quinolone inhibitors of the HCV NS5B polymerase. Bioorganic Med. Chem. Lett. 2012, 22, 300–304.
- Farooqi, S.I.; Arshad, N.; Channar, P.A.; Perveen, F.; Saeed, A.; Larik, F.A.; Javeed, A. Synthesis, theoretical, spectroscopic and electrochemical DNA binding investigations of 1, 3, 4-thiadiazole derivatives of ibuprofen and ciprofloxacin: Cancer cell line studies. J. Photochem. Photobiol. B Biol. 2018, 189, 104–118.
- Gao, L.-Z.; Xie, Y.-S.; Li, T.; Huang, W.-L.; Hu, G.-Q. . Yao Xue Xue Bao 2014, 49, 1694–1698.
- Wang, G.; Duan, N.; Cao, T.; Wen, X.; Yin, J.; Wang, W.; Xie, S.; Huang, W.; Hu, G. Antitumor fluoroquinolone C3-isostere derivatives(I)—Synthesis and activity of bis-oxadiazole methyl-sulfide derivatives. Yingyong Huaxue 2012, 29, 769–774.
- Feng, H.; Feng, C.-J. CoMFA Model of Anti-tumor Activity for Fluoroquinolon-3-yl s-Triazole Sulfide-ketone Derivatives and Implications for Molecular Design. Chin. J. Struct. Chem. 2021, 40, 703–710.
- Shaharyar, M.; Ali, M.A.; Abdullah, M.-M. Synthesis and antiproliferative activity of 1--6-fluoro-3--1,3,4-oxadiazol-2-yl-7-piperazino-1,4-dihydro-4-quinolinone derivatives. Med. Chem. Res. 2007, 16, 292–299.
- Jing, Y.; Zhao, H.; Wu, S.; Ni, L.; Yan, Q.; Hu, G. Preparation of OFLOXACIN Thiosemicarbazone Derivatives as Antitumor Agents. CN Patent CN104592252A, 6 May 2015.
- Jing, Y.; Wu, S.; Ni, L.; Yan, Q.; Gao, L.; Xie, Y.; Hu, G. Preparation of Cyclopropyl Fluoroquinolone C-3 S-Triazole Thioether Ketone Semicarbazone Compounds as Antitumor Agents. CN Patent CN104628702A, 20 May 2015.
- Zhang, H.; Liu, J.; Li, D.; Zhao, Y. Preparation of Lomefloxacin Derivatives for Treatment of Cancer. CN Patent CN109400631A, 1 March 2019.
- Lu, L.; Zhang, H.; Wang, H.; Hu, G. Preparation of Lomefloxacin Derivatives for Treatment of Cancer. CN Patent CN109400627A, 1 March 2019.
- Li, Y.; Liang, J.; Zhou, J.; Hu, G. Bis-fluoroquinolone oxadiazole carbazide N-methyl-Ciprofloxacin derivative, its preparation method and application as antitumor agents. CN Patent CN109369677A, 22 February 2019.
- Feng, Y.; Jiang, Y.; Geng, S.; Hu, G. Bis-Fluoroquinolone Oxadiazole Urea N-Acetyl-Ciprofloxacin Derivative, Its Preparation Method and Application. CN Patent CN109336902A, 15 February 2019.
- Jiang, Y.; Liu, Q.; Shao, X.; Hu, G. Preparation of Bis-Fluoroquinolone Oxadiazole Urea Derivative as Antitumor Drug. CN Patent CN109369676A, 22 February 2019.
- Cen, S.; Yang, L.; Li, X.; Hu, G. Preparation Method of Rufloxacin-Containing Bis-Fluoroquinolone Oxadiazole Urea Derivative Applied to Antitumor Drug. CN Patent CN109438482A, 8 March 2019.
- Hu, G.; Zhang, C.; Sun, J.; Wang, N.; Shen, R. Preparation Method and Application of Bis-Fluoroquinolone Thiadiazole Urea-series N-Acetyl-Ciprofloxacin Derivative. CN Patent CN109761999A, 17 May 2019.
- Chandramouli; Shivanand, M.R.; Nayanbhai, T.B.; Bheemachari; Udupi, R.H. Synthesis and biological screening of certain new triazole Schiff bases and their derivatives bearing substituted benzothiazole moiety. J. Chem. Pharm. Res. 2012, 4, 1151–1159.
