Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Laura Sabatino | -- | 1886 | 2023-06-15 11:11:03 | | | |
| 2 | Catherine Yang | Meta information modification | 1886 | 2023-06-16 03:30:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sabatino, L. Thyroid Hormone Signaling. Encyclopedia. Available online: https://encyclopedia.pub/entry/45634 (accessed on 27 July 2026).
Sabatino L. Thyroid Hormone Signaling. Encyclopedia. Available at: https://encyclopedia.pub/entry/45634. Accessed July 27, 2026.
Sabatino, Laura. "Thyroid Hormone Signaling" Encyclopedia, https://encyclopedia.pub/entry/45634 (accessed July 27, 2026).
Sabatino, L. (2023, June 15). Thyroid Hormone Signaling. In Encyclopedia. https://encyclopedia.pub/entry/45634
Sabatino, Laura. "Thyroid Hormone Signaling." Encyclopedia. Web. 15 June, 2023.
Copy Citation
Thyroid hormones (TH) perform a plethora of actions in numerous tissues and induce an overall increase in metabolism, with an augmentation in energy demand and oxygen expenditure. Oxidants are required for normal thyroid-cell proliferation, as well as for the synthesis of the main hormones secreted by the thyroid gland, triiodothyronine (T3) and thyroxine (T4).
thyroid hormones
oxidative stress
antioxidants
Nrf2
cardioprotection
1. Molecular Aspects of Thyroid-Hormone Signaling
The thyroid hormones (THs) include the prohormone thyroxine (T4) and the biologically active form triiodothyronine (T3) and regulate a wide range of genes, intervening in many physiological processes, such as cell growth, development, differentiation, and survival [1]. They are synthesized in the thyroid follicles after the iodination of thyroglobulin (Tg) by thyroid peroxidase (TPO) [2].
Largely in the form of T4, THs are released in the circulation, where they are mostly bound to transport proteins and reach the peripheral tissues, where the 5′-monodeiodinases (DIO1 and DIO2) catalyze T4 to T3 activation [2]. A third monodeiodinase (DIO3) has been described in the cells and mediates T4 conversion to metabolically inactive reverse T3 (rT3) [2].
