Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vittoriano Della Corte | -- | 5436 | 2023-06-07 12:48:44 | | | |
| 2 | Alfred Zheng | Meta information modification | 5436 | 2023-06-08 04:51:26 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Della Corte, V.; Pacinella, G.; Todaro, F.; Pecoraro, R.; Tuttolomondo, A. Physiologic Effects of Natriuretic Peptides. Encyclopedia. Available online: https://encyclopedia.pub/entry/45289 (accessed on 10 August 2026).
Della Corte V, Pacinella G, Todaro F, Pecoraro R, Tuttolomondo A. Physiologic Effects of Natriuretic Peptides. Encyclopedia. Available at: https://encyclopedia.pub/entry/45289. Accessed August 10, 2026.
Della Corte, Vittoriano, Gaetano Pacinella, Federica Todaro, Rosaria Pecoraro, Antonino Tuttolomondo. "Physiologic Effects of Natriuretic Peptides" Encyclopedia, https://encyclopedia.pub/entry/45289 (accessed August 10, 2026).
Della Corte, V., Pacinella, G., Todaro, F., Pecoraro, R., & Tuttolomondo, A. (2023, June 07). Physiologic Effects of Natriuretic Peptides. In Encyclopedia. https://encyclopedia.pub/entry/45289
Della Corte, Vittoriano, et al. "Physiologic Effects of Natriuretic Peptides." Encyclopedia. Web. 07 June, 2023.
Copy Citation
Natriuretic peptides are a complex and interesting network of molecules playing pleiotropic effects on many organs and tissues, ensuring the maintenance of homeostasis mainly in the cardiovascular system and regulating the water–salt balance. Natriuretic peptides (NPs) and their receptors play several physiological roles in maintaining and ensuring homeostasis in the human body by performing pleiotropic actions on multiple organs and target sites. Various other mechanisms have been characterized in other organs, which were made possible by the specific and individual interactions between each natriuretic peptide and the corresponding receptor in a particular site of action.
natriuretic peptide
cardiovascular system
nervous system
musculoskeletal system
1. Introduction
The family of natriuretic peptides (NPs) represents a group of peptides mainly characterized by the amino acid structure consisting of a peptide ring with a circular structure and by activating the same family of receptors with guanylyl cyclase activity [1][2][3]. In addition to the well-known atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP) in the cardiovascular field, it can include other members, some of which have been known for some time, such as C-type natriuretic peptide (CNP) and urodilatin (URO), and others of more recent discovery such as dendroaspis natriuretic peptide (DNP) and guanylin peptides (GPs) [1][2][3]. NPs are represented in multiple organs and tissues, and the actions they mediate therefore involve different systems. This is the first literature review which, in addition to reporting the main characteristics of the well-known NPs, has also included other family members such as urodilatin, DNP, and guanylin peptides, as well as mentioning the most recent discoveries in this field concerning linear fragments derived from NPs and positive allosteric modulators of the NPs receptors.
2. Physiologic Effects of Natriuretic Peptides
What has been discussed so far suggests that NPs and their receptors play several physiological roles in maintaining and ensuring homeostasis in the human body by performing pleiotropic actions on multiple organs and target sites (Figure 1). The well-known effects of NPs are at the level of the cardiovascular system and bone tissue. Still, over the years, various other mechanisms have been characterized in other organs, which were made possible by the specific and individual interactions between each natriuretic peptide and the corresponding receptor in a particular site of action (Figure 2).
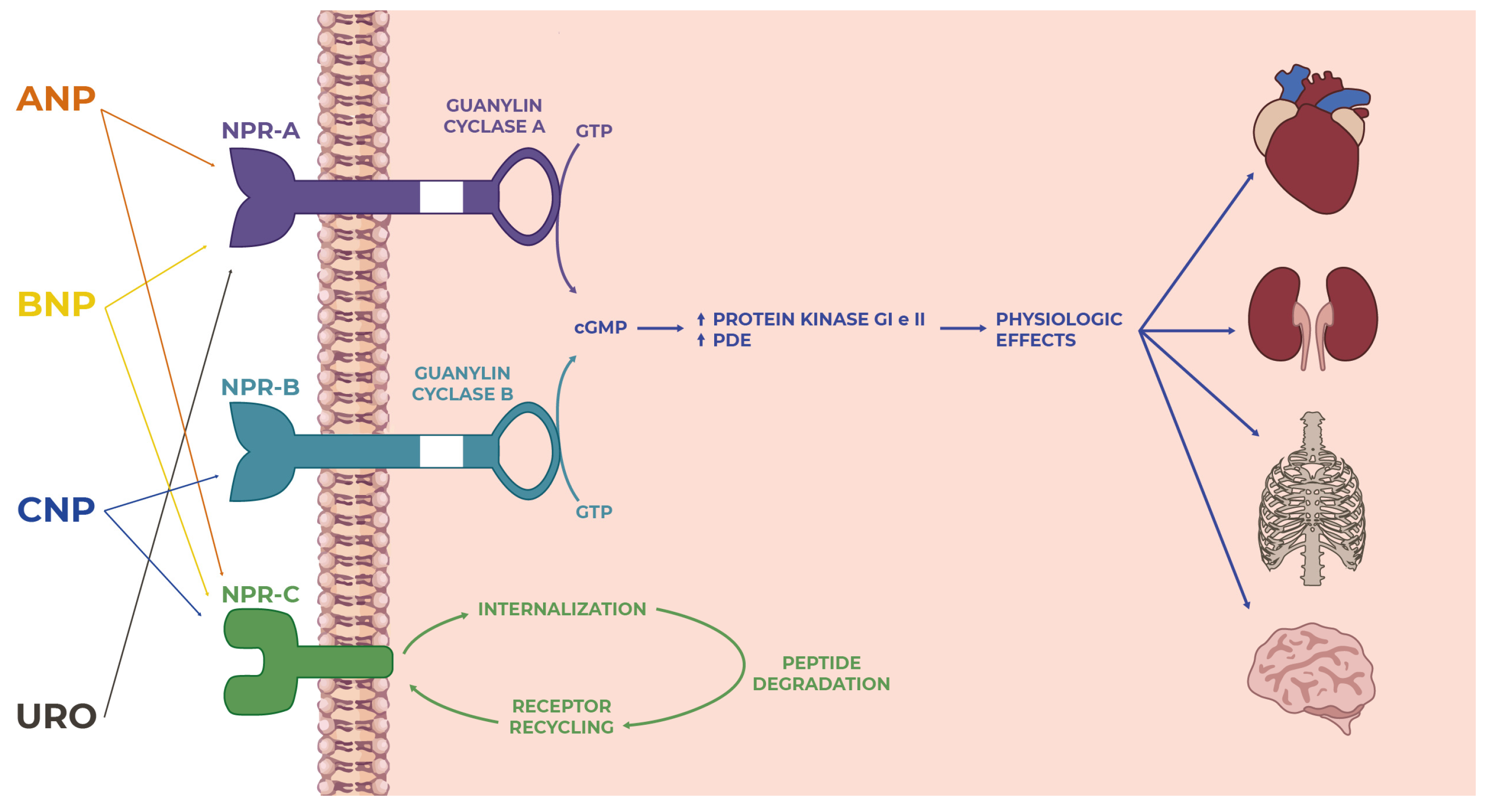
Figure 1. Overview of the natriuretic peptide family: from ligand–receptor interactions to intracellular signaling to actions on targets in many tissues and organs where they perform several functions.