- Hu, G.Q.; Hou, L.L.; Yang, Y.; Yi, L.; Xie, S.Q.; Wang, G.Q.; Duan, N.N.; Chao, T.Y.; Wen, X.Y.; Huang, W. Long Synthesis and antitumor evaluation of fluoroquinolone C3 fused heterocycles (II): From triazolothiadiazines to pyrazolotriazoles. Chin. Chem. Lett. 2011, 22, 804–806.
- Chao, W.; Feng, C.-J. QSAR Studies on the inhibitory activity of levofloxacin-thiadiazole HDACi Conjugates to histone deacetylases. Chin. J. Struct. Chem. 2018, 37, 1679–1688.
- Wan, Z.; Sun, R. Jing QSAR and molecular docking study on the biological activity of levofloxacin and thiodiazole histone deacetylase inhibitors. Chem. Rev. Lett. 2020, 3, 12–18.
- Sun, Y.; Xu, Q.; Hou, L.; Yue, X.; Wu, Z.; Huang, W.; Xie, S.; Hu, G. Synthesis and antitumor activities of fluoroquinolone C-3 isosteres(IV):s-triazole-oxadiazole methylsulfide Mannich-base derivatives. Zhongguo Yaoke Daxue Xuebao 2014, 45, 39–42.
- Xu, Q.; Hou, L.; Wu, Z.; Yue, X.; Hu, G.; Huang, W. Synthesis and antitumor activity of fluoroquinolone C-3 isostere III: S-triazole oxadiazole methylsulfide derivatives from pefloxacin. Zhongguo Yaoke Daxue Xuebao 2013, 44, 511–514.
- Hu, G.; Wang, G.; Duan, N.; Wen, X.; Cao, T.; Xie, S.; Huang, W. Design, synthesis and antitumor activities of fluoroquinolone C-3 heterocycles (IV): S-triazole Schiff–Mannich bases derived from ofloxacin. Acta Pharm. Sin. B 2012, 2, 312–317.
- Xie, Y.; Gao, L.; Yan, Q.; Wu, S.; Ni, L.; Liu, Y.; Huang, W.; Hu, G. Synthesis and antitumor activity of fluoroquinolon-3-yl s-triazole sulfide ketone thiosemicarbazone derivatives of ofloxacin. Yingyong Huaxue 2016, 33, 25–31.
- Hu, G.; Wu, X.; Wang, X.; Zhang, Z.; Xie, S.; Huang, W.; Zhang, H. Synthesis and antitumor activity of C3 heterocyclic-substituted fluoroquinolone derivatives (I): Ciprofloxacin aminothiodiazole Schiff-bases. Yaoxue Xuebao 2008, 43, 1112–1115.
- Hu, G.; Ye, Q.; Yu, Z.; Chen, Y.; Liu, H.; Li, J.; Kang, J.; Huang, F. Levorotatory Fluoroquinolone-C3 Diazole Derivatives as Antitumor Agents and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of Cancer. China. CN102391287A, 28 March 2012.
- Xie, S.; Chen, Y.; Wang, G.; Duan, N.; Wen, X.; Cao, T.; Yin, J.; Wang, W.; Hu, G.; Huang, W. Synthesis and antitumor evaluation of s-triazolothiadiazines and pyrazolo s-triazoles derived from ciproxacin. Yaoxue Xuebao 2012, 47, 66–71.
- Hu, G.; Zhang, Z.; Wang, H.; Wu, X.; Wang, X.; Du, G.; Xie, S.; Huang, W.; Zhang, H. Synthesis and antitumor activity of fluoroquinolon-3-yl fused heterocyclic systems (III): S-triazolothiadiazinone derivatives derived enrofloxacin. Huaxue Xuebao 2009, 67, 2592–2596.
- Wu, S.-m.; Yan, Q.; Ni, L.-l.; Xie, Y.-s.; Gao, L.-z.; Liu, Y.-j.; Huang, W.-l.; Hu, G.-q. Synthesis and antitumor activity of fluoroquinolone C-3 fused heterocyclic thiazolotriazole derivatives (VI). Zhongguo Yaoxue Zazhi 2016, 51, 353–357.
- Li, Y.; Zhang, C.; Huang, W.; Chen, C.; Hu, G. Synthesis and Antitumor Activities of C-3 Thiazolotriazole Unsaturated Ketone Derivatives of Levofloxacoin. Chin. J. Appl. Chem. 2019, 36, 671–676.
- You, Q.; He, X.; Li, Z.; Chen, X.; Li, Y.; Li, H.; Zhang, W. Preparation of Quinolone Derivatives as Antitumor Agents. CN Patent CN1473827A, 11 February 2004.