The signaling of TH in the target cells is highly complex and finely regulated [3]. Transporters of TH mediate the uptake of TH and, once inside the cell, TH can mediate genomic effects in the nucleus, binding to specific receptors (TRs), which directly interact with responding elements (TREs) in the target promoters, thus regulating the transcription of specific genes (Figure 1) [4]. Two genes encoding TRs have been described, THRA and THRB, codifying for TRα and TRβ, respectively, and these two isoforms are differently expressed during embryonic development and in adult life [1][5].
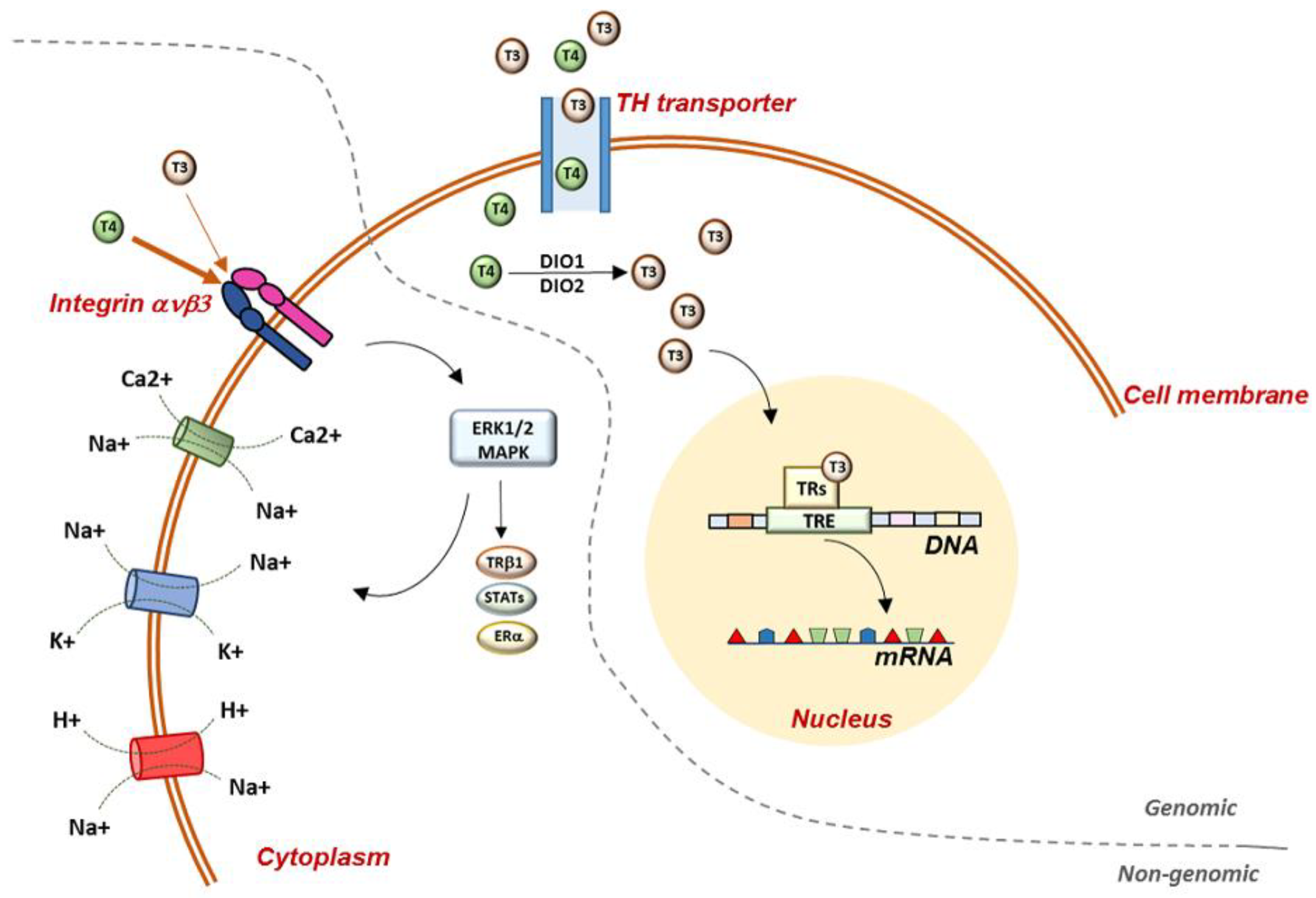
Figure 1. Representation of genomic and non-genomic actions of TH in the cell. Genomic actions begin at the plasma membrane and THs enter the cell through specific TH transporters. Once in the cell, T4 is converted to T3 by D1 and D2 deiodinases and T3 enters the nucleus, where it binds to specific receptors, which mediate the interaction with the DNA. Non-genomic mechanisms require the mediation of integrin ανβ3, which has a higher binding affinity for T4 than T3. Once in the cell, THs activate several MAPK-mediated signaling pathways. At the plasma-membrane level, TH regulate glucose transporter, Na+/K+-ATPase, Na+/H+-exchanger, Ca2+-ATPase, and the Na+-sensitive amino-acid transporter. TH: thyroid hormones; T3: triiodothyronine; T4: thyroxine; TRs: thyroid-hormone receptors; TRE: thyroid-responsive elements; DIO1: deiodinase 1; DIO2: deiodinase 2; STATs: signal transducer and activator of transcription 1α and 3; ERα: estrogen receptor α.
2. Thyroid Hormone Excess, Oxidative Stress, and Nrf2 Activation
Diseases of THs are strongly associated with oxidative stress, and while, on one hand, oxidants interfere with the synthesis, activity, and metabolism of hormones, the reverse condition is also possible, and TH can regulate the antioxidant levels in cells.
