Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bongjin Lee | -- | 1589 | 2023-06-04 10:52:59 | | | |
| 2 | Jason Zhu | Meta information modification | 1589 | 2023-06-05 03:22:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lee, I.; Lee, B. Cysteine Modification in Aurora Kinase A. Encyclopedia. Available online: https://encyclopedia.pub/entry/45170 (accessed on 27 July 2026).
Lee I, Lee B. Cysteine Modification in Aurora Kinase A. Encyclopedia. Available at: https://encyclopedia.pub/entry/45170. Accessed July 27, 2026.
Lee, In-Gyun, Bong-Jin Lee. "Cysteine Modification in Aurora Kinase A" Encyclopedia, https://encyclopedia.pub/entry/45170 (accessed July 27, 2026).
Lee, I., & Lee, B. (2023, June 04). Cysteine Modification in Aurora Kinase A. In Encyclopedia. https://encyclopedia.pub/entry/45170
Lee, In-Gyun and Bong-Jin Lee. "Cysteine Modification in Aurora Kinase A." Encyclopedia. Web. 04 June, 2023.
Copy Citation
Aurora kinase A (AURKA), which is a member of serine/threonine kinase family, plays a critical role in regulating mitosis. AURKA has drawn much attention as its dysregulation is critically associated with various cancers, leading to the development of AURKA inhibitors, a new class of anticancer drugs. As the spatiotemporal activity of AURKA critically depends on diverse intra- and inter-molecular factors, including its interaction with various protein cofactors and post-translational modifications, each of these pathways should be exploited for the development of a novel class of AURKA inhibitors other than ATP-competitive inhibitors.
Aurora kinase A (AURKA)
redox-active molecules
1. Two Distinct Pathways of AURKA Activation
Spatiotemporal regulation of AURKA’s activity is multifactorial. A growing body of evidences suggests the following two main pathways that active AURKA: (i) the phosphorylation of conserved Thr288 residing on the activation segment or the (ii) interaction with co-factor proteins (e.g., TPX2) that induces the structural rearrangement competent for the phosphotransfer activity [1][2][3][4][5][6][7][8][9][10]. Although the phosphorylation of Thr288 and TPX2 binding synergistically activate AURKA in vitro [6][11], two distinct pathways seem to work independently in an intracellular environment [12][13][14][15]. As discussed in detail below, biochemical, biophysical, structural, and cellular evidences have shown that the redox modification of the cysteine residues of AURKA affects both the phosphorylation state of Thr288 and the conformation state of structural elements critical for the kinase activity, leading to the regulation of AURKA activity.
2. H2O2 Induced Oxidative Modification of Cysteine
Hydrogen peroxide (H2O2) is a major ROS member that can act as a signaling molecule in many biological systems [16]. Endogenously, H2O2 can be produced as a result of [17] metabolic reactions such as respiration, and is implicated in many redox signaling pathways [18][19]. Furthermore, H2O2 levels have also been implicated in the regulation of mitosis by directly oxidizing various kinases and phosphatases involved in the cell cycle regulation [20][21][22][23]. Considering the variable intracellular H2O2 level in multiple stages of the cell cycle [24][25], cells can be considered to have critical ways of sensing intracellular H2O2 levels and responding to them. For example, high levels of H2O2 have been shown to induce the cell cycle arrest, and relatively low levels of H2O2 are required for mitotic entry [17][24][26].
So far, several lines of evidence have shown that H2O2-induced regulation of the AURKA activity, possibly through direct oxidation of Cys290 present in the activation segment of the kinase domain. In mammalian cells, the oxidative stress induced by the additional H2O2 results in the mitotic delay and abnormal mitotic spindle formation [17]. As the spindle formation is the process that is mainly governed by the kinase activity of AURKA, these observations led to the hypothesis that H2O2 regulates the activity of AURKA [27][28]. Indeed, the phosphorylation level of Thr288 of AURKA was significantly elevated when the mammalian cells were treated with H2O2, suggesting that the hyperphosphorylation of AURKA induces mitotic delay and abnormal spindle formation [17].