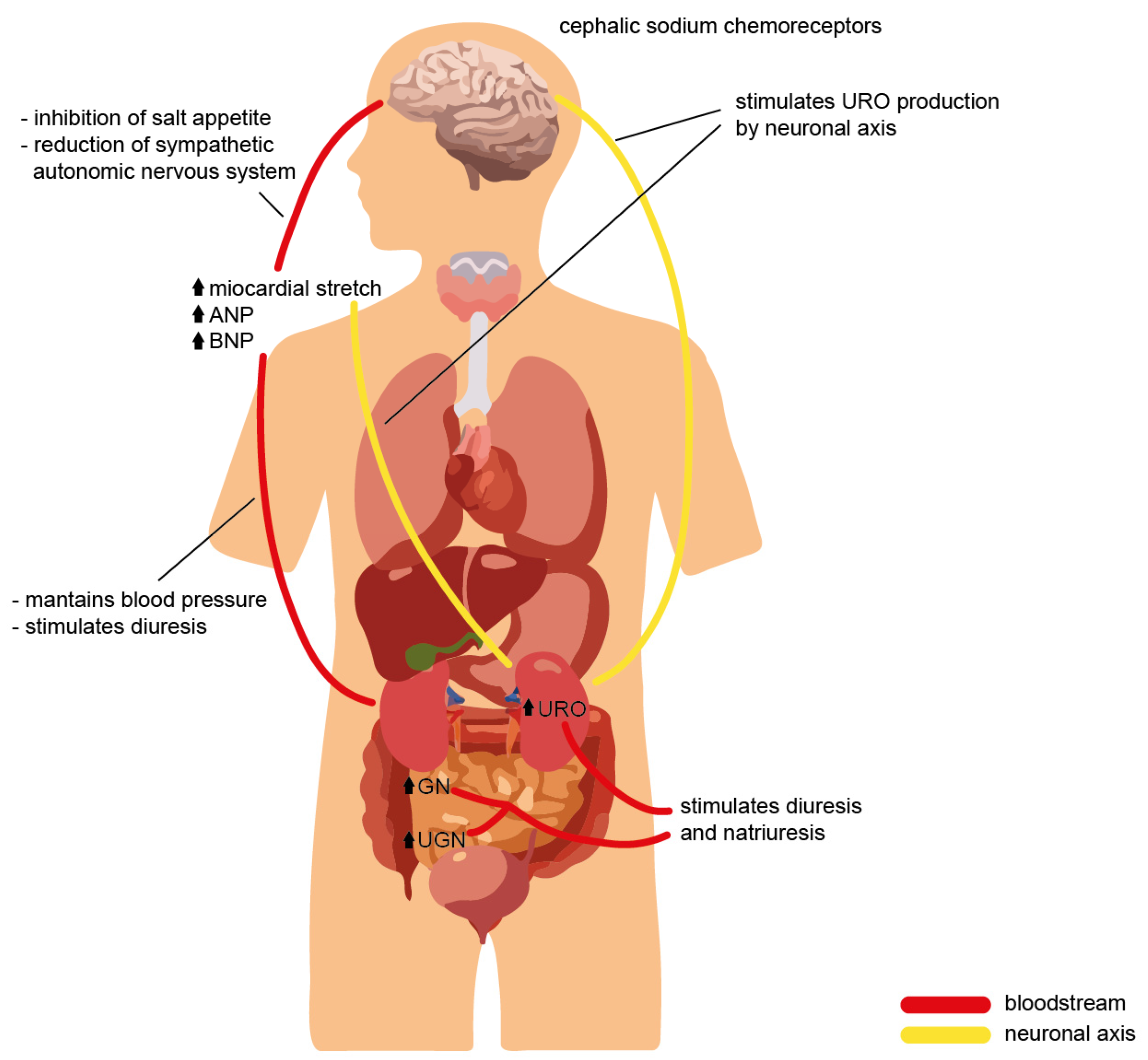
Figure 2. Cross-talk of the natriuretic peptide family during volume overload, with evidence of the effects on different organs and the physiological role required to ensure homeostasis.
Knowledge of the physiological effects of NPs has been a cornerstone for modern medicine, as it has made it possible, conversely, to identify the role played by these actors in the pathogenesis of cardiovascular disease and heart failure: dysregulation of the complex natriuretic peptide machinery is crucial in the onset of the diseases described above, so much so that in laboratory diagnostics they play an ever-increasing role.
2.1. Natriuretic Peptide Effects on Cardiovascular System
The action of NPs on the cardiovascular system has been known for several years: the NPR-A, via activation by its endogenous ligands ANP and BNP, possesses beneficial biological properties such as blood pressure regulation, natriuresis, suppression of adverse remodeling, and inhibition of the renin–angiotensin–aldosterone system, with ANP representing the most biologically active of the two cardiac-derived peptides in activating the NPR-A [4].
The interaction of ANP with NPR-A promotes the activation of the homeostatic regulation of blood pressure, such that in laboratory mice lacking ANP or with suppression of its receptor, a 20–40 mmHg increased blood pressure was observed compared with control mice [5]. In contrast, blood pressure was found to be significantly reduced in transgenic mice with an overexpression of ANP and BNP [6].
It appears that not all of the NPs play a decisive role in blood pressure regulation: infusion of CNP results in acute reductions in blood pressure values, but in mice with a deletion of the CNP gene or the NPR-B receptor, no alterations in blood pressure regulation were reported, thus confirming that the CNP/NPR-B complex is not diriment for blood pressure control [7].
The effects on blood pressure regulation mediated by NPR-A depend on multiple mechanisms: increased urinary sodium excretion and consequent increase in diuresis, counteracting renin–angiotensin–aldosterone system (RAAS) activity, vasodilation, and increased endothelial permeability. The understanding of these processes was derived from the observation that animals undergoing atrial extract infusion incurred rapid elimination of water and sodium; in more detail, the researchers understood that the presence of NPR-A in addition to ANP was necessary to achieve the pressure-lowering effect because, in mice with deletion of the NPR-A gene, infusion of large amounts of ANP or BNP or sudden volemic expansion did not cause any natriuretic and/or diuretic responses [8]. In addition to its diuretic and natriuretic actions, NPR-A also mediates a vasodilatory process that, in acute conditions, permits the regulation of blood pressure values: infusion of ANP and BNP in laboratory animals with reduced NPR-A gene expression at the level of vessel smooth muscle does not result in the vasodilatory response that was observed under physiological conditions. However, in chronic blood pressure regulation, this process does not play such a significant role [9]. Regarding the anti-RAAS activity, Cataliotti et al. showed that in congestive heart failure patients, furosemide and BNP had favorable cardiovascular hemodynamic actions compared with furosemide alone, probably because the suppression of the RAAS may have prevented the anti-natriuretic actions of angiotensin II on the proximal tubule and of aldosterone on the distal tubule [10]. Moreover, Siragy et al. showed that in uninephrectomized conscious dogs, intrarenal ANP prevents the angiotensin II-induced decrement urinary sodium excretion and urine flow rates, thereby highlighting how ANP may play an important role in the escape from the sodium-retaining action of intrarenal angiotensin II [11].
The role of NPs in the pathogenesis of arterial hypertension has been described by interesting works. Belluardo et al. determined the levels of BNP and NT-proBNP in subjects with mild, moderate, and severe hypertension comparing them with healthy subjects. They demonstrated how in subjects with mild hypertension, BNP was unchanged while NT-proBNP was found to be significantly reduced compared with the controls. As severity increased in humans with mild-to-severe hypertension, both BNP and NT-proBNP levels were increased while not being affected by the presence of left ventricular hypertrophy. The lack of activation of BNP, together with the reduction of NT-proBNP in mild hypertension, may represent an impaired response of the BNP system in the early phase of hypertension. The later activation of both forms of BNP may be a late compensatory effect, as it correlates with the severity of hypertension rather than cardiac hypertrophy [12]. Furthermore, Macheret et al. demonstrated the existence of an impaired production and/or release of Nt-proBNP along with a concomitant reduction of BNP and NT-proBNP in the early stages of hypertension, with a significant elevation observed only in stage 2 hypertension. Moreover, they simultaneously demonstrated a lack of compensatory ANP elevation in advanced hypertension [13].
