Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexandar M. Zhivkov | -- | 3921 | 2023-06-03 14:55:30 | | | |
| 2 | Alfred Zheng | Meta information modification | 3921 | 2023-06-05 05:58:36 | | | | |
| 3 | Alexandar M. Zhivkov | + 15 word(s) | 3936 | 2023-07-30 09:43:40 | | | | |
| 4 | Alfred Zheng | -15 word(s) | 3921 | 2023-07-31 07:30:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zhivkov, A.M.; Popov, T.T.; Hristova, S.H. Types of Hydrogels for Bearing Anticancer Chemotherapeutics. Encyclopedia. Available online: https://encyclopedia.pub/entry/45163 (accessed on 23 July 2026).
Zhivkov AM, Popov TT, Hristova SH. Types of Hydrogels for Bearing Anticancer Chemotherapeutics. Encyclopedia. Available at: https://encyclopedia.pub/entry/45163. Accessed July 23, 2026.
Zhivkov, Alexandar M., Trifon T. Popov, Svetlana H. Hristova. "Types of Hydrogels for Bearing Anticancer Chemotherapeutics" Encyclopedia, https://encyclopedia.pub/entry/45163 (accessed July 23, 2026).
Zhivkov, A.M., Popov, T.T., & Hristova, S.H. (2023, June 03). Types of Hydrogels for Bearing Anticancer Chemotherapeutics. In Encyclopedia. https://encyclopedia.pub/entry/45163
Zhivkov, Alexandar M., et al. "Types of Hydrogels for Bearing Anticancer Chemotherapeutics." Encyclopedia. Web. 03 June, 2023.
Copy Citation
The advantages of hydrogels as a depot for local application of medicinal substances are due to their tunable physicochemical properties, biocompatibility and the possibility for controllable degradation; due to that, they are intensively investigated as local drug delivery systems. A hydrogel can be defined as a quasi-solid body composed of a three-dimensional (3D) network of hydrophilic macromolecules and water.
hydrogels
nanoparticles
thermo-reversible physical hydrogels
irreversible chemical hydrogels
biodegradability of hydrogels
1. Introduction
Cancer is a socially significant disease that is the second leading cause of death worldwide after cardiovascular diseases. For instance, in 2020, almost 10 million people died of oncological disease [1]. In developed countries, the incidence rate of cancer is steadily increasing (around 3–5% per year), this is a silent pandemic [1][2]. Conventionally, chemotherapy and/or radiotherapy are used in clinical practice for the treatment of neoplasms, but some cancer types are resistant to them and because of this, it is necessary to search for new treatment approaches [3][4]. The main problem is that the chemotherapeutics attack all dividing cells, including those of the immune system, which lead to immunosuppression and as a result, there is risk for the patient to die from a banal bacterial or viral infection [5][6]. This problem can be mitigated if chemotherapeutics are administered locally so that their concentration is high in cancer tissue and low in healthy tissue [7][8]. Another problem is that the intracellular concentration of chemotherapeutics fluctuates depending on the frequency of administration (injectable or oral), and this requires the administration of higher concentrations, at which the toxic effect is particularly strong. To avoid this problem, a chemotherapeutic depot should be used so that its concentration over time is maintained at an optimal therapeutic level [9]. For this purpose, different types of chemotherapeutic carriers, such as nanoparticles [10][11][12], hydrogels [13][14], composed nanoparticle–hydrogels [15], micelles [16] and liposomes [17], can be used.
Some problems concerning anticancer therapy can be solved by using hydrogel as a local depot for chemotherapeutics, in particular in the treatment of neoplasms with a superficial localization such as skin cancer, for oral administration in gastric or colon cancer, or of intra-body tumors by injectable hydrogels [18][19][20]. Local administration of effective but highly toxic drugs, such as platinum derivatives, can provide concentrations high enough for effective treatment and minimize their toxic effect on healthy tissues [21]. In addition, the use of gel-depot ensures a uniform penetration of chemotherapeutic agents into the cancer tissue over time, avoiding particularly toxic peak concentrations [22].
To perform its role as a depot, the hydrogel must be able to retain the chemotherapeutic agent and release it gradually as its concentration in the cancer tissue decreases [23]. Chemotherapeutic molecules can be incorporated into the free state, linked by covalent bonds to a biodegradable gel network, or adsorbed onto nano- or micrometer-sized particles (composite gels) [24]. One major advantage of hydrogels is that no convection (caused by different temperatures or concentration gradients) occurs in them, but only the diffusion of molecules is possible, which is, however, slowed down due to the high viscosity caused by the structuring of water molecules into associates around the filaments of the gel network (which are much larger than dynamic nanoassociates in pure water) [25].