In contrast, when the purified recombinant AURKA was treated with H2O2, its overall kinase activity of AURKA (as measured in terms of the phosphorylation level of fluorescent-tagged substrate peptide) was decreased, while the phosphorylation level of Thr288 remained almost unchanged [29]. The mutational analysis further confirmed that the Cys290 was the main site for H2O2-induced oxidative modifications, and the inhibitory effect of H2O2 on AUKRA activity was reversed by almost equimolar concentrations of the reducing agent dithiothreitol (DTT), suggesting a reversible inhibitory effect of H2O2 on AURKA activity [29]. Interestingly, adding relatively higher amounts of DTT (~100 mM) to the Xenopus laevis (X. laevis) egg extract system, a powerful tool for studying the cell cycle at the molecular level, inhibited the phosphorylation of Thr295 in the X. laevis AURKA (equivalent to Thr288 of human AURKA) [30]. These observations suggest the presence of indirect or cell cycle-specific signaling pathways that lead to elevated Thr288 phosphorylation levels and AURKA activation. As the phosphorylation state of Thr288 alone does not fully reflect the enzymatic activity of the kinase, direct measurements of kinase activity of AURKA in addition to Thr288 phosphorylation level at the specific intracellular localization and timing in cells would aid confirming the interplay of diverse AURKA cofactors that regulates the activity.
3. Structural Transitions Induced upon Covalent Modification of Cysteine in AURKA with Coenzyme A
Coenzyme A (CoA) is a fundamental metabolic cofactor that participates in numerous biological metabolic processes [31]. It particularly plays a central role and functions as an obligate cofactor in energy and fatty acid metabolic pathways [32]. As CoA contains a thiol group, it can interact with other cellular thiols to form a disulfide bond and can also covalently modify protein thiols in cysteine or methionine amino acids [31]. Covalent modification with CoA (CoAlation) of cysteine residues plays a role in post-translational modification, which can lead to altered enzyme activity, protein-protein interactions, and localization [31][33][34]. Interestingly, it has been reported that CoA and its derivatives regulate the activity of several protein kinases, such as PKC (protein kinase C), CaMKII, and AURKA through direct activation or inhibition [35][36]. Several CoAlated structures of AURKA have enabled a deeper understanding of CoA-mediated regulation of AURKA activity at the structural and molecular levels.
The previous study reported by Tsuchiya et al. revealed the detailed biophysical and structural basis of the CoA-mediated AURKA inhibition [37]. Kinome-wide screening of CoA against various protein kinases revealed that CoA specifically inhibits the catalytic activity of AURKA. Mass spectrometric and mutational analyses confirmed that the CoA molecule covalently modified the Cys290, and the modification of Cys290 decreased AURKA phosphotransferase activity towards myelin basic protein substrate in a dose dependent manner [37]. The crystal structure of CoAlated Cys290 AURKA elucidated the structural basis of the inhibitory effect of Cys290 CoAlation (Figure 1). In the crystal structure, the 3′-phospho-ADP moiety of CoA was bound to the ATP-binding pocket of the kinase domain of AURKA, suggesting that the CoA can occupy the ATP-binding pocket and thus compete with cellular ATP (Figure 1b). The pantetheine moiety of CoA stretches toward the catalytic activation segment of AURKA, allowing the sulfhydryl group to form a disulfide bond with the Cys290 located in the vicinity of Thr288.
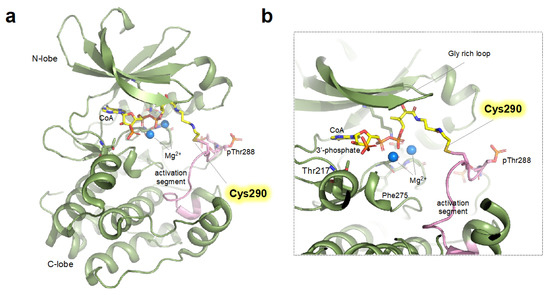
Figure 1. Structure of monomeric CoAlated AURKA structure (a) Crystal structure (shown as ribbon representation) of AURKA bound to CoA (PDB ID: 6I2U) displaying monomeric structure (See main text for details). (b) Close-up view of the active site (ATP binding pocket) of CoAlated AURKA. Cys290 forming covalent bonds with CoA is shown as yellow stick and activation segment is colored in pink.