- Hu, G.; Yang, Y.; Yi, L.; Wang, X.; Zhang, Z.; Xie, S.; Huang, W. Design, synthesis and antitumor action of C3/C3 bis-fluoroquinolones linked-cross 2,5-oxadiazole. Yaoxue Xuebao 2010, 45, 1012–1016.
- Hu, G.; Xie, S.; Hou, L.; Sun, M.; Zhang, J.; Yang, Y.; Yi, L. Preparation of 1,3,4-Oxadiazole Linked Fluoroquinolone Dimers for Treating Neoplasm and Microbial Infection. CN Patent CN101643471A, 10 February 2010.
- Hu, G.-q.; Hou, L.-l.; Wang, G.-q.; Duan, N.-n.; Wen, X.-y.; Cao, T.-y.; Yin, J.; Wang, W.; Xie, S.-q.; Huang, W.-l. Design, synthesis and antitumor activity of fluoroquinolone C-3 heterocycles: Bis-oxadiazole methylsulfide derivatives derived from ciprofloxacin. Yaoxue Xuebao 2012, 47, 1017–1022.
- Hu, G.-q.; Wang, G.-q.; Duan, N.-n.; Wen, X.-y.; Cao, T.-y.; Xie, S.-q.; Huang, W.-l. Design, Synthesis and Antitumor Activity of Fluoroquinolone C3 Heterocyclic Bis-oxadiazole Methylsulfide Derivatives Derived from Levofloxacin. Chem. Res. Chin. Univ. 2012, 28, 980–984.
- Hu, G.-Q.; Yang, Y.; Yi, L.; Wang, G.-Q.; Duan, N.-N.; Wen, X.-Y.; Cao, T.-Y.; Xie, S.-Q.; Huang, W.-L. Design, synthesis and antitumor activity of C3/C3 bis-fluoroquinolones cross-linked with triazolothiadiazole. Acta Pharm. Sin. B 2011, 1, 172–177.
- Hu, G.Q.; Zhang, Z.Q.; Xie, S.Q.; Huang, W. Long Synthesis and antitumor evaluation of C3/C3 fluoroquinolone dimers (I), tethered with a fused heterocyclic s-triazolothiadiazole. Chin. Chem. Lett. 2010, 21, 661–663.
- Hu, G.; Xie, S.; Hou, L.; Sun, M.; Zhang, J.; Yang, Y.; Yi, L. Preparation of 1,2,4-Triazolothiadiazole Linked Fluoroquinolone Dimers for Treating Tumor and Antimicrobial Infection. CN Patent CN101648962A, 17 February 2010.
- Li, T.; Gao, L.-z.; Xie, Y.-s.; Hu, G.-q.; Huang, W.-l. Synthesis and antitumor activity fluoroquinolon-3-yloxadiazole sulfanylacetylhydrazone derivatives. Zhongguo Yaoxue Zazhi 2014, 49, 2206–2209.
- Xu, Q.-j.; Hou, L.-l.; Wu, Z.-f.; Yue, X.-b.; Xie, S.-q.; Huang, W.-l.; Hu, G.-q. Synthesis and antitumor evaluation of fluoroquinolone C3 s-triazole oxadiazole methylsulfide derivatives of ofloxacin. Zhongguo Yaoxue Zazhi 2014, 49, 609–612.
- Li, K.; Cai, Y.; Zang, F.; Hu, G. Levofloxacin-Containing Bis-Fluoroquinolone Oxadiazole Carbamide Derivative Useful in Treatment of Cancer and Its Preparation. CN Patent CN109369674A, 22 February 2019.
- Hou, L.; Zhang, C.; Hu, G.; Sun, J.; Wang, N.; Shen, R. Bis-Fluoroquinolone Thiadiazole Urea-Series Fleroxacin Derivative Preparation Method and Application as Antitumor Agents. CN Patent CN109761998A, 17 May 2019.
- Hou, L.; Du, L.; Li, Y.; Hu, G.; Sun, J.; Zhang, C.; Shen, R.; Wang, N. Preparation of Fluoroquinolone 1,3,4-Thiadiazole Urea Rufloxacin Derivatives Useful for the Treatment of Cancer. CN Patent CN109762005A, 17 May 2019.
- Hou, L.; Hu, G.; Zhang, C.; Sun, J.; Wang, N.; Shen, R. Process for Preparation and Application of Bis-Fluoroquinolone Thiadiazole Urea-Series N-Methyl Lomefloxacin Derivatives. CN Patent CN109761997A, 17 May 2019.