A major disadvantage of using hydrogels as a depot for anticancer chemotherapeutics is that most of them are poorly soluble in water [7]. For this reason, incorporation of a chemotherapeutic in the molecular state is inefficient, but can be rectified by mixing a suspension of crystals with the aqueous solution of the polymer (in sol-state before gelation). Another approach to incorporate hydrophobic chemotherapeutics is to use non-homogeneous gels with a hydrophilic network containing hydrophobic domains that arise under certain conditions or are hydrophobic segments of the block copolymer chain [26]. Even for relatively well water-soluble chemotherapeutics, hydrogels do not have significant adsorption capacity because the gel network is composed of linear macromolecules that lack a significant surface area (unlike nanoparticles) necessary for physical adsorption via noncovalent interactions [27]. One possible solution is gels with pH-dependent polymer unit charges that can electrostatically bind oppositely charged chemotherapeutic molecules and release them upon pH change, but this approach faces the following difficulty, that most often, both components have no ionizable groups with pKa in the physiological pH-range and in addition, the pH changes only minimally (in cancer tissue, the pH is lowered by lactic acid accumulation as cells are energized by glycolysis rather than by oxidative phosphorylation). A specific case is that of the covalent binding of a chemotherapeutic, but this is only applicable if the gel network is degraded, for example by enzymes after injection into cancer tissues [28].
Another approach is the incorporation into the gel of nano- or micrometer-sized particles with adsorbed chemotherapeutic molecules that are gradually released spontaneously (according to the equilibrium constant and their local concentration), or induced (e.g., by thermal or photothermal action). The high surface area/mass ratio of the nanoparticles provides a sufficiently large physical adsorption, and their electrical charge (intrinsic or added by the chemical modification of their surface) causes pH-dependent electrostatic adsorption during gel preparation and desorption into the cancer tissue. For adsorption of hydrophobic chemotherapeutics, the particle surface can be chemically modified to become hydrophobic, and after adsorption, coated with a surfactant (low molecular weight or polymeric) so that the composite particles become water-soluble for incorporation into the hydrophilic gel network [28].
Recent studies have demonstrated that nanoparticles interact with biological media (blood plasma, intra- and extracellular liquid) by forming a protein corona on their surface [29][30]. This alters the physicochemical properties of nanoparticles and should be considered when nanoparticles carrying chemotherapeutics are applied in cancer therapy [31]. The effect of the protein corona depends strongly on the type of plasma proteins (especially the proteins with the highest concentration, such as albumin, globulins—mainly IgG, fibrinogen, apolipoproteins, transferrin, complement factors, etc.) that cover the nanoparticles when they enter the blood after resorption upon systemic administration: enteral (peroral, sublingual, rectal) or parenteral (intravenous, intramuscular, subcutaneous, etc.). The structure and amount of plasma proteins can vary depending on the patient’s comorbidities (as many diseases and conditions can lead to changes in plasma proteomics), family background (genetic factors), lifestyle, geographical factors, etc. It has been proven that people with different diseases and medical conditions (such as diabetes, hypercholesterolemia, rheumatism, hemophilia A and B, thalassemia, breast cancer, hemodialysis, pregnancy, smoking, etc.) form different “personalized protein coronas” [32][33][34]. Consistent with the concept of personalized medicine, the “personalized protein corona effect” must be taken into account when chemotherapeutic nanoparticles are applied in human medicine.
2. Types of Hydrogels
A hydrogel can be defined as a quasi-solid body composed of a three-dimensional (3D) network of hydrophilic macromolecules and water. The weight content of the polymer is drastically less (down to two-three orders of magnitude) than that of the water included in the gel, but should be sufficient to form intermolecular bonds (cross-linking), depending on which types of gels can be divided into physical gels with non-covalent (hydrogen, electrostatic and van-der-Waals forces: London dispersion, permanent and charge-induced dipoles) bonds, and chemicals with covalent intermolecular cross-linking. Convection is absent in hydrogels because the water molecules are structured around the strands of the gel network; only diffusion is possible, allowing them to be used for the delayed release of incorporated drug substances. Hydrogels made from natural polymers of animal and plant origin, such as collagen (a linear protein with a triple polypeptide helix) or amylum (a carbohydrate with a highly branched chain), have been known for centuries, while chemically synthesized ones were developed after 1960 [35]. The advantages of hydrogels as a depot for local application of medicinal substances are due to their tunable physicochemical properties, biocompatibility and the possibility for controllable degradation; due to that, they are intensively investigated as local drug delivery systems [36][37]. Since anticancer chemotherapeutics are poorly or practically insoluble in water, hydrogels forming hydrophobic domains that serve as a sink for various hydrophobic drugs are of particular importance [38].