In consideration of the evidence such as those just mentioned, the efforts of the researchers are concentrated precisely on the NP system to find a new and more effective treatment of arterial hypertension. Since ANP is the most relevant component of the family that is able to modulate blood pressure and contribute to hypertension development, it is therefore expected that ANP-based therapeutic approaches may give a significant contribution to the development of efficacious therapies against hypertension. Since native ANP cannot be administered due to its short half-life, several approaches were attempted over the years to overcome the difficulties inherent to ANP instability. These approaches included ANP recombinant and fusion peptides, inhibition of ANP degradation by neprilysin inhibition, gene therapy, and designer peptides. For example, Chen et al. showed the BP-lowering properties of a novel ANP-based compound (MANP [mutant ANP]) in humans. MANP is an ANP mimetic retaining the 28 amino acids of ANP but including a novel 12 amino acid extension to the carboxy-terminal end. The latter confers a greater resistance to enzymatic degradation by both NEP (neprilysin) and IDE (insulin-degrading enzyme) and reduces removal by NP clearance receptors. The study by Chen et al. demonstrated the natriuretic and aldosterone suppression properties of MANP, as well as a good degree of tolerability, and a long lasting and dose-dependent BP-lowering effect, with no relevant side effects during the 24 h of observation [14].
Speaking of endothelial permeability, the ANP/NPR-A system plays an essential function with a direct impact on the regulation of circulating blood volume: in murine models, the decreased endothelial expression of the NPR-A gene induced by genetic engineering techniques, led to hypertension, increased plasma volume, and reduced clearance of albumin from the vascular tree [15].
In the cardiovascular system, NPs also have the role of regulating myocardial hypertrophy and cardiac remodeling by inhibiting them; on this point, it is essential to specify that hypertrophy and remodeling are not the immediate and direct responses of exposure to high blood pressure, rather they constitute the results of specific alterations in the molecular pathways of which the NPs are vital players.
The ANP/NPR-A system regulates myocardial hypertrophy, whereas both ANP/BNP/NPR-A and CNP/NPR-B contribute to cardiac remodeling. Although long-standing hypertension can promote the onset of myocardial hypertrophy, as mentioned above, the latter is mainly dependent on the ANP/NPR-A system. In mice deficient for NPR-A, hypertrophy is much more pronounced than in the controls with regular receptor gene expression, suggesting that the absence of NPR-A plays a role in promoting the processes that trigger it. Confirming the above, NPR-A knockout mice underwent cardiac enlargement even when treated with antihypertensive drugs from birth [16]. Again, mice with reduced expression of NPR-A at the level of cardiomyocytes developed moderate myocardial hypertrophy even though they were moderately hypotensive [17].
The ANP/NPR-A interaction oversees the control of myocardial hypertrophy as the scientific literature has extensively documented; the BNP/NPR-A interaction regulates the mechanism of fibrosis: deletion of the BNP gene results in normal pressure values and regular cardiac volumes in mice, but can also generate ventricular fibrosis especially if the animal has received pressure overload [18].
In NPR-A knockout mice, echocardiographic and histologic findings showed hypertrophy and interstitial fibrosis; these effects, however, were more attenuated in mice with a simultaneous deletion of the gene of angiotensin II receptor 1A (AT1A receptor), or in which an AT1a receptor antagonist was administered. In mice with an exclusive deletion of NPR-A, administration of antihypertensives such as 6-hydroxydopamine resulted in blood pressure values that were comparable to those of mice with a blockade of the AT1A receptor (through either gene deletion or pharmacological inhibition), but did not result in any effects relating to limiting fibrosis and myocardial hypertrophy as NPR-A, as mentioned above, directly regulates hypertrophy and fibrosis independently of blood pressure [19]. These data show that the response of the cardiomyocytes of NPR-A knockout mice to angiotensin II is enhanced, and results in hypertrophy and interstitial fibrosis, again confirming the role of NPR-A as an inhibitor of the pathological processes of myocardial hypertrophy and cardiac interstitial fibrosis.
More specifically, to establish the role of NPR-A in the mechanisms of fibrosis and hypertrophy, scientists have previously crossed transgenic mice that overexpressed NPR-A with NPR-A knockout mice, obtaining mice that expressed NPR-A in the heart: these animals had similar blood pressure and heart rates to the NPR-A knockout mice, but had smaller cardiomyocytes; consequently, the levels of ANP and its mRNA at the cardiac and systemic levels were lower due to the expression of NPR-A than in the NPR-A knockout mice [20]. Conversely, in mice with a selective deletion of the cardiomyocyte NPR-A gene, serum ANP levels were found to be higher than in the wild-type mice. Moreover, blood pressure was 7–10 mmHg lower due to the NPs’ effects on both renal and vascular districts. Still, these animals’ cardiomyocyte response to pressure overload resulted in significant hypertrophy and systolic dysfunction [21].
In addition to cardiomyocytes, in the heart, NPs act on fibroblasts: the activation of NPR-A and NPR-B causes an increase in cGMP production, limits the Ang II-induced increase in preproendothelin-1 expression, and restricts the proliferation of fibroblasts themselves; by regulating the growth and proliferation of these cells, it is clear how the natriuretic peptide system governs the cardiac fibrosis pathway [22].
Additionally, the relationship between ANP and cardiac mass previously observed in animal models has also confirmed in humans: reduced circulating levels of ANP are associated with more pronounced cardiac hypertrophy in subjects with essential hypertension. Indeed, in subjects with an allelic variant of the ANP gene promoter in which the serum levels of this natriuretic peptide were reduced, hypertrophy and cardiac mass were found to be at greater levels [23]. Furthermore, in diseases such as obesity and metabolic syndrome, ANP levels are lower than in healthy individuals, probably due to both increased clearance and reduced synthesis; based on the above, an inverse–proportionality relationship between ANP levels and cardiac mass has been reported in these subjects, with pronounced myocardial hypertrophy [24]. Interestingly Cataliotti et al. showed that in patients with end-stage renal disease, elevation of the plasma BNP concentration is more specifically related to left ventricular hypertrophy compared with the other NP levels independent of congestive heart failure [25]. Similarly, Zoccali et al. found that in dialysis patients, BNP and ANP were the strongest independent correlates of the left ventricular mass index, independent predictors of ejection fraction, and predicted overall and cardiovascular mortality [26]. Based on this evidence, Cataliotti et al. used the myocardium-tropic adeno-associated virus serotype 9 (AAV9) vector to achieve continuously enhanced cardiac rat proBNP expression. In spontaneously hypertensive rats, a single systemic administration of the AAV9 vector allowed long-term cardiac BNP overexpression, resulting in reductions in both systolic and diastolic BP for 9 months after injection, proving that the long-term BNP gene delivery prevented the development of hypertensive heart disease in this population of rats [27].
In conclusion, activation of NPR-A by ANP blocks myocardial hypertrophy, whereas activation of the receptor by BNP inhibits fibrosis.
The CNP/NPR-B complex, on the other hand, appears to play a role in cardiac remodeling. In mice with induced cardiac ischemia, CNP infusion blocked postischemic remodeling, thereby inhibiting the cascade of events that drive cardiac enlargement and the onset of congestive heart failure [28].
Furthermore, in CNP knockout mice subjected to pressure overload by abdominal aortic constriction, a series of detrimental cardiac changes were observed, such as LV dilatation, a reduction in ejection fraction, and increased hypertrophy and fibrosis. The same structural and functional alterations resulted in NPR-C knockout mice, suggesting that this receptor is actively implicated in the beneficial effects mediated by CNP. Moreover, a recent study suggested the role of CNP/NPR-C pathway enhancements in improving HF [29].
Concerning vascular effects, in vitro studies had previously documented that ANP can promote angiogenesis and endothelial cell proliferation [30].