Depending on the tissue demand, in normal conditions, a baseline level of oxidants is necessary to preserve cell homeostasis, and this number of oxidants is generally low in most tissues. When the oxidants exceed the ability of the cells to remove the oxidant surplus, oxidative stress arises. Thus, the role of oxidants in cells depends mainly on their initial concentrations, which determine the downstream cellular responses.
The THs are key regulators of cellular metabolism, and several studies found that in hyperthyroidism, the augmented metabolic demand promotes the synthesis of chemical energy by mitochondrial oxidation-reduction reactions, thus increasing the oxidant levels in the cell and inducing lower antioxidant ability [6][7].
The available data indicate that TH administration increases H2O2 generation by mitochondria in rat tissues, and this event is often associated with increased rates of oxygen consumption in target tissues, such as the liver, kidneys, heart, and skeletal muscles, where the need for metabolic capacity is higher [8][9]. Variability in the antioxidant response leading to an imbalance in oxidant clearance was observed in the tissues of hyperthyroid-induced animals, and further variations were appreciable, according to the age and the characteristics of the animals undergoing TH treatment [10].
Hyperthyroidism and thyrotoxicosis have been associated with the activation of Nrf2 signaling in TH target tissues. More specifically, in rat livers, T3 administration led to a rapid and transient cytosol-to-nuclear translocation of Nrf2, and it was hypothesized that the increase in oxidative status induced by T3 administration may inactivate Keap1-mediated ubiquination/degradation and expand Nrf2 nuclear-pool availability [11].
Several studies hypothesized that Nrf2 activation is triggered by mitogen-activated protein kinases (MAPKs) produced by T3-induced oxidants; however, the exact role of MAPKs and the underlying molecular mechanism remain poorly defined [11]. On the other hand, some other studies evaluated the hypothesis that the direct phosphorylation of Nrf2 by MAPKs contributes little to the modulation of Nrf2 activity and suggested that MAPKs mainly regulate the Nrf2 signaling pathway through indirect mechanisms [12].
In the last decades, several studies have provided new approaches to detailing the interactions between the TH system and mitochondrial compartments and to elucidating the effects of TH on electron-transport complexes and the existing relationship with oxidative metabolism [13]. Recently, it was demonstrated that respiratory complexes are organized in higher-order structures, called supercomplexes, which guarantee the major stabilization of the assembly and better control over oxidant production in the electron-transport chain, thanks to the better accessibility of substrates necessary for enzymatic reactions [14][15]. Moreover, the discovery of supercomplexes represents an important step forward in the study of the functional and structural properties of the mitochondrial respiratory chain, even though their functional advantages and their possible pathophysiological involvement in TH disease are far from being fully understood.
In an experimental model of hyperthyroidism, it was found that more than 58% of mitochondria were swollen, and that their cristae were radially oriented towards the center of organelles [16]. Alterations in mitochondrial morphology can slightly reduce the efficiency of phosphorylation, whereas the TH-induced increase in mitochondrial respiratory complexes explains the increase in respiratory rate [17].
In normal conditions, Nrf2 affects the mitochondrial membrane potential, fatty-acid oxidation, the availability of substrates for respiration (NADH and FADH2/succinate), and ATP synthesis. In conditions of stress, Nrf2 activation counteracts oxidant production in mitochondria via the transcriptional upregulation of uncoupling protein 3 and influences mitochondrial biogenesis by maintaining adequate levels of NRF1 and PGC-1α, as well as by promoting purine-nucleotide biosynthesis in rapidly growing cells [18].
The Nrf2 plays an important role in the maintenance of mitochondrial homeostasis and structural integrity. This is especially true in conditions of oxidative, electrophilic, and inflammatory stress, when the request for cytoprotective responses is crucial for the survival of the cell and the organism. The effects on mitochondria are among the principal protective mechanisms mediated by Nrf2. Diseases of the THs, analogously to many other pathological conditions, are characterized by oxidative stress, inflammation, and mitochondrial dysfunction as essential components of their pathogenesis. Therefore, Nrf2’s possible involvement holds promise for disease prevention and treatment.