2.1. Thermo-Reversible Physical Hydrogels
Such are the classical gels of natural and synthetic hydrophilic polymers, which, after heating and cooling, form a hydrogel that can be repeatedly destroyed and re-formed by thermal action, respectively, when raised above and lowered below the critical gelling temperature. The initial heating is necessary to break the intra-molecular bonds defining the native conformation of the macromolecule, e.g., the triple helix of collagen, and transition the polymer chain to a random coil state, which creates the conditions for the formation of random bonds with adjacent chains forming the gel network upon cooling.
The phase state of the polymer solution is determined by the concentration of the polymer and the properties of the solvent (temperature, pH, ionic composition, water-soluble low molecular weight organic compounds such as alcohols, etc.) [39]. The interactions (intra- or intermolecular) between the polymer units are determined by the thermodynamic properties of the polymer and the solvent (mainly water molecules in the case of hydrogels) expressed by Gibbs free energy ΔG = ΔH − TΔS (enthalpy ΔH and entropy ΔS at temperature T), with the three components of ΔG (polymer–polymer, polymer–water and water–water) determining the temperature range of the hydrogel existence. The balance of forces depends on the ability to form hydrogen bonds (electrostatic H-atom sharing between two electronegative atoms such as O and N, O…H-O in particular) and the presence of whole (Coulomb) and partial (polarized covalent bonds) electric charges, that associate water molecules and determine negative enthalpy ΔH values for hydrophilic groups, or the inability for such bonds; in the last case, only van-der-Waals forces are operative and the orientational order of water molecules around the hydrophobic groups increases (lowering the entropy term ΔS).
The chemical nature of the polymer and the composition of the solvent define a temperature range limited by low and high critical solution temperatures (LCSTs and HCSTs), beyond which the chains are in a random coil conformation (then, the polymer chain occupies 1–3% of the volume of the globule) [40]. As the solvent quality deteriorates, in particular by temperature variation, the size of the polymer globule (defined by the averaged values of the radius of gyration Rg = 〈Rg2〉1/2 or the distance h = 〈h2〉1/2 between the ends of a linear chain) decreases due to the dominance of polymer–polymer over polymer–water interactions, and the polymer chains may even collapse into a globule. When the polymer concentration is high enough, intermolecular bonds are formed in addition to intramolecular bonds, leading to the formation of a gel network in the temperature range ΔT = HCST − LCST; the gel breaks down to a polymer solution when the temperature exceeds the upper threshold (ΔT > HCST). Polymer network formation in the gel-forming ΔT temperature range is due to two factors acting in sequence: (a) interweaving of the polymer chains in an unfolded conformation (random coil at T > HCST or T < LCST) due to the high concentration of the polymer solution (the average distance between adjacent chains is less than their gyration radius) and (b) shrinkage of the chains due to solvent deterioration as the temperature increases (at HCST > T > LCST), when polymer–polymer interactions predominate.
The balance of forces for some polymers in aqueous solution defines a state of sol at room temperature or below (T < 25 °C), a region of gel existence at physiological temperatures (T ≈ 37 °C) and reversibly breaking down to a polymer solution when heated above. This offers the advantageous option of injecting the polymer solution into the cancer tissue where the gel forms; such thermosensitive gels are termed injectable [41][42][43][44]; an example of such a polymer is poly(N-isopropylacrylamide) [45]. The gel–sol transition at T > HCST allows the gel to be destroyed if necessary, for example by photothermal irradiation with infrared light.
A second type of thermo-reversible physical hydrogels is presented by the chemically synthesized block copolymers with a chain composed of segments with different affinities to water molecules. The choice of copolymers of a different chemical nature in the synthesis provides additional possibilities to achieve suitable physicochemical properties in aqueous solution. For the injectable gels, block copolymers have undoubted advantages over homogeneous (linear or branched) chain polymers, since the choice of the length of the hydrophobic segments allows for achieving a suitable gelation temperature (solution at room temperature and gel at physiological temperature), and the length, flexibility and charge of the hydrophilic segments determine the pH-dependent gelation and an acceptably high in sol-state viscosity, which is an additional problem in injection.