ANP binding to NPR-A is a key signaling pathway regulating normal homeostatic blood pressure. This was previously demonstrated in mice lacking ANP or its receptor NPR-A, which were found to have blood pressures that were elevated 20–40 mmHg, compared to control mice [31]. The link between NPR-A and blood pressure in mice is particularly strong, as Smithies and colleagues demonstrated that NPR-A copy number is inversely related to blood pressure in a remarkably linear manner [32]. Conversely, blood pressures in transgenic mice overexpressing ANP or BNP were substantially decreased [6]. Although the infusion of supraphysiological levels of CNP into animals acutely reduced blood pressure [33], mice lacking functional CNP or NPR-B are normotensive [7], suggesting that the CNP/NPR-B pathway is not a fundamental regulator of basal blood pressure in mice. NPR-A-dependent decreases in blood pressure are achieved through natriuresis and diuresis, vasorelaxation, increased endothelium permeability, and antagonism of the renin–angiotensin system [1].
It should indeed be noted that many of the effects described so far can be traced back to in vitro trials, and that the reality of the human body is quite different and much more articulated; the insights into the role of NPs are significant, but more information needs to be gained from in vivo trials to understand what occurs under physiological conditions and, even more interestingly, in the pathological conditions on each target district.
2.2. Natriuretic Peptide Effects on the Nervous System
While NPs were initially discovered in cardiac myocytes, the extensive distribution of NPs and their receptors in the brain of animal species have been repeatedly reported. NPs and their receptors were shown to be present across various neuronal structures, glial cells, and cerebral vessels. Notably, findings from animal studies suggest that NP may regulate neuroplasticity, blood-brain barrier integrity, neuro-inflammation, and memory function [34].
The complete understanding of the effects of NPs certainly also comes from assessing the distribution of receptors in different areas of the brain and the specific functions of these receptors: NPR-C is well represented in all areas of the central nervous system since its action here is mainly to block glial cell growth and proliferation [35].
NPR-A, on the other hand, is mainly located in the areas close to the third ventricle of the brain, which are not isolated from the peripheral circulation by the blood-brain barrier: this allows the receptors to bind to NPs that were produced in the central nervous system and those produced peripherally; this receptor, unlike the previous one, primarily mediates the sense of water drinking and salt appetite, with direct and preferential effects on blood pressure [36].
NPR-B is distributed mainly in the hypothalamus and other rostral regions of the brain, with regulatory effects on the sympathetic nervous system and vegetative functions.
Therefore, people have observed how NPs are produced and act at the level of the CNS. As far as the circulating NPs are concerned, although they cannot cross the blood-brain barrier (BBB), they exert their action on selected sites in the central nervous system by reaching targets outside the BBB: the median eminence of the hypothalamus or the area postrema. The NPs ANP, BNP, and CNP are produced in the brain: biologically active substances that regulate blood pressure and vasoactive amines such as endothelin, noradrenaline, and vasopressin (not angiotensin II), which induce the production of these molecules in hypothalamic neuron cultures [37].
The effects of NPs at the brain level are complementary to those on other targets, with a reinforcing impact on the latter: the inhibition of salt appetite and stimulation of the sense of thirst stimulate natriuretic and diuretic actions at the kidney level [38]. In addition, other reports show that NPs exhibit a central effect by limiting the production and release of vasopressin with direct actions on the hypothalamic nuclei and, in other cases slowing down ACTH production by acting on the pituitary gland, and thus modulating all the regulatory effects of blood pressure and circulating blood flow downstream [39].
Even more interestingly, NPs regulate the tone of the sympathetic autonomic nervous system in a negative direction, thus promoting a reduction in blood pressure. Moreover, in mouse models with genetic forms of essential hypertension, blocking the action of NPs on the nucleus of the solitary tract further increases the pressure values, thus suggesting a hypotensive effect at this site [40].
All of the studies cited so far have been conducted on animals. However, there is also evidence available showing the involvement of NPs in the human brain. For example, van der Weerd et al. showed that both NP and NPR genes are ubiquitously expressed throughout the brain in healthy humans. Furthermore, NP and NPR are present in various cellular structures, including neurons, astrocyte-like networks, and cerebral vessels. The abundance of NP and NPR in human brains suggests NP involvement across multiple brain functions. Moreover, human evidence indicates that higher plasma levels of NPs are associated with dementia and accelerated cognitive decline. Although this link was mainly attributed to cardiovascular pathologies, recent findings suggest plasma NPs are associated with cognitive decline independent of cardiovascular disease [34].
A neuroprotective effect of NPs seems to be related to cGMP. As people have also seen, NPRs are guanylate cyclase receptors, and numerous pieces of evidence show how cGMP, regardless of its source, plays a vital role in brain physiology. Its actions are significant regarding modulations of long-term synaptic activity changes in the hippocampus, amygdala, cerebellum (by pre-synaptic transmitter release), and post-synaptic functions via activation of different protein kinase G (PKG) isoforms. Recent studies have demonstrated a neuroprotective role of NPs and the cGMP signaling pathway. An increase in intracellular cGMP concentration protects neurons against excitotoxic, metabolic, as well as oxidative damage and N-methyl d-aspartate (NMDA)-induced neurotoxicity [41].
All this evidence suggests a possible role of NPs in the function and metabolism of nervous tissue and as a possible link between the kidneys, cardiovascular, and nervous systems. Further studies will be needed to clarify all these aspects.
2.3. Natriuretic Peptide Effects on Renal System
The natriuretic effect of NPs occurs through their flow-regulating actions at the renal level and through a direct tubular effect. The mechanism of natriuresis, once triggered, lasts longer than changes in renal hemodynamics. This is because the two effects are distinct and differentially regulated.
NPs promote the contraction of the efferent arteriole and vasodilation of the afferent arteriole, thereby generating increased blood flow in glomerular capillaries and consequently increased glomerular filtration [42]. Moreover, at the mesangial level, activation of NPs receptors promotes, through increased cGMP concentration, relaxation of mesangial cells with an increased available filtration surface area [43].
Several studies have shown that plasma concentrations of NPs not capable of increasing the glomerular filtration rate are, however, able to bring about a natriuretic effect through direct actions at the level of the renal tubules: urodilatin is an example of a natriuretic peptide produced at this site that acts by a paracrine mechanism, complementary to the systemic effects of the other NPs that have already described [44].
The complexity of the renal action of NPs is mainly due to the variety of targets at which they act: at the level of the proximal convoluted tubule NPs block angiotensin II-dependent sodium and water transport [45], at cortical collecting ducts inhibit the vasopressin-dependent transit of water [46], and at the level of the medullary collecting ducts inhibit sodium reabsorption through increased cGMP production [47].
Practical applications of this knowledge have made it possible to observe that the infusion of ANP and BNP in humans at doses slightly higher than the physiological plasma concentrations of these NPs causes increases in both diuresis and natriuresis independently of changes in blood pressure. Moreover, in the trials in which it was assessed, the result was a reduction in renin concentration and angiotensin II-mediated aldosterone secretion [48].
CNP similarly reduces aldosterone secretion, but its action on blood pressure and water and sodium excretion is lower than with other NPs [49].
People have already seen how CNP is present in both the genitourinary and renal tracts [50].
Particularly, the presence of both the peptide and its receptor in human kidneys suggests a role for CNP as a renal autocrine and/or paracrine factor. To date, however, the pathophysiological role of CNP in the human kidney remain unclear. In anesthetized rats and normal humans, CNP has been shown to be mildly diuretic and natriuretic, although other reports have demonstrated that intrarenal arterial administration of CNP has no effect on the urine volume or urine sodium excretion [51]. With this in mind, Cataliotti et al. assessed plasma CNP concentrations and urinary CNP excretion in patients with nephrotic syndrome. First of all, they confirmed the production of CNP in normal human kidneys and its localization by both in situ hybridization and immunohistochemistry [51].
Moreover, they found increased plasma CNP and urinary CNP excretion in nephrotic syndrome, the latter being significantly reduced by the low-protein diet, whereas plasma CNP remained unchanged. These findings demonstrate that CNP metabolism is altered in nephrotic syndrome, and supports the hypothesis that the increase in intrarenal release and production of CNP can be partially offset by a restriction in protein intake [51].