3. Thyroid Hormones and Their Antioxidant Role in Cardioprotection: Nrf2 Mediation
Experimental studies showed the negative effects of TH-altered metabolism on cardiac function, cell protection, and mitochondrial function, whereas the reversibility of these conditions restores the euthyroid state, suggesting that TH exert an important cardioprotective role [19].
Oxidative stress is a determining factor in the pathological progression of cardiac diseases, and excess of oxygen species may occur when oxygen supply is limited, such as during cardiac ischemia. In these conditions, oxidants can provoke irreversible damage by oxidation-membrane phospholipids, proteins, and DNA [20]. Subsequently, the heart reacts with a remodeling process that starts as a compensatory event characterized by the hypertrophy of surviving myocytes and the fibrosis of non-myocyte components, but soon involves the activation of the neuroendocrine and inflammatory systems and leads to decompensation and heart failure [21]. In particular, the progression to heart failure is associated with a progressive compromise of mitochondrial respiratory activity and a reduction in its capacity to produce ATP, which, in turn, leads to secondary dysregulation and altered Ca2+ handling and energy deficiency [22].
In both clinical settings and experimental studies of acute myocardial infarction, the reduction in circulating T3 levels (low-T3 syndrome) is one of the principal alterations observed and correlates with intense pro-inflammatory and stress responses [23]. The low-T3 state induces several important molecular, biochemical, and histological changes in the myocardium [24] and, for a long time, low T3 has been considered part of a beneficial adaptive mechanism aiming to reduce cardiac energy expenditure. However, clinical and experimental data demonstrated that low T3 is a strong prognostic predictor of short-term and long-term mortality [25][26], and that constant and low-level T3 administration allow the normalization of the hormone in the serum, attenuate myocardial damage, reduce remodeling, and prevent oxidative stress, with the final effect of improving cardiac function [27][28].
Many Nrf2-regulated enzymes are involved in the pathogenesis of cardiovascular diseases and may act as specific markers of the progression towards heart failure. These genes include antioxidant-related genes [29], stress-response genes [30], and genes limiting the inflammatory processes and conferring protection against ischemia/reperfusion events [31].
Coronary artery disease and ischemic heart disease are the most prevalent causes of mortality worldwide, and post-myocardial infarction hypertrophy, fibrosis, and apoptosis are the major events driving the progression towards heart failure. Coronary interventions and revascularization initially provide benefits after acute myocardial infarction; however, ischemia/reperfusion injury occurring during revascularization may worsen general cardiac conditions due to oxidant formation and inflammatory infiltration [31][32][33][34]. In this context, Nrf2 has been demonstrated to play a central role in cardiac protection, through the regulation of a broad spectrum of target genes [35][36].
Mouse models of Nrf2 overexpression or Nrf2 knockouts have been widely used to characterize Nrf2’s role in cardiac pathological contexts. In mice with constitutively active Nrf2 cardiac overexpression, beyond the increased expression of antioxidant genes, some hypertrophic genes (i.e., genes for natriuretic peptides A and B) are also stimulated, increasing the risk of developing pathological cardiac remodeling [37]. By contrast, Nrf2-KO mice have a marked exposure to oxidative insult and oxidative-stress-associated pathologies [38]. Although some antioxidant gene expression is still appreciable in Nrf2-KO mice, it is not sufficient to compensate for oxidative stress and cardiac hypertrophy due to acute exercise stress, leading to cardiac dysfunction [38][39]. Figure 2 reports a schematic representation of the main cardiac phenotypes associated with Nrf2 over expression or lack of expression.