The structural prerequisite for sufficient activation of AURKA is the presence of several structural elements such as “DFG-in”, “α-Helix in”, and the salt bridge between catalytically important Lys162 and Glu181 [5]; interestingly, the CoA-bound AURKA harbors these hallmarks of the active conformation. However, covalent modification of the Cys290 resides in the “P + 1 loop”, a portion of the activation segment that constitutes a binding site for substrate peptide, would sterically constrain the P + 1 loop, leading to impaired geometry unsuited for substrate binding (Figure 1b) [38][39]. Furthermore, the covalent binding of CoA resulted in the loss of hydrogen bonding between Arg255 (the central residue of the catalytic HRD motif) and phosphorylated Thr288, which would further impair the ideal geometry for the catalysis [40]. Interestingly, the binding of TPX2, a major cofactor of the AURKA, almost completely blocks the inhibitory effect of CoA [37]. This indicates that, in the presence of TPX2 (i.e., spindle pool of AURKA), the inhibitory effects of CoA are expected to be significantly limited. Whether the other major cofactors (e.g., CEP192 or Bora) also block the inhibitory effect of CoAlation of cysteine should be further determined.
Although the study by Tsuchiya et al. detailed the structural and biophysical basis for CoA- mediated AURKA inhibition, the effect of CoAlation on AURKA does not seem straightforward, as other Cys290 CoAlated structures have shown the structural implication of CoAlation for the AURKA activation (Figure 2) [30]. A study by Lim et al. reported a CoA-bound structure displaying an activation segment-swapped homodimer, a more catalytically competent conformation (Figure 2). Contrary to the monomer structure reported by Tsuchiya et al. this structure contained a homodimer and CoA molecules covalently bounded to the Cys290 residues of neighboring monomers, suggesting that the formation of the Cys290-CoA covalent bond may facilitate stabilization of the activation segment swapped dimeric structure, which would consequently promote the autophosphorylation of Thr288 (Figure 2b). Whether the CoAlation leads to the formation of dimeric structure in solution needs further investigation. As the authors used a TPX2-fused chimeric protein, with the N-terminal AURKA residues (at positions 1–115) replaced with TPX2 (at positions 7–20), the structure might reflect that of the distinct CoA-bound AURKA in the presence of TPX2.
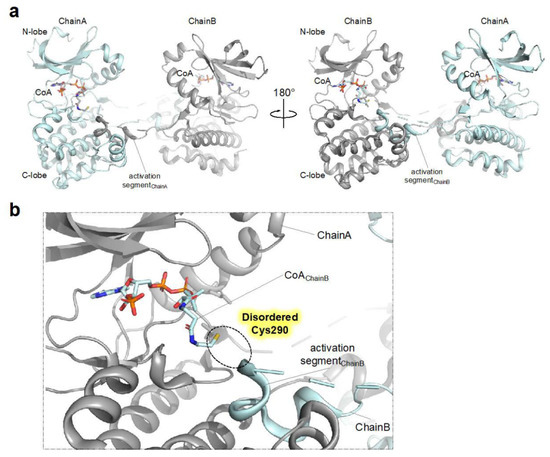
Figure 2. Structure of dimeric CoAlated AURKA structure (a) Crystal structure (shown as ribbon representation) of AURKA bound to CoA (PDB ID: 6XKA) displaying dimeric structure. (b) Close-up view of the active site (ATP binding pocket) of CoAlated AURKA. As the structure contains only one molecule in the asymmetric unit, Chain B denotes the AURKA molecule present in the neighboring asymmetric unit, not in the same unit. Also, the portion of the activation segment containing Cys290 is disordered and therefore not modeled in the structure.
Due to the flexible nature of the kinase activation segment, several distinct structural populations can exist in solution, and relatively minor conformations that are prone to crystallize can be captured in crystal structure. Therefore, concluding solely from the crystal structure could often be misleading. Other biophysical and cellular experimental approaches reflecting more physiological conditions would complement the information obtained from the crystal structure analyses and aid the comprehensive understanding of CoA-mediated regulation of AURKA.
References
- Bayliss, R.; Sardon, T.; Vernos, I.; Conti, E. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol. Cell 2003, 12, 851–862.
- Kufer, T.A.; Silljé, H.H.; Körner, R.; Gruss, O.J.; Meraldi, P.; Nigg, E.A. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J. Cell Biol. 2002, 158, 617–623.
- Wittmann, T.; Wilm, M.; Karsenti, E.; Vernos, I. TPX2, A novel xenopus MAP involved in spindle pole organization. J. Cell Biol. 2000, 149, 1405–1418.
- Nolen, B.; Taylor, S.; Ghosh, G. Regulation of protein kinases: Controlling activity through activation segment conformation. Mol. Cell 2004, 15, 661–675.