Despite the demonstrated diuretic and natriuretic effects of ANP and BNP, the first indications that these NPs may not be the physiological regulators of Na + excretion arise from a study in which the left atrial pressure was elevated in two groups of dogs, one normal and the other with denervated hearts. Elevated atrial pressure increased circulating ANP in similar amounts in each group of dogs. However, only normal dogs developed increased diuresis and natriuresis during these experiments [52], indicating that the renal response induced by atrial distension required intact cardiac nerves and not just the release of ANP. A study evaluating the circadian rhythm of ANP in plasma found no relationship between blood sodium and circulating ANP levels [53].
Urodilatin, the only natriuretic peptide produced in the kidney, induces the diuretic and natriuretic effects at lower doses than those required for the other NPs, most likely due to a greater resistance to the activity of endogenous endopeptidases; this feature could make the use of this molecule more convenient than the other NPs for therapeutic purposes in clinical practice, beyond only trials conducted for scientific research [54].
After isolating the urodilatin natriuretic peptide from human urine, more data have been generated supporting the view that neither ANP or BNP, but urodilatin appears to be the natriuretic peptide responsible for renal manipulation of sodium. Drummer and colleagues found it interesting that not ANP in plasma but urodilatin in urine was found to be correlated closely with natriuretic responses observed following acute saline infusion. The same group demonstrated that natriuresis following the ingestion of meals with different salt concentrations was accompanied by an increase in urodilatin excretion rather than an increase in circulating ANP [55]. In addition, a negative correlation was observed between plasma ANP and renal sodium excretion during left atrial distension in dogs with cardiac denervation [56]. This body of evidence suggests that it is urodilatin, rather than the other NPs that is primarily involved in the physiological regulation of renal sodium excretion. On the contrary, due to the rapid secretion of ANP and BNP as a response to several cardiovascular stimuli, and due to the numerous effects on the cardiovascular system, it seems reasonable to postulate that the primary target of ANP and BNP is the cardiovascular system and not the kidneys.
The importance of renal effects, and possible therapeutic implications related to the use of NPs, emerged from a trial that evaluated the effects in animal models, associated with the administration of a natriuretic peptide antagonist on A and B receptors termed HS-142-1 [57]: in both healthy animals and animals in which heart failure was experimentally induced, the drug was able to inhibit diuresis and natriuretic peptide-mediated natriuresis, elevate vascular resistance in the renal circulation, and increase the concentration of renin, aldosterone, and catecholamines [58]. In addition, in cirrhotic rats with ascites and diabetic rats, this drug caused a reduction in glomerular filtration by reducing blood flow to the kidney: this suggests that disruption of the natriuretic peptide system may play a role in the onset of these diseases [59].
Finally, as it have already reported, GN and UGN could represent an endocrine axis between the intestine and kidney, stimulating natriuresis and diuresis in response to a salty meal.
2.4. Natriuretic Peptide Effects on Musculoskeletal System
NPs also have a function on bone growth, long thought to be minor but now increasingly better characterized: experiments carried out on animal models using transgenic or knockout mice have made it possible to describe the role of NPs, their receptors, and the intracellular molecular signaling pathways they trigger. The precondition for understanding the role played by NPs on bone growth is a brief description of the mechanisms of embryogenesis and the development of bone segments.
In vertebrates, bone genesis occurs through two mechanisms. The first is that of “intramembranous ossification” (which, for example, concerns the flat bones of the skull) in which mesenchymal cells undergo a process of differentiation into osteoblasts that begin to produce bone matrix from ossification centers; there is no strong evidence from studies to date that suggest NPs play an active role in this mechanism specifically. The other mechanism is “endochondral ossification” and concerns the genesis of the long bones of the skeleton and vertebrae. In this process, mesenchymal cells group to form the primordial bone, at the center of which some of these cells differentiate into chondrocytes, which, through specific molecular regulatory pathways, undergo proliferation, hypertrophy, calcification, and finally apoptosis to be ultimately replaced by bone marrow [60].
Surrounding this central zone, mesenchymal cells differentiate into perichondrium cells, becoming osteoblasts and replacing part of the cartilage with bone tissue. In the future development and homeostasis of the bone in question, a key role is played by the chondrocytes, also derived from the differentiation of mesenchymal cells: as the bone develops, the chondrocytes in the central zone differentiate into hypertrophic chondrocytes, and the hypertrophic chondrocytes that have aged further in the periphery contribute to the mineralization of the bone by undergoing apoptosis; therefore, the maintenance of bone is ensured by the dynamism of these cell populations [60].
In the regulation of these mechanisms, several molecules play a crucial role, including Indian hedgehog (IHH), parathyroid hormone-related peptide (PTHrP), and members of the bone morphogenetic protein (BMP) family. Chondrocytes produce IHH and promote these cells’ differentiation towards later maturational stages, while PTHrP is expressed in the joint perichondrium and acts on proliferating and prehypertrophic chondrocytes [61][62].
Through genetic engineering techniques, transgenic or knockout mice were constituted in various animal models to study the role of each of these intracellular signaling pathways. It was found that interference with several of these molecular pathways results in profound and significant alterations in bone growth and development [63][64]
The role of NPs in bone growth and development began to be understood in 1988 when ANP was seen to inhibit DNA synthesis in epiphyseal plate cell cultures in avian tibiae, including through inhibiting the PTHrP-mediated pathway [65].
Later, Hagiwara et al. demonstrated that in mouse chondrocyte cultures, NPR-B was expressed, and that its most characteristic ligand (CNP) was found to significantly inhibit DNA synthesis [66]. Despite what in vitro studies have shown, namely that NPs acting on their receptors limited bone growth, transgenic mouse models that overexpressed BNP at the liver revealed disproportionate bone growth [67]. The researchers, in this case, hypothesized that BNP acts on NPR-B by promoting bone growth, but less intensively than CNP, which in vivo stimulates and promotes bone growth and skeletal development.
Further evidence for the role of stimulation on bone growth in vivo was derived from animal models in which a mutation had been chemically induced in the NPR-C gene or animals with a deletion of this gene, in which skeletal overgrowth was observed [68][69]. As the main role of this receptor consists of cleaving NPs, the defects in NPR-C function observed in these animals caused an excess of NPs in bone and accounted for their excessive growth.
In CNP knockout mice who developed a form of dwarfism in which chondrocytes of the proliferative and hypertrophic zone were less represented, inducing the transgenic expression of CNP at the chondrocyte level corrected this bone growth defect, indicating a paracrine action of CNP at the bone tissue level as well [70].
Selective deletions of ANP, BNP, and NPR-A genes do not result in skeletal defects, proving that the CNP/NPR-B system plays a crucial role in bone growth and homeostasis.
Another critical protagonist in this context is musclin, also termed osteocrin (OSTN), as it was initially identified as a secretory peptide from muscle and bone [71]. The preprocessed mature form of mouse OSTN consists of 130 amino acids. Its carboxy terminus contains tandem NP-like sequences separated by polybasic amino acids, presumably cleaved by peptidases. Therefore, musclin can be considered a member of the NP family without the “ring” that is the common feature of ANP, BNP, and CNP, and is mainly produced by skeletal muscles and osteoblasts [72].
In skeletal muscle, musclin improves insulin-dependent glucose metabolism and enhances physical endurance by promoting mitochondrial biogenesis through NP-induced cGMP production due to NPR-C blockade [73]. The recent significant discovery made was that this peptide binds NPR-C competitively and efficaciously inhibits NPR-C-mediated NP degradation, thereby increasing NP levels, with a consequent reduction in BP and enhanced protective activities in many tissues, including the heart. Musclin was found to attenuate cardiac dysfunction and myocardial fibrosis by augmenting the CNP/NPR-B-stimulated cross-talk of cGMP and cAMP in cardiomyocytes, and by inhibiting p38 MAP kinase (MAPK) signaling through the activation of PKG in cardiac fibroblasts. Furthermore, musclin mRNA levels in skeletal muscle were increased by physical activity and, on the contrary, were markedly downregulated in biopsies from patients suffering from HF with sarcopenia or cachexia [74]. This evidence suggests that musclin could act as a possible bridge between sarcopenia and cachexia, which are highly prevalent in advanced states of HF [75], as well as the progression of HF itself. Furthermore, it could represent the biological basis of the vicious circle between reduced physical activity, decreased muscle mass, and HF in older patients [29].