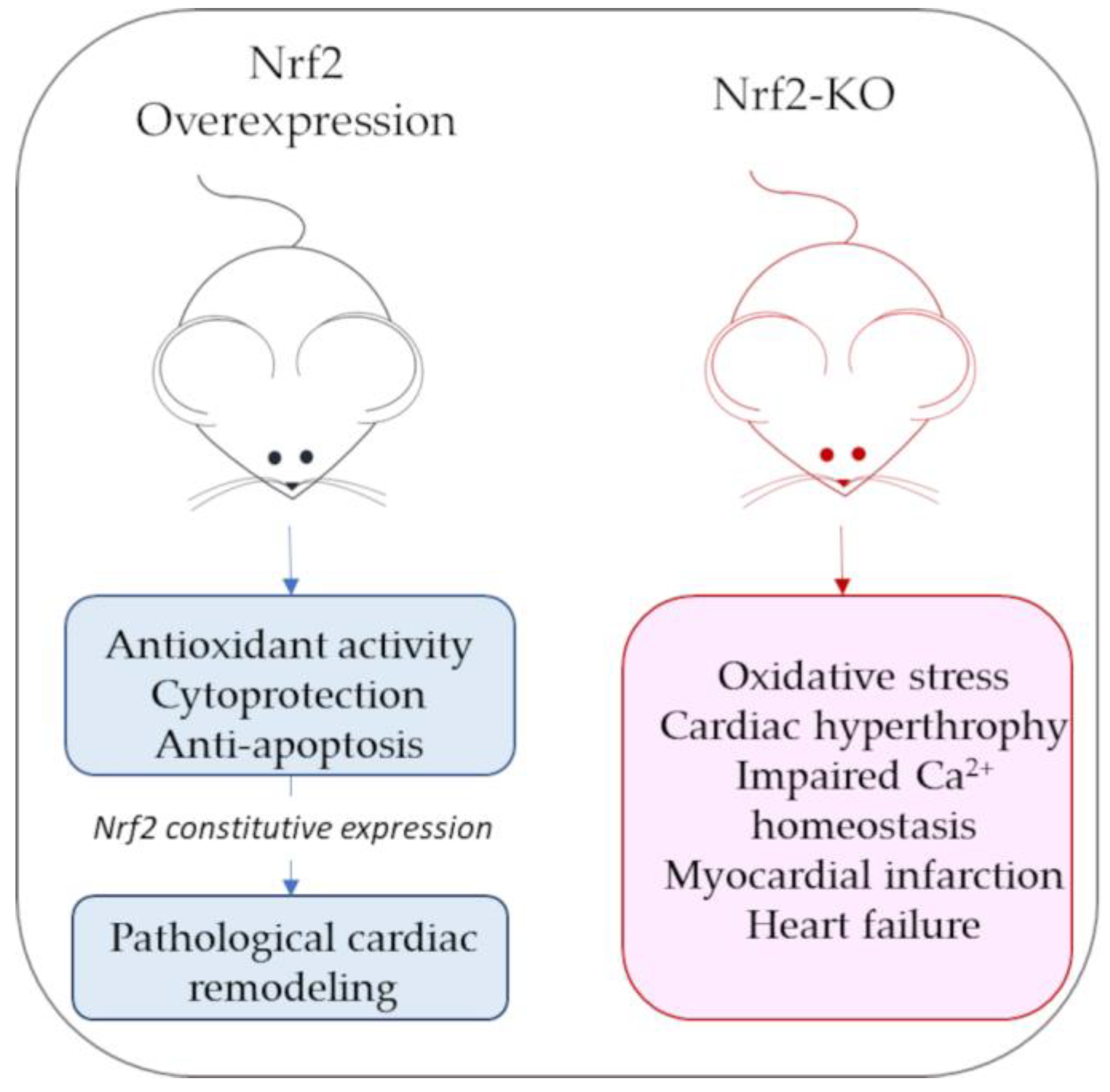
Figure 2. Schematic representation of main cardiac phenotypes associated with Nrf2 overexpression or lack of expression (Nrf-KO mice).