- Zorba, A.; Buosi, V.; Kutter, S.; Kern, N.; Pontiggia, F.; Cho, Y.-J.; Kern, D. Molecular mechanism of Aurora A kinase autophosphorylation and its allosteric activation by TPX2. eLife 2014, 3, e02667.
- Eyers, P.A.; Erikson, E.; Chen, L.G.; Maller, J.L. A novel mechanism for activation of the protein kinase Aurora A. Curr. Biol. 2003, 13, 691–697.
- Ruff, E.F.; Muretta, J.M.; Thompson, A.R.; Lake, E.W.; Cyphers, S.; Albanese, S.K.; Hanson, S.M.; Behr, J.M.; Thomas, D.D.; Chodera, J.D. A dynamic mechanism for allosteric activation of Aurora kinase A by activation loop phosphorylation. eLife 2018, 7, e32766.
- Cyphers, S.; Ruff, E.F.; Behr, J.M.; Chodera, J.D.; Levinson, N.M. A water-mediated allosteric network governs activation of Aurora kinase A. Nat. Chem. Biol. 2017, 13, 402–408.
- Bertolin, G.; Sizaire, F.; Herbomel, G.; Reboutier, D.; Prigent, C.; Tramier, M. A FRET biosensor reveals spatiotemporal activation and functions of aurora kinase A in living cells. Nat. Commun. 2016, 7, 1–16.
- Lake, E.W.; Muretta, J.M.; Thompson, A.R.; Rasmussen, D.M.; Majumdar, A.; Faber, E.B.; Ruff, E.F.; Thomas, D.D.; Levinson, N.M. Quantitative conformational profiling of kinase inhibitors reveals origins of selectivity for Aurora kinase activation states. Proc. Natl. Acad. Sci. USA 2018, 115, E11894–E11903.
- Dodson, C.A.; Bayliss, R. Activation of Aurora-A kinase by protein partner binding and phosphorylation are independent and synergistic. J. Biol. Chem. 2012, 287, 1150–1157.
- Joukov, V.; De Nicolo, A.; Rodriguez, A.; Walter, J.C.; Livingston, D.M. Centrosomal protein of 192 kDa (Cep192) promotes centrosome-driven spindle assembly by engaging in organelle-specific Aurora A activation. Proc. Natl. Acad. Sci. USA 2010, 107, 21022–21027.
- Hammond, D.; Zeng, K.; Espert, A.; Bastos, R.N.; Baron, R.D.; Gruneberg, U.; Barr, F.A. Melanoma-associated mutations in protein phosphatase 6 cause chromosome instability and DNA damage owing to dysregulated Aurora-A. J. Cell Sci. 2013, 126, 3429–3440.
- Toya, M.; Terasawa, M.; Nagata, K.; Iida, Y.; Sugimoto, A. A kinase-independent role for Aurora A in the assembly of mitotic spindle microtubules in Caenorhabditis elegans embryos. Nat. Cell Biol. 2011, 13, 708–714.
- Dutertre, S.; Cazales, M.; Quaranta, M.; Froment, C.; Trabut, V.; Dozier, C.; Mirey, G.; Bouché, J.-P.; Theis-Febvre, N.; Schmitt, E. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2–M transition. J. Cell Sci. 2004, 117, 2523–2531.
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728.
- Wang, G.-F.; Dong, Q.; Bai, Y.; Yuan, J.; Xu, Q.; Cao, C.; Liu, X. Oxidative stress induces mitotic arrest by inhibiting Aurora A-involved mitotic spindle formation. Free. Radic. Biol. Med. 2017, 103, 177–187.
- Veal, E.; Day, A. Hydrogen peroxide as a signaling molecule. Antioxid. Redox Signal. 2011, 15, 147–151.
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The role of hydrogen peroxide in redox-dependent signaling: Homeostatic and pathological responses in mammalian cells. Cells 2018, 7, 156.
- Lim, J.M.; Lee, K.S.; Woo, H.A.; Kang, D.; Rhee, S.G. Control of the pericentrosomal H2O2 level by peroxiredoxin I is critical for mitotic progression. J. Cell Biol. 2015, 210, 23–33.
- Rhee, S.G. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883.
- Kwon, J.; Lee, S.-R.; Yang, K.-S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424.
- Deshpande, N.N.; Sorescu, D.; Seshiah, P.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Griendling, K.K. Mechanism of hydrogen peroxide-induced cell cycle arrest in vascular smooth muscle. Antioxid. Redox Signal. 2002, 4, 845–854.