The most common copolymers are linear chain copolymers of the ABA or BAB type, where A and B are hydrophilic and hydrophobic polymers, respectively. In these, the formation of a physical gel is governed by the same factors as in the homogeneous polymers discussed above: high polymer concentration and chain shrinkage as the solvent quality deteriorates, in particular by temperature change. Since hydrophobicity is determined by the total area of atomic groups unable to form hydrogen bonds, increasing the length of the polymer chain segments made of hydrophobic groups, such as methylene groups (-CH2-), allows for reaching the gel-state at 37 °C and sol-state at T ≤ 20 °C.
The most commonly used hydrophilic polymer is oxipolyethylene (-CH2-CH2-O-)n), which, depending on the molecular mass M, is referred to as polyethylene glycol (PEG, low molecular weight, M ≤ 3000 g/mol), synthesized without catalyst, electrically neutral) or polyethylene oxide (PEO, high molecular weight, synthesized with a complex metal-organic catalyst and therefore containing bound Ca2+ or Zn2+ ions that impart a weak positive charge to the chain [46]). The presence of an oxygen atom in the chain backbone determines the high flexibility of the PEG/PEO chain due to increased conformational freedom around the C–O–C bonds, and hydrophilicity is determined by the strong hydration of the oxygen atoms in the ethylene oxide units [47]. The most commonly used block copolymers are ABA and BAB of (A) hydrophilic PEG and (B) hydrophobic polypropylene glycol (PPG, (-CH2-CH(CH3)-O-)n), and its additional methylene group (-CH3) determines the hydrophobicity of the PPG segments. Some hydrophobic chain polymers used for the synthesis of block copolymers are shown on Figure 1.
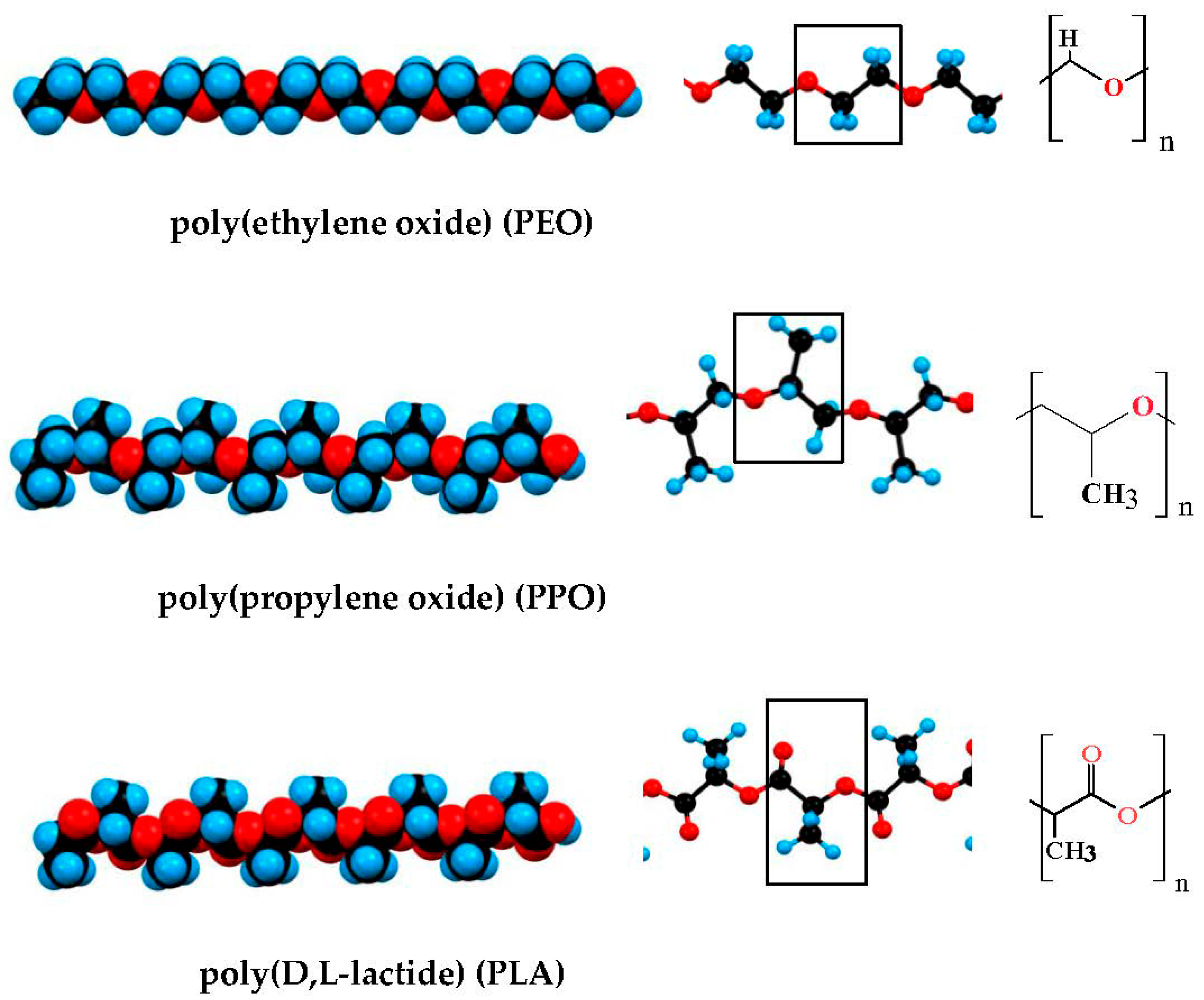
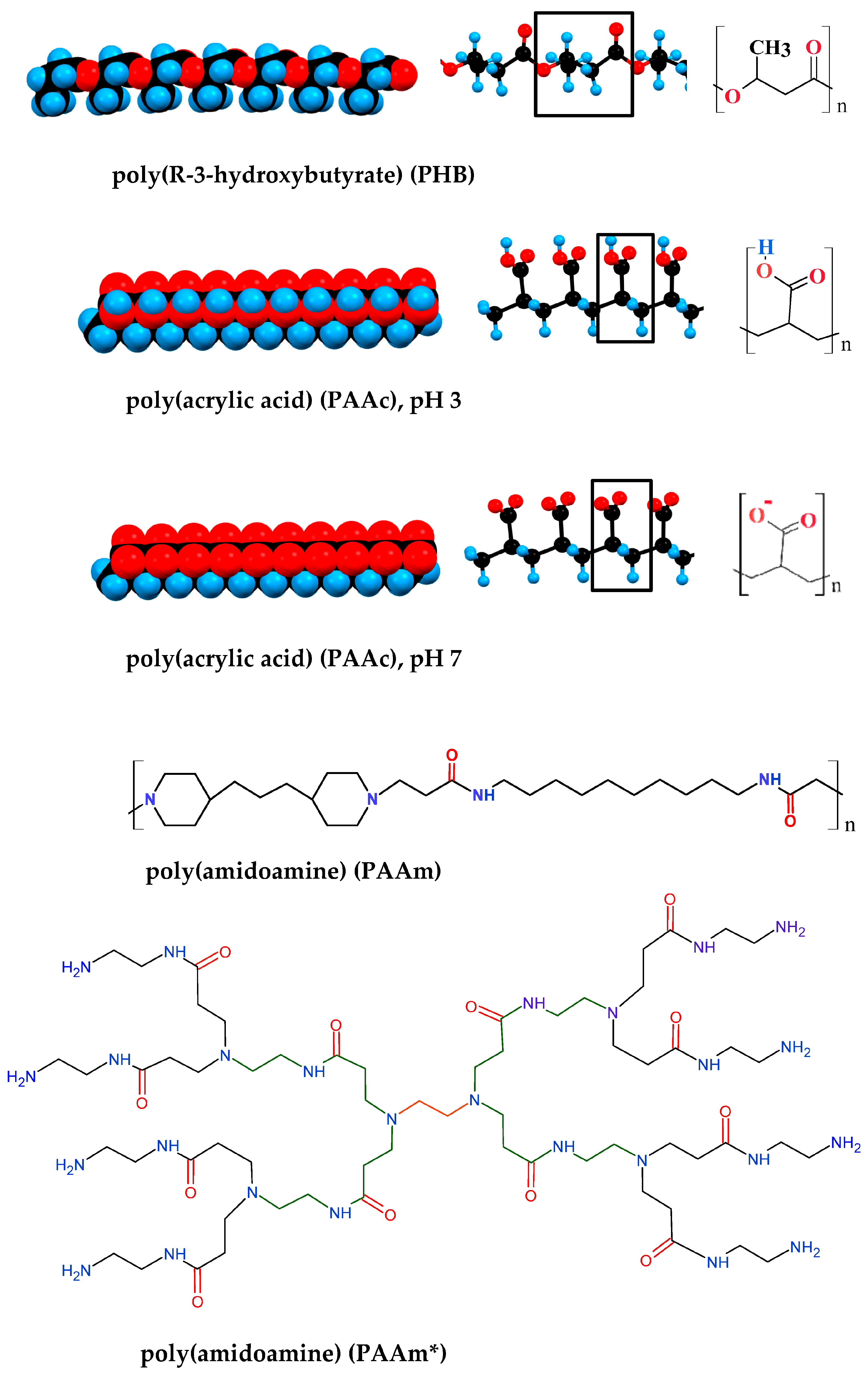
Figure 1. Polymers most commonly used for composition hydrogels with included nanoparticles carrying different chemotherapeutics. * dendrimer.