The Nrf2 affects cell survival through some mediators, such as the anti-apoptotic proteins Bcl-2 and the heme oxygenase-1 (HO-1), a stress protein with antioxidant, anti-apoptotic, anti-thrombotic, and anti-inflammatory properties [40], and it is therefore considered a reliable marker of oxidative stress [41]. During ischemia/reperfusion, Nrf2’s dissociation from Keap1 is encouraged and Nrf2 translocation to the cardiomyocyte nucleus increases, thus increasing antioxidant responses [42]. The stimulation of Nrf2 in cardioprotection is associated with the activation of the pro-survival pathway phosphoinositide 3-kinase (PI3K)/Akt kinase, which is considered a key factor in many aspects of cardiac physiology, such as cell survival, contractility, and electrophysiology [43]. Moreover, the PI3K/Akt pathway is considered to be involved in T3 protection against ischemic injury, both in vivo and in vitro. In fact, in H2O2-treated cardiomyocytes, pre-treatment with T3 stimulates PI3K and Akt signaling through their phosphorylation [42][43], and in a mouse experimental model, TH-replacement therapy restores myocardial function after ischemia/reperfusion injury [44]. The levels of Nrf2 increase in response to T3 treatment, suggesting the pivotal role of this factor in the mediation of T3’s protective function in cardiomyocytes [42]. Moreover, HO-1, which is regulated by Nrf2, is also augmented after T3 treatment in vitro, supporting resistance to oxidative stress and mitochondrial biogenesis [18].
References
- Oetting, A.; Yen, P.M. New insights into thyroid hormone action. Best. Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 193–208.
- Citterio, C.E.; Targovnik, H.M.; Arvan, P. The role of thyroglobulin in thyroid hormonogenesis. Nat. Rev. Endocrinol. 2019, 15, 323–338.
- Cheng, S.-Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170.
- Lazar, M.A. Thyroid hormone action: A binding contract. J. Clin. Investig. 2003, 112, 497–499.
- Brent, G.A. Mechanisms of thyroid hormoneaction. J. Clin. Investig. 2012, 122, 3035–3043.
- Mancini, A.; Di Segni, C.; Raimondo, S.; Olivieri, G.; Silvestrini, A.; Meucci, E.; Currò, D. Thyroid Hormones, Oxidative Stress, and Inflammation. Mediat. Inflamm. 2016, 2016, 6757154.
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553.
- Asayama, K.; Kato, K. Oxidative muscular injury and its relevance to hyperthyroidism. Free Radic. Biol. Med. 1990, 8, 293–303.
- Dekkers, J.C.; van Doornen, L.J.; Kemper, H.C. The role of antioxidant vitamins and enzymes in the prevention of exercise-induced muscle damage. Sports Med. 1996, 21, 213–238.
- Venditti, P.; Di Meo, S. Thyroid hormone-induced oxidative stress. Cell. Mol. Life Sci. 2006, 63, 414–434.
- Romanque, P.; Cornejo, P.; Valdés, S.; Videla, L.A. Thyroid hormone administration induces rat liver Nrf2 activation: Suppression by N-acetylcysteine pretreatment. Thyroid 2011, 21, 655–662.
- Sun, Z.; Huang, Z.; Zhang, D.D. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS ONE 2009, 4, e6588.
- Venediktova, N.; Solomadin, I.; Nikiforova, A.; Starinets, V.; Mironova, G. Functional State of Rat Heart Mitochondria in Experimental Hyperthyroidism. Int. J. Mol. Sci. 2021, 22, 11744.
- Letts, J.A.; Sazanov, L.A. Clarifying the supercomplex: The higher-order organization of the mitochondrial electron transport chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808.
- Lobo-Jarne, T.; Ugalde, C. Respiratory chain supercomplexes: Structures, function and biogenesis. Semin. Cell. Dev. Biol. 2018, 76, 179–190.