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of late G1/S phase transition and APCCdh1 by reactive oxygen species. Mol. Cell. Biol. 2006, 26, 4701–4711.
- Goswami, P.C.; Sheren, J.; Albee, L.D.; Parsian, A.; Sim, J.E.; Ridnour, L.A.; Higashikubo, R.; Gius, D.; Hunt, C.R.; Spitz, D.R. Cell cycle-coupled variation in topoisomerase IIα mRNA is regulated by the 3′-untranslated region: Possible role of redox-sensitive protein binding in mRNA accumulation. J. Biol. Chem. 2000, 275, 38384–38392.
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009, 69, 2647–2654.
- Hochegger, H.; Hégarat, N.; Pereira-Leal, J. Aurora at the pole and equator: Overlapping functions of Aurora kinases in the mitotic spindle. Open Biol. 2013, 3, 120185.
- Marumoto, T.; Zhang, D.; Saya, H. Aurora-A—A guardian of poles. Nat. Rev. Cancer 2005, 5, 42–50.
- Byrne, D.P.; Shrestha, S.; Galler, M.; Cao, M.; Daly, L.A.; Campbell, A.E.; Eyers, C.E.; Veal, E.A.; Kannan, N.; Eyers, P.A. Aurora A regulation by reversible cysteine oxidation reveals evolutionarily conserved redox control of Ser/Thr protein kinase activity. Sci. Signal. 2020, 13, eaax2713.
- Lim, D.C.; Joukov, V.; Rettenmaier, T.J.; Kumagai, A.; Dunphy, W.G.; Wells, J.A.; Yaffe, M.B. Redox priming promotes Aurora A activation during mitosis. Sci. Signal. 2020, 13, eabb6707.
- Gout, I. Coenzyme A, protein CoAlation and redox regulation in mammalian cells. Biochem. Soc. Trans. 2018, 46, 721–728.
- Leonardi, R.; Zhang, Y.-M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153.
- Baković, J.; Yu, B.Y.K.; Silva, D.; Chew, S.P.; Kim, S.; Ahn, S.-H.; Palmer, L.; Aloum, L.; Stanzani, G.; Malanchuk, O. A key metabolic integrator, coenzyme A, modulates the activity of peroxiredoxin 5 via covalent modification. Mol. Cell. Biochem. 2019, 461, 91–102.
- Tsuchiya, Y.; Zhyvoloup, A.; Baković, J.; Thomas, N.; Yu, B.Y.K.; Das, S.; Orengo, C.; Newell, C.; Ward, J.; Saladino, G. Protein CoAlation and antioxidant function of coenzyme A in prokaryotic cells. Biochem. J. 2018, 475, 1909–1937.
- McCoy, F.; Darbandi, R.; Lee, H.C.; Bharatham, K.; Moldoveanu, T.; Grace, C.R.; Dodd, K.; Lin, W.; Chen, S.-I.; Tangallapally, R.P. Metabolic activation of CaMKII by coenzyme A. Mol. Cell 2013, 52, 325–339.
- Ford, D.A.; Horner, C.C.; Gross, R.W. Protein kinase C acylation by palmitoyl coenzyme A facilitates its translocation to membranes. Biochemistry 1998, 37, 11953–11961.
- Tsuchiya, Y.; Byrne, D.P.; Burgess, S.G.; Bormann, J.; Baković, J.; Huang, Y.; Zhyvoloup, A.; Yu, B.Y.K.; Peak-Chew, S.; Tran, T. Covalent Aurora A regulation by the metabolic integrator coenzyme A. Redox Biol. 2020, 28, 101318.
- Levinson, N.M. The multifaceted allosteric regulation of Aurora kinase A. Biochem. J. 2018, 475, 2025–2042.
- Guimarães, C.R.; Rai, B.K.; Munchhof, M.J.; Liu, S.; Wang, J.; Bhattacharya, S.K.; Buckbinder, L. Understanding the impact of the P-loop conformation on kinase selectivity. J. Chem. Inf. Model. 2011, 51, 1199–1204.
- La Sala, G.; Riccardi, L.; Gaspari, R.; Cavalli, A.; Hantschel, O.; De Vivo, M. HRD motif as the central hub of the signaling network for activation loop autophosphorylation in Abl kinase. J. Chem. Theory Comput. 2016, 12, 5563–5574.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
05 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No