For medical practice, it is important that the sol–gel transition (inducted by the jump from room temperature to 37 °C) does not take place in the needle while the polymer solution is injected by syringe, this can be avoided by using pH as a second factor required for gel network formation (sol-state at pH ≤ 5 and gel-state at pH 7.4) [19][48][49][50]. For this purpose, block-copolymers with hydrophobic segments containing chargeable groups with a constant pKa allows to alter the degree of ionization at transfer to pH 7.4, and by that, the hydrophobicity are used. pH-dependence can be achieved by introducing charged groups into the polymer units with an appropriate dissociation constant (then the hydrophilic segments behave as a polyelectrolyte).
Polymers with carboxyl groups (pKa ≈ 4) are suitable for this purpose because the pH-dependent dissociation (COOH ↔ COO−) emerges in the pH range 3–5, so that at a low pH, the polymer chain is electrically neutral and at pH 7.4, it is negatively charged. The presence of closely disposed ionizable groups (each unit of the chain can carry one or more COO− groups that leads to a high liner charge density) results in a shift of pKa towards the alkaline region due to the increased local concentration of H3O+ cations and to an anticooperative effect: the curve of the degree of ionization as a function of pH becomes flatter. An example of such a polymer is carboxymethyl cellulose (CMC), which is produced by the chemical modification of natural cellulose (poly-1,4-D-glucose by its chemical nature) at a degree of substitution DS = 0.8–1.2 of the hydroxylic groups of the glucose units with methyl-carboxyl groups (–CH2COOH). At degree of substitution DS ≈ 1 (one charge per glucose unit) and pH 7, the high density of negatively charged groups COO− leads to condensation of counterions from the medium (for instance Na+ cations), which reduce the effective charge of the chain. A feature of CMC is the high chain rigidity, which is caused by the highly constrained conformational freedom between adjacent glucose units due to the β-1,4-linkage between them (the rotation round C–O–C bonds between the C-1 and C-4 atoms is impossible). This is in contrast to the natural polymer amylose (chemically identical to cellulose), with a flexible chain due to α-configuration at C1 atom in the α-1,4-linkage that allows for rotation between the glucose units.
Other chargeable polymers are those congaing groups of tertiary amines that can obtain a pH-dependent positive charge: ≡N ↔ ≡NH+. Such polymers are convenient for injectable gels because the ionization constant of ≡N groups allows for altering their charge in the physiological pH range. An example of such thermo- and pH-dependent injectable gels is the triblock copolymer PAAm-PEG-PAAm [51]. At 20 °C and pH 6.8, the polymer is in a solution-state, but becomes a gel at the physiological 37 °C and pH 7.4 (viscosity increases with more than five orders of magnitude (form 0.1 to 104.7 Pa·s) at a polymer concentration of 12.5 wt%. The pH is the most important factor for the sol–gel phase transition: at 25 °C, the relatively small decrease of the H+-concentration from pH 6.8 to pH 7.4 leads to a drastic increase in the viscosity with four orders of magnitude, while the temperature increases from 25 to 37 °C—with only a half order. These parameters are achieved using the appropriate polymer concentration and amphiphilic structure of the triblock copolymer: two long-chain hydrophobic PAAm (poly(amidoamine) segments with dual (pH and temperature) functionality and a medial hydrophilic PEG segments. The pH and temperature increase caused the growth of the hydrophobicity of PAAm segments because of the pH-determined partial deionization of the ≡NH+ groups (half of them are charged at pH 7.4 and 20 °C), and additionally by a thermos-induced shift of their ionization constant from pKa 7.4 to pKa 6.8 at temperature increasing from 20 to 40 °C. The increased hydrophobicity of PAAm segments leads to the formation of a gel network by van-der-Waals contacts between the approaching chain parts of the next macromolecules, which are entangled with the segments of adjacent polymer chains (due to the high concentration of the polymer solution).