- Venediktova, N.I.; Mashchenko, O.V.; Talanov, E.Y.; Belosludtseva, N.V.; Mironova, G.D. Energy metabolism and oxidative status of rat liver mitochondria in conditions of experimentally induced hyperthyroidism. Mitochondrion 2020, 52, 190–196.
- Venediktova, N.I.; Pavlik, L.L.; Belosludtseva, N.V.; Khmil, N.V.; Murzaeva, S.V.; Mironova, G.D. Formation of lamellar bodies in rat liver mitochondria in hyperthyroidism. J. Bioenerg. Biomembr. 2018, 50, 289–295.
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 2008, 103, 1232–1240.
- Mastorci, F.; Sabatino, L.; Vassalle, C.; Pingitore, A. Cardioprotection and Thyroid Hormones in the Clinical Setting of Heart Failure. Front. Endocrinol. 2020, 10, 927.
- Kutala, V.K.; Khan, M.; Angelos, M.G.; Kuppusamy, P. Role of oxygen inpostischemic myocardial injury. Antioxid. Redox Signal. 2007, 9, 1193–1206.
- Hill, M.F.; Singal, P.K. Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am. J. Pathol. 1996, 148, 291–300.
- Huss, J.M.; Kelly, D.P. Mitochondrial energy metabolism in heart failure: A question of balance. J. Clin. Investig. 2005, 115, 547–555.
- Kimur, T.; Kotajima, N.; Kanda, T.; Kuwabara, A.; Fukumura, Y.; Kobayashi, I. Correlation of circulating interleukin-10 with thyroid hormone in acute myocardial infarction. Res. Commun. Mol. Pathol. Pharmacol. 2001, 110, 53–58.
- Pantos, C.; Mourouzis, I.; Saranteas, T.; Brozou, V.; Galanopoulos, G.; Kostopanagiotou, G.; Cokkinos, D.V. Acute T3 treatment protects the heart against ischemia-reperfusion injury via TRα1 receptor. Mol. Cell. Biochem. 2011, 353, 235–241.
- Iervasi, G.; Pingitore, A.; Landi, P.; Raciti, M.; Ripoli, A.; Scarlattini, M.; L’Abbate, A.; Donato, L. Low-T3 syndrome: A strong prognostic predictor of death in patients with heart disease. Circulation 2003, 107, 708–713.
- Pantos, C.; Mourouzis, I.; Markakis, K.; Tsagoulis, N.; Panagiotou, M.; Cokkinos, D.V. Long-term thyroid hormone administration reshapes left ventricular chamber and improves cardiac function after myocardial infarction in rats. Basic Res. Cardiol. 2008, 103, 308–318.
- Pantos, C.; Mourouzis, I.; Tsagoulis, N.; Markakis, K.; Galanopoulos, G.; Roukounakis, N.; Perimenis, P.; Liappas, A.; Cokkinos, D.V. Thyroid hormone at supra-physiological dose optimizes cardiac geometry and improves cardiac function in rats with old myocardial infarction. J. Physiol. Pharmacol. 2009, 60, 49–56.
- de Castro, A.L.; Tavares, A.V.; Campos, C.; Fernandes, R.O.; Siqueira, R.; Conzatti, A.; Bicca, A.M.; Fernandes, T.R.; Sartório, C.L.; Schenkel, P.C.; et al. Cardioprotective effects of thyroid hormones in a rat model of myocardial infarction are associated with oxidative stress reduction. Mol. Cell. Endocrinol. 2014, 391, 22–29.
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207.
- Itoh, K.; Ishii, T.; Wakabayashi, N.; Yamamoto, M. Regulatory mechanisms of cellular response to oxidative stress. Free Radic. Res. 1999, 31, 319–324.
- Zhang, H.; Liu, Y.; Cao, X.; Wang, W.; Cui, X.; Yang, X.; Wang, Y.; Shi, J. Nrf2 Promotes Inflammation in Early Myocardial Ischemia-Reperfusion via Recruitment and Activation of Macrophages. Front. Immunol. 2021, 12, 763760.