References
- Potter, L.R.; Yoder, A.R.; Flora, D.R.; Antos, L.K.; Dickey, D.M. Natriuretic peptides: Their structures, receptors, physiologic functions and therapeutic applications. In Handbook of Experimental Pharmacology; Schmidt, H.H.H.W., Hofmann, F., Stasch, J.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 191, pp. 341–366. ISBN 9783540689607.
- Sinđić, A.; Schlatter, E. Mechanisms of action of uroguanylin and guanylin and their role in salt handling. Nephrol. Dial. Transplant. 2006, 21, 3007–3012.
- Richards, A.M.; Lainchbury, J.G.; Nicholls, M.G.; Cameron, A.V.; Yandle, T.G. Dendroaspis natriuretic peptide: Endogenous or dubious? Lancet 2002, 359, 5–6.
- Sangaralingham, S.J.; Kuhn, M.; Cannone, V.; Chen, H.H.; Burnett, J.C. Natriuretic peptide pathways in heart failure: Further therapeutic possibilities. Cardiovasc. Res. 2023, 118, 3416–3433.
- John, S.W.M.; Krege, J.H.; Oliver, P.M.; Hagaman, J.R.; Hodgin, J.B.; Pang, S.C.; Flynn, T.G.; Smithies, O. Genetic Decreases in Atrial Natriuretic Peptide and Salt-Sensitive Hypertension. Science 1995, 267, 679–681.
- Ogawa, Y.; Itoh, H.; Tamura, N.; Suga, S.; Yoshimasa, T.; Uehira, M.; Matsuda, S.; Shiono, S.; Nishimoto, H.; Nakao, K. Molecular cloning of the complementary DNA and gene that encode mouse brain natriuretic peptide and generation of transgenic mice that overexpress the brain natriuretic peptide gene. J. Clin. Investig. 1994, 93, 1911–1921.
- Tamura, N.; Doolittle, L.K.; Hammer, R.E.; Shelton, J.M.; Richardson, J.A.; Garbers, D.L. Critical roles of the guanylyl cyclase B receptor in endochondral ossification and development of female reproductive organs. Proc. Natl. Acad. Sci. USA 2004, 101, 17300–17305.
- Kishimoto, I.; Dubois, S.K.; Garbers, D.L. The heart communicates with the kidney exclusively through the guanylyl cyclase-A receptor: Acute handling of sodium and water in response to volume expansion. Proc. Natl. Acad. Sci. USA 1996, 93, 6215–6219.
- Holtwick, R.; Gotthardt, M.; Skryabin, B.; Steinmetz, M.; Potthast, R.; Zetsche, B.; Hammer, R.E.; Herz, J.; Kuhn, M. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc. Natl. Acad. Sci. USA 2002, 99, 7142–7147.
- Cataliotti, A.; Boerrigter, G.; Costello-Boerrigter, L.C.; Schirger, J.A.; Tsuruda, T.; Heublein, D.M.; Chen, H.H.; Malatino, L.S.; Burnett, J.C., Jr. Brain Natriuretic Peptide Enhances Renal Actions of Furosemide and Suppresses Furosemide-Induced Aldosterone Activation in Experimental Heart Failure. Circulation 2004, 109, 1680–1685.
- Siragy, H.M.; Lamb, N.E.; Rose, C.E.; Peach, M.J.; Carey, R.M. Angiotensin II modulates the intrarenal effects of atrial natriuretic peptide. Am. J. Physiol.-Ren. Physiol. 1988, 255, F545–F551.
- Belluardo, P.; Cataliotti, A.; Bonaiuto, L.; Giuffrè, E.; Maugeri, E.; Noto, P.; Orlando, G.; Raspa, G.; Piazza, B.; Babuin, L.; et al. Lack of activation of molecular forms of the BNP system in human grade 1 hypertension and relationship to cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1529–H1535.
- Macheret, F.; Heublein, D.; Costello-Boerrigter, L.C.; Boerrigter, G.; McKie, P.; Bellavia, D.; Mangiafico, S.; Ikeda, Y.; Bailey, K.; Scott, C.G.; et al. Human Hypertension Is Characterized by a Lack of Activation of the Antihypertensive Cardiac Hormones ANP and BNP. J. Am. Coll. Cardiol. 2012, 60, 1558–1565.
- Chen, H.H.; Wan, S.-H.; Iyer, S.R.; Cannone, V.; Sangaralingham, S.J.; Nuetel, J.; Burnett, J.C., Jr. First-in-Human Study of MANP: A Novel ANP (Atrial Natriuretic Peptide) Analog in Human Hypertension. Hypertension 2021, 78, 1859–1867.
- Sabrane, K.; Kruse, M.N.; Fabritz, L.; Zetsche, B.; Mitko, D.; Skryabin, B.V.; Zwiener, M.; Baba, H.A.; Yanagisawa, M.; Kuhn, M. Vascular endothelium is critically involved in the hypotensive and hypovolemic actions of atrial natriuretic peptide. J. Clin. Investig. 2005, 115, 1666–1674.
- Knowles, J.W.; Esposito, G.; Mao, L.; Hagaman, J.R.; Fox, J.E.; Smithies, O.; Rockman, H.A.; Maeda, N. Pressure-independent enhancement of cardiac hypertrophy in natriuretic peptide receptor A–deficient mice. J. Clin. Investig. 2001, 107, 975–984.
- Patel, J.B.; Valencik, M.L.; Pritchett, A.M.; Burnett, J.J.C.; McDonald, J.A.; Redfield, M.M. Cardiac-specific attenuation of natriuretic peptide A receptor activity accentuates adverse cardiac remodeling and mortality in response to pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H777–H784.
- Tamura, N.; Ogawa, Y.; Chusho, H.; Nakamura, K.; Nakao, K.; Suda, M.; Kasahara, M.; Hashimoto, R.; Katsuura, G.; Mukoyama, M.; et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc. Natl. Acad. Sci. USA 2000, 97, 4239–4244.
- Li, Y.; Kishimoto, I.; Saito, Y.; Harada, M.; Kuwahara, K.; Izumi, T.; Takahashi, N.; Kawakami, R.; Tanimoto, K.; Nakagawa, Y.; et al. Guanylyl Cyclase-A Inhibits Angiotensin II Type 1A Receptor-Mediated Cardiac Remodeling, an Endogenous Protective Mechanism in the Heart. Circulation 2002, 106, 1722–1728.
- Kishimoto, I.; Rossi, K.; Garbers, D.L. A genetic model provides evidence that the receptor for atrial natriuretic peptide (guanylyl cyclase-A) inhibits cardiac ventricular myocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 2703–2706.
- Holtwick, R.; van Eickels, M.; Skryabin, B.V.; Baba, H.A.; Bubikat, A.; Begrow, F.; Schneider, M.D.; Garbers, D.L.; Kuhn, M. Pressure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J. Clin. Investig. 2003, 111, 1399–1407.
- Cao, L.; Gardner, D.G. Natriuretic Peptides Inhibit DNA Synthesis in Cardiac Fibroblasts. Hypertension 1995, 25, 227–234.
- Rubattu, S.; Bigatti, G.; Evangelista, A.; Lanzani, C.; Stanzione, R.; Zagato, L.; Manunta, P.; Marchitti, S.; Venturelli, V.; Bianchi, G.; et al. Association of Atrial Natriuretic Peptide and Type A Natriuretic Peptide Receptor Gene Polymorphisms with Left Ventricular Mass in Human Essential Hypertension. J. Am. Coll. Cardiol. 2006, 48, 499–505.