In the review [13] the sol–gel phase transition of PAAm-PEG-PAAm copolymer (described in Refs. [19][51]) is explained by the transition of PAAc segments from a hydrophilic to hydrophobic state at an increase in pH from low to high values (pH 3.0 → pH 7.4). However, this explanation is incorrect because PAAm is wrongly written as PAAc (poly(acrylic acid) instead (poly(amidoamine); the confusion comes from the fact that in the literature, as least three polymers with quite different structures are designed by the abbreviation PAA (Figure 1). Furthermore, the assertion that a hydrophilic polymer segment with acid groups, such as (poly(acrylic acid), became hydrophobic at ionization (COOH ↔ COO−), is principally erroneous, since upon dissociation of a proton from the carboxyl groups they have become even more hydrophilic as the partial charge of the oxygen atom (due to the high electron-affinity, leading to the polarization of the C–O and C–H covalent bonds) becomes a whole (Coulomb) charge, and this leads to an even stronger orientation of the dipole H2O molecules, the closest of which are strongly electrostatically bonded to this oxygen atom and form hydrogen bonds with the second layer water molecules, i.e., when the acidity of the polymer solution is reduced from pH 3 to pH 7, the hydrophilic segments become even more hydrophilic, but not hydrophobic.
2.2. Irreversible Chemical Hydrogels
This type of hydrogel is produced by the covalent cross-linking of chains of one type of hydrophilic polymer or with chains of another type (copolymers) of hydrophilic or hydrophobic polymer, resulting in a thermo-irreversible gel network. The pore size depends on the length of the polymer chains and the polymer concentration. An example of a small pore hydrophilic gel is poly(acrylamide) (PAM) (used in biochemical studies for the electrophoresis of proteins denatured with the surfactant sodium dodecyl sulfate), with small pores that do not allow for the translational movement of globular proteins in a native conformation.
When the gel network is a co-polymer of hydrophilic and hydrophobic filaments (amphiphilic gel), if the latter have sufficient length, they adopt a collapsed ball conformation—domains—in which hydrophobic chemotherapeutics can be embedded as free molecules or adsorbed onto nanoparticles with a hydrophobic surface [52]. An example of one such gel is a co-polymer of hydrophilic poly(acrylamide) (PAM) and hydrophobic poly(methacrylate) (PMC) [53].
There are several polymerization techniques: free-radical, esterification and photo-polymerization. Free-radical polymerization is only acceptable for externally applied gels, as this type of polymerization always leaves a residual free monomer radical that is highly toxic to tissues. Photopolymerization is used as a second stage of the polymerization of a hydrogel previously formed by free-radical polymerization, so a co-network between poly(methacrylate) (MA) chains crosslinked by ethylene glycol dimethacrylate (EGDMA) is obtained [54].
In Table 1, the most commonly used polymers for hydrogels are given.
Table 1. Polymers used for hydrogels. The abbreviations repeat those used by the authors of the cited references.