- Hou, L.; Du, L.; Li, Y.; Hu, G.; Sun, J.; Zhang, C.; Shen, R.; Wang, N. Process for Preparation and Application of Bis-Fluoroquinolone Thiadiazole Urea-Based N-Methyl Moxifloxacin Derivative. CN Patent CN109678890A, 26 April 2019.
- Hu, G.; Sun, J.; Zhang, C.; Wang, N.; Shen, R. Preparation of Bis-Fluoroquinolone Thiadiazole Urea-Based Pefloxacin Derivatives as Antitumor Agents. CN Patent CN109678882A, 26 April 2019.
- Hu, G.; Sun, J.; Zhang, C. Preparation of Bis-Fluoroquinolone Thiadiazole Urea-Based Ofloxacin Derivatives as Antitumor Agents. CN Patent CN109678881A, 26 April 2019.
- Hu, G.; Sun, J.; Zhang, C. Bis-Fluoroquinolone Thiadiazole Urea-Based Levofloxacin Derivative Useful in Treatment of Cancer and Its Preparation. CN Patent CN109678889A, 26 April 2019.
- Hou, L.; Li, Y.; Hu, G.; Sun, J.; Zhang, C.; Shen, R.; Wang, N. Bis-Fluoroquinolone Thiadiazole Urea-Based N-Methyl Gatifloxacin Derivative Useful in Treatment of Cancer and Its Preparation. CN Patent CN109678885A, 26 April 2019.
- Cen, S.; Geng, S.; Yang, L.; Hu, G. N-Methyl Moxifloxacin-Containing Bis-Fluoroquinolone Oxadiazole Urea Derivative Useful in Treatment of Cancer and Its Preparation. CN Patent CN109438481A, 8 March 2019.
- Hu, G.; Feng, Y.; Liu, J.; Zhao, Y. Preparation Method of N-methyl Gatifloxacin-Containing Bis-Fluoroquinolone Oxadiazole Urea Derivative Applied to Antitumor Drug. CN Patent CN, 109438472A, 8 March 2019.
- Wang, S.; Mao, Y.; Lu, L.; Hu, G. Preparation of Fluoroquinolone Oxadiazole Urea Fleroxacin Derivatives Useful for the Treatment of Cancer. CN Patent CN109400626A, 1 March 2019.
- Liu, Q.; Shao, X.; Mao, Y.; Hu, G. Bifluoroquinolone Oxadiazole Urea Pefloxacin Derivative Useful in Treatment of Cancer and Its Preparation. CN Patent CN109369675A, 22 February 2019.
- You, Q.-D.; Li, Z.-Y.; Huang, C.-H.; Yang, Q.; Wang, X.-J.; Guo, Q.-L.; Chen, X.-G.; He, X.-G.; Li, T.-K.; Chern, J.-W. Discovery of a Novel Series of Quinolone and Naphthyridine Derivatives as Potential Topoisomerase I Inhibitors by Scaffold Modification. J. Med. Chem. 2009, 52, 5649–5661.
- Zhang, S.-l.; Wei, Y.-x.; Li, Q.; Sun, H.-p.; Peng, H.; You, Q.-d. Pharmacophore-based drug design and biological evaluation of novel ABCB1 inhibitors. Chem. Biol. Drug Des. 2013, 81, 349–358.
- Pyrih, A.; Berninger, M.; Gzella, A.; Lesyk, R.; Holzgrabe, U. Synthesis and evaluation of antitrypanosomal activity of some thiosemicarbazide derivatives of 1-butyl-6-fluoro-7-morpholino-4-oxo-1,4-dihydroquinoline-3-carboxylic acid. Synth. Commun. 2018, 48, 1883–1891.
- Lihumis, H.S.; Al Talebi, Z.A.; Khaleel, A.K. Synthesis and Identification of Some New Heterocyclic Compounds for Levofloxacin Drug Derivatives with Evaluating of Their Biological Efficiency and Antioxidant Activity. J. Med. Chem. Sci. 2022, 5, 596–606.
- Ujan, R.; Saeed, A.; Channar, P.A.; Larik, F.A.; Abbas, Q.; Alajmi, M.F.; El-Seedi, H.R.; Rind, M.A.; Hassan, M.; Raza, H.; et al. Drug-1,3,4-Thiadiazole Conjugates as Novel Mixed-Type Inhibitors of Acetylcholinesterase: Synthesis, Molecular Docking, Pharmacokinetics, and ADMET Evaluation. Molecules 2019, 24, 860.
More
Information
Subjects:
Chemistry, Medicinal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
621
Revisions:
2 times
(View History)
Update Date:
19 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No