- Rubattu, S.; Sciarretta, S.; Ciavarella, G.M.; Venturelli, V.; De Paolis, P.; Tocci, G.; De Biase, L.; Ferrucci, A.; Volpe, M. Reduced levels of N-terminal-proatrial natriuretic peptide in hypertensive patients with metabolic syndrome and their relationship with left ventricular mass. J. Hypertens. 2007, 25, 833–839.
- Cataliotti, A.; Malatino, L.S.; Jougasaki, M.; Zoccali, C.; Castellino, P.; Giacone, G.; Bellanuova, I.; Tripepi, R.; Seminara, G.; Parlongo, S.; et al. Circulating Natriuretic Peptide Concentrations in Patients with End-Stage Renal Disease: Role of Brain Natriuretic Peptide as a Biomarker for Ventricular Remodeling. Mayo Clin. Proc. 2001, 76, 1111–1119.
- Zoccali, C.; Mallamaci, F.; Benedetto, F.A.; Tripepi, G.; Parlongo, S.; Cataliotti, A.; Cutrupi, S.; Giacone, G.; Bellanuova, I.; Cottini, E.; et al. Cardiac Natriuretic Peptides Are Related to Left Ventricular Mass and Function and Predict Mortality in Dialysis Patients. J. Am. Soc. Nephrol. 2001, 12, 1508–1515.
- Cataliotti, A.; Tonne, J.M.; Bellavia, D.; Martin, F.L.; Oehler, E.A.; Harders, G.E.; Campbell, J.M.; Peng, K.-W.; Russell, S.J.; Malatino, L.S.; et al. Long-Term Cardiac pro-B-Type Natriuretic Peptide Gene Delivery Prevents the Development of Hypertensive Heart Disease in Spontaneously Hypertensive Rats. Circulation 2011, 123, 1297–1305.
- Wang, Y.; De Waard, M.C.; Sterner-Kock, A.; Stepan, H.; Schultheiss, H.-P.; Duncker, D.J.; Walther, T. Cardiomyocyte-restricted over-expression of C-type natriuretic peptide prevents cardiac hypertrophy induced by myocardial infarction in mice. Eur. J. Heart Fail. 2007, 9, 548–557.
- Sarzani, R.; Allevi, M.; Di Pentima, C.; Schiavi, P.; Spannella, F.; Giulietti, F. Role of Cardiac Natriuretic Peptides in Heart Structure and Function. Int. J. Mol. Sci. 2022, 23, 14415.
- Kuhn, M.; Völker, K.; Schwarz, K.; Carbajo-Lozoya, J.; Flögel, U.; Jacoby, C.; Stypmann, J.; van Eickels, M.; Gambaryan, S.; Hartmann, M.; et al. The natriuretic peptide/guanylyl cyclase–A system functions as a stress-responsive regulator of angiogenesis in mice. J. Clin. Investig. 2009, 119, 2019–2030.
- Oliver, P.M.; Fox, J.E.; Kim, R.; Rockman, H.A.; Kim, H.-S.; Reddick, R.L.; Pandey, K.N.; Milgram, S.L.; Smithies, O.; Maeda, N. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc. Natl. Acad. Sci. USA 1997, 94, 14730–14735.
- Oliver, P.M.; John, S.W.M.; Purdy, K.E.; Kim, R.; Maeda, N.; Goy, M.F.; Smithies, O. Natriuretic peptide receptor 1 expression influences blood pressures of mice in a dose-dependent manner. Proc. Natl. Acad. Sci. USA 1998, 95, 2547–2551.
- Clavell, A.L.; Stingo, A.J.; Wei, C.M.; Heublein, D.M.; Burnett, J.C. C-type natriuretic peptide: A selective cardiovascular peptide. Am. J. Physiol. 1993, 264 Pt 2, R290–R295.
- Mahinrad, S.; Bulk, M.; van der Velpen, I.; Mahfouz, A.; van Roon-Mom, W.; Fedarko, N.; Yasar, S.; Sabayan, B.; van Heemst, D.; van der Weerd, L. Natriuretic Peptides in Post-mortem Brain Tissue and Cerebrospinal Fluid of Non-demented Humans and Alzheimer’s Disease Patients. Front. Neurosci. 2018, 12, 864.
- Levin, E.R.; Frank, H.J. Natriuretic peptides inhibit rat astroglial proliferation: Mediation by C receptor. Am. J. Physiol. 1991, 261 Pt 2, R453–R457.
- Langub, M.C.; Dolgas, C.M.; Watson, R.E.; Herman, J.P. The C-Type Natriuretic Peptide Receptor is the Predominant Natriuretic Peptide Receptor mRNA Expressed in Rat Hypothalamus. J. Neuroendocr. 1995, 7, 305–309.
- Huang, W.; Lee, D.; Yang, Z.; Copolov, D.L.; Lim, A.T. Norepinephrine stimulates immunoreactive (ir) atrial natriuretic peptide (ANP) secretion and pro-ANP mRNA expression from rat hypothalamic neurons in culture: Effects of alpha 2-adrenoceptors. Endocrinology 1992, 130, 2426–2428.
- Blackburn, R.E.; Samson, W.K.; Fulton, R.J.; Stricker, E.M.; Verbalis, J.G. Central oxytocin and ANP receptors mediate osmotic inhibition of salt appetite in rats. Am. J. Physiol. Integr. Comp. Physiol. 1995, 269, R245–R251.
- Samson, W.K. Natriuretic peptides: A family of hormones. Trends Endocrinol. Metab. 1992, 3, 86–90.
- Yang, R.H.; Jin, H.K.; Wyss, J.M.; Chen, Y.F.; Oparil, S. Pressor effect of blocking atrial natriuretic peptide in nucleus tractus solitarii. Hypertension 1992, 19, 198–205.
- Della Corte, V.; Tuttolomondo, A.; Pecoraro, R.; Pinto, A. Chronic hyponatremia in a patient with renal salt wasting and without cerebral disease: Relationship between RSW, risk of fractures and cognitive impairment. Intern. Emerg. Med. 2018, 13, 1167–1171.
- Marin-Grez, M.; Fleming, J.T.; Steinhausen, M. Atrial natriuretic peptide causes pre-glomerular vasodilatation and post-glomerular vasoconstriction in rat kidney. Nature 1986, 324, 473–476.
- Stockand, J.D.; Sansom, S.C. Regulation of filtration rate by glomerular mesangial cells in health and diabetic renal disease. Am. J. Kidney Dis. 1997, 29, 971–981.
- Schulz-Knappe, P.; Forssmann, K.; Herbst, F.; Hock, D.; Pipkorn, R.; Forssmann, W.G. Isolation and structural analysis of “Urodilatin”, a new peptide of the cardiodilatin-(ANP)-family, extracted from human urine. Klin. Wochenschr. 1988, 66, 752–759.
- Harris, P.J.; Thomas, D.; Morgan, T.O. Atrial natriuretic peptide inhibits angiotensin-stimulated proximal tubular sodium and water reabsorption. Nature 1987, 326, 697–698.
- Dillingham, M.A.; Anderson, R.J. Inhibition of Vasopressin Action by Atrial Natriuretic Factor. Science 1986, 231, 1572–1573.
- Zeidel, M.L. Regulation of collecting duct Na+ reabsorption by ANP 31–67. Clin. Exp. Pharmacol. Physiol. 1995, 22, 121–124.
- Wijeyaratne, C.N.; Moult, P.J. The effect of alpha human atrial natriuretic peptide on plasma volume and vascular permeability in normotensive subjects. J. Clin. Endocrinol. Metab. 1993, 76, 343–346.
- Hunt, P.J.; Richards, A.M.; Espiner, E.A.; Nicholls, M.G.; Yandle, T.G. Bioactivity and metabolism of C-type natriuretic peptide in normal man. J. Clin. Endocrinol. Metab. 1994, 78, 1428–1435.