- Takemura, G.; Nakagawa, M.; Kanamori, H.; Minatoguchi, S.; Fujiwara, H. Benefits of reperfusion beyond infarct size limitation. Cardiovasc. Res. 2009, 83, 269–276.
- Eefting, F.; Rensing, B.; Wigman, J.; Pannekoek, W.J.; Liu, W.M.; Cramer, M.J.; Lips, D.J.; Doevendans, P.A. Role of apoptosis in reperfusion injury. Cardiovasc. Res. 2004, 61, 414–426.
- Sun, Y. Myocardial repair/remodelling following infarction: Roles of local factors. Cardiovasc. Res. 2009, 81, 482–490.
- Zhou, S.; Sun, W.; Zhang, Z.; Zheng, Y. The role of Nrf2-mediated pathway in cardiac remodeling and heart failure. Oxid. Med. Cell. Longev. 2014, 2014, 260429.
- Xu, B.; Zhang, J.; Strom, J.; Lee, S.; Chen, Q.M. Myocardial ischemic reperfusion induces de novo Nrf2 protein translation. Biochim. Biophys. Acta 2014, 1842, 1638–1647.
- Shanmugam, G.; Narasimhan, M.; Tamowski, S.; Darley-Usmar, V.; Rajasekaran, N.S. Constitutive activation of Nrf2 induces a stable reductive state in the mouse myocardium. Redox Biol. 2017, 12, 937–945.
- Muthusamy, V.R.; Kannan, S.; Sadhaasivam, K.; Gounder, S.S.; Davidson, C.J.; Boeheme, C.; Hoidal, J.R.; Wang, L.; Rajasekaran, N.S. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic. Biol. Med. 2012, 52, 366–376.
- Gutiérrez-Cuevas, J.; Galicia-Moreno, M.; Monroy-Ramírez, H.C.; Sandoval-Rodriguez, A.; García-Bañuelos, J.; Santos, A.; Armendariz-Borunda, J. The Role of NRF2 in Obesity-Associated Cardiovascular Risk Factors. Antioxidants 2022, 11, 235.
- Loboda, A.; Jazwa, A.; Grochot-Przeczek, A.; Rutkowski, A.J.; Cisowski, J.; Agarwal, A.; Jozkowicz, A.; Dulak, J. Heme oxygenase-1 and the vascular bed: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 1767–1812.
- Das, A.; Gopalakrishnan, B.; Voss, O.H.; Doseff, A.I.; Villamena, F.A. Inhibition of ROS-induced apoptosis in endothelial cells by nitrone spin traps via induction of phase II enzymes and suppression of mitochondria-dependent pro-apoptotic signaling. Biochem. Pharmacol. 2012, 84, 486–497.
- Zeng, B.; Liu, L.; Liao, X.; Zhang, C.; Ruan, H. Thyroid hormone protects cardiomyocytes from H2O2-induced oxidative stress via the PI3K-AKT signaling pathway. Exp. Cell Res. 2019, 380, 205–215.
- Rota, M.; Boni, A.; Urbanek, K.; Padin-Iruegas, M.E.; Kajstura, T.J.; Fiore, G.; Kubo, H.; Sonnenblick, E.H.; Musso, E.; Houser, S.R.; et al. Nuclear targeting of Akt enhances ventricular function and myocyte contractility. Circ. Res. 2005, 97, 1332–1341.
- Mourouzis, I.; Mantzouratou, P.; Galanopoulos, G.; Kostakou, E.; Roukounakis, N.; Kokkinos, A.D.; Pantos, C. Dose-dependent effects of thyroid hormone on post-ischemic cardiac performance: Potential involvement of Akt and ERK signalings. Mol. Cell. Biochem. 2012, 363, 235–243.
More
Information
Subjects:
Endocrinology & Metabolism
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Revisions:
2 times
(View History)
Update Date:
16 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No