- Davidson, N.C.; Barr, C.S.; Struthers, A.D. C-Type Natriuretic Peptide. An endogenous inhibitor of vascular angiotensin-converting enzyme activity. Circulation 1996, 93, 1155–1159.
- Cataliotti, A.; Giordano, M.; De Pascale, E.; Giordano, G.; Castellino, P.; Jougasaki, M.; Costello, L.C.; Boerrigter, G.; Tsuruda, T.; Belluardo, P.; et al. CNP production in the kidney and effects of protein intake restriction in nephrotic syndrome. Am. J. Physiol.-Ren. Physiol. 2002, 283, F464–F472.
- Goetz, K.L.; Wang, B.C.; Geer, P.G.; Leadley, R.J.; Reinhardt, H.W. Atrial stretch increases sodium excretion independently of release of atrial peptides. Am. J. Physiol. 1986, 250 Pt 2, R946–R950.
- Leppäluoto, J.; Ruskoaho, H. Atrial natriuretic peptide, renin activity, aldosterone, urine volume and electrolytes during a 24-h sleep-wake cycle in man. Acta Physiol. Scand. 1990, 139, 47–53.
- Saxenhofer, H.; Raselli, A.; Weidmann, P.; Forssmann, W.G.; Bub, A.; Ferrari, P.; Shaw, S.G. Urodilatin, a natriuretic factor from kidneys, can modify renal and cardiovascular function in men. Am. J. Physiol. 1990, 259 Pt 2, F832–F838.
- Drummer, C.; Franck, W.; Heer, M.; Forssmann, W.G.; Gerzer, R.; Goetz, K. Postprandial natriuresis in humans: Further evidence that urodilatin, not ANP, modulates sodium excretion. Am. J. Physiol.-Ren. Physiol. 1996, 270, F301–F310.
- Goetz, K.; Drummer, C.; Zhu, J.L.; Leadley, R.; Fiedler, F.; Gerzer, R. Evidence that urodilatin, rather than ANP, regulates renal sodium excretion. J. Am. Soc. Nephrol. 1990, 1, 867–874.
- Morishita, Y.; Sano, T.; Kase, H.; Yamada, K.; Inagami, T.; Matsuda, Y. HS-142-1, a novel nonpeptide atrial natriuretic peptide (ANP) antagonist, blocks ANP-induced renal responses through a specific interaction with guanylyl cyclase-linked receptors. Eur. J. Pharmacol. Mol. Pharmacol. 1992, 225, 203–207.
- Wada, A.; Tsutamoto, T.; Matsuda, Y.; Kinoshita, M. Cardiorenal and neurohumoral effects of endogenous atrial natriuretic peptide in dogs with severe congestive heart failure using a specific antagonist for guanylate cyclase-coupled receptors. Circulation 1994, 89, 2232–2240.
- Zhang, P.L.; Mackenzie, H.S.; Troy, J.L.; Brenner, B.M. Effects of an atrial natriuretic peptide receptor antagonist on glomerular hyperfiltration in diabetic rats. J. Am. Soc. Nephrol. 1994, 4, 1564–1570.
- Erlebacher, A.; Filvaroff, E.H.; Gitelman, S.E.; Derynck, R. Toward a molecular understanding of skeletal development. Cell 1995, 80, 371–378.
- Chung, U.-I.; Kronenberg, H.M. Parathyroid hormone-related peptide and Indian hedgehog. Curr. Opin. Nephrol. Hypertens. 2000, 9, 357–362.
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of Rate of Cartilage Differentiation by Indian Hedgehog and PTH-Related Protein. Science 1996, 273, 613–622.
- Minina, E.; Wenzel, H.M.; Kreschel, C.; Karp, S.; Gaffield, W.; McMahon, A.P.; Vortkamp, A. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 2001, 128, 4523–4534.
- Karp, S.J.; Schipani, E.; St-Jacques, B.; Hunzelman, J.; Kronenberg, H.; McMahon, A.P. Indian Hedgehog coordinates endochondral bone growth and morphogenesis via Parathyroid Hormone related-Protein-dependent and -independent pathways. Development 2000, 127, 543–548.
- Pines, M.; Hurwitz, S. The Effect of Parathyroid Hormone and Atrial Natriuretic Peptide on Cyclic Nucleotides Production and Proliferation of Avian Epiphyseal Growth Plate Chondroprogenitor Cells. Endocrinology 1988, 123, 360–365.
- Hagiwara, H.; Sakaguchi, H.; Itakura, M.; Yoshimoto, T.; Furuya, M.; Tanaka, S.; Hirose, S. Autocrine regulation of rat chondrocyte proliferation by natriuretic peptide C and its receptor, natriuretic peptide receptor-B. J. Biol. Chem. 1994, 269, 10729–10733.
- Suda, M.; Ogawa, Y.; Tanaka, K.; Tamura, N.; Yasoda, A.; Takigawa, T.; Uehira, M.; Nishimoto, H.; Itoh, H.; Saito, Y.; et al. Skeletal overgrowth in transgenic mice that overexpress brain natriuretic peptide. Proc. Natl. Acad. Sci. USA 1998, 95, 2337–2342.
- Matsukawa, N.; Grzesik, W.J.; Takahashi, N.; Pandey, K.N.; Pang, S.; Yamauchi, M.; Smithies, O. The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. Proc. Natl. Acad. Sci. USA 1999, 96, 7403–7408.
- Jaubert, J.; Jaubert, F.; Martin, N.; Washburn, L.L.; Lee, B.K.; Eicher, E.M.; Guénet, J.-L. Three new allelic mouse mutations that cause skeletal overgrowth involve the natriuretic peptide receptor C gene (Npr3). Proc. Natl. Acad. Sci. USA 1999, 96, 10278–10283.
- Chusho, H.; Tamura, N.; Ogawa, Y.; Yasoda, A.; Suda, M.; Miyazawa, T.; Nakamura, K.; Nakao, K.; Kurihara, T.; Komatsu, Y.; et al. Dwarfism and early death in mice lacking C-type natriuretic peptide. Proc. Natl. Acad. Sci. USA 2001, 98, 4016–4021.
- Nishizawa, H.; Matsuda, M.; Yamada, Y.; Kawai, K.; Suzuki, E.; Makishima, M.; Kitamura, T.; Shimomura, I. Musclin, a Novel Skeletal Muscle-derived Secretory Factor. J. Biol. Chem. 2004, 279, 19391–19395.
- Thomas, G.; Moffatt, P.; Salois, P.; Gaumond, M.-H.; Gingras, R.; Godin, E.; Miao, D.; Goltzman, D.; Lanctôt, C. Osteocrin, a Novel Bone-specific Secreted Protein That Modulates the Osteoblast Phenotype. J. Biol. Chem. 2003, 278, 50563–50571.
- Subbotina, E.; Sierra, A.; Zhu, Z.; Gao, Z.; Koganti, S.R.K.; Reyes, S.; Stepniak, E.; Walsh, S.A.; Acevedo, M.R.; Perez-Terzic, C.M.; et al. Musclin is an activity-stimulated myokine that enhances physical endurance. Proc. Natl. Acad. Sci. USA 2015, 112, 16042–16047.
- Szaroszyk, M.; Kattih, B.; Martin-Garrido, A.; Trogisch, F.A.; Dittrich, G.M.; Grund, A.; Abouissa, A.; Derlin, K.; Meier, M.; Holler, T.; et al. Skeletal muscle derived Musclin protects the heart during pathological overload. Nat. Commun. 2022, 13, 149.
- Zhang, Y.; Zhang, J.; Ni, W.; Yuan, X.; Zhang, H.; Li, P.; Xu, J.; Zhao, Z. Sarcopenia in heart failure: A systematic review and meta-analysis. ESC Heart Fail. 2021, 8, 1007–1017.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
08 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No