| № | Polymer | Abbreviation |
|---|---|---|
| 1 | gelatin | G |
| 2 | hyaluronic acid | HA |
| 3 | alginate | ALG |
| 4 | chitosan | CS |
| 5 | dextran | DEX |
| 6 | oleopolyol | OA |
| 7 | poly(N-isopropylacrylamide) | PNIPAm |
| 8 | Poly (N, N-diethyl acrylamide) | PDEA |
| 9 | polyacrylonitrile-polyamide | PAN-PA |
| 10 | poly(acrylic acid) | PAAc |
| 11 | poly(amidoamine) | PAAm |
| 12 | poly(methacrylic acid) | PMAA |
| 13 | poly(N-isopropylacrylamide-co-acrylamide | PNIPAAm |
| 14 | poly(N-vinylpyrrolidone) | PVPON |
| 15 | poly(N-isopropylacrylamide-co-acrylamide) | PNIPAAm-co-AAm |
| 16 | poly(β-aminoester urethane) | PAEU |
| 17 | acrylamide-methylenebisacrylamide-green tea | AM-MBA-GT |
| 18 | poly(polypropylene glycol) | PPG |
| 19 | poly(ethylene glycol) | PEG |
| 20 | methoxypoly(ethylene glycol) | mPEG |
| 21 | polyethyleneimine | PEI |
| 22 | carboxymethyl cellulose | CMC |
| 23 | poly lactic-co-glycolic acid | PLGA |
| 24 | poly(ethylene glycol) -oleic acid | OA-PEG |
| 25 | prepare aminated guar gum | AGG |
| 26 | polyethyleneimine | PEI |
| 27 | poly vinyl alcohol | PVA |
| 28 | poly(N-isopropylacrylamide) | PNIPAM |
| 29 | poly(β-amino ester) | PBAE |
| 30 | poly(N-isopropylacrylamide-co-maleic anhydride)]@strach | PNIPAAm-co-MA @starch |
| 31 | poly(ethylene glycol)-block-poly(N-isopropylacrylamide- co-maleic anhydride)2-graft-poly(ethylene glycol) | PEG-b-(PNIPAAm- co-PMA)2-g-PEG |
| 32 | N,N′-(dimethylamino)ethyl methacrylate-co-maleic anhydride | DMAEMA-co-MA |
| 33 | poly(N-isopropylacrylamide-co-itaconic anhydride)- | P(NIPAAm-co-IA)-PEG |
| 34 | poly[(2-succinyloxyethylmethacrylate)-b-(N-isopropyl acrylamide)-b-dimethylaminoethylmethacrylate) |
P(SEMA-b-NIPAM- b-DMAEMA) |
| 35 | glycidylmethacrylate-grafted-maleated cyclodextrin | P(GMA-g-MACD) |
| 36 | poly(D,L-lactide-co-glycolide)-b-poly(ethylene glycol)-b-poly(D,L-lactide-co-glycolide) | PLGA-PEG-PLGA |
| 37 | sodium alginate- poly(acrylamide- co-N-vinylcaprolactam-co-acrylamidoglycolic acid) |
SA-PAVA |
| 38 | poly (N-vinyl pyrrolidone/dextran) | PVP-DEX |
| 39 | Strychnos potatorum L. (SPL) polysaccharide-based dual-responsive semi-IPN-type | SPL-DMA |
| 40 | N-fluorenylmethoxycarbonyl-di-phenylalanine | Fmoc-FF |
| 41 | poly(N-isopropylacrylamide-co-acrylamide) | NIPAAm-co-AAm |
| 42 | poly(N-isopropyl-acrylamide-acrylic acid) | PNA |
2.3. Biodegradability of Hydrogels
The biodegradability of the hydrogels has an essential role in the increasing use of hydrogels as a drug delivery system. In the term “biodegradability”, all types of in vivo degradation are included: from simple hydrolysis to enzymatically catalyzed degradation. In the biodegradation process, the polymers that form the hydrogels are degraded to monomers by breaking chemical bonds. There are three main mechanisms of biodegradation [55]: (a) Solubilization. A great number of water-soluble polymers are determined as biodegradable due to their ability to dissolve in water. Such polymers are dextran (DEX), poly(ethylene glycol) (PEG), poly(ethylene oxide) (PEO), polyvinyl alcohol (PVA), etc. [56][57]; (b) Hydrolysis. Another mechanism of biodegradation of the polymers is the hydrolysis of ester bonds between the monomers with the formation of an alcohol and a carboxylic acid, as well as amide bonds between the monomers with the formation of an amide and a carboxylic acid. Chemical hydrolysis is characteristic for polylactic-co-glycolic acid (PLGA), poly(amidoamine) (PAA) poly(ethylene glycol)-oleic acid (OA-PEG), poly(β-amino ester) (PBAE), etc. [58]; (c) Enzymatic degradation. In the human body, there are special enzymes—hydrolases (class III enzymes), which catalyze hydrolytic bonds (carbon–oxygen (C–O), carbon–nitrogen (C–N), carbon–carbon (C–C), phosphorus–nitrogen (P–N) bonds, etc.) and cleavage involving water. For example, hyaluronic acid (HA) is degraded by the hydrolase hyaluronidase [59]. Other polymers that undergo enzymatic hydrolysis and/or pH-sensitive hydrolysis/gelation are chitosan, gelatin, etc. [60][61].
Some polymers used for the composition of hydrogels cannot undergo degradation. Such non-biodegradable polymers are cellulose derivatives (for instance: carboxymethyl cellulose, CMC), poly(methacrylate) (PMA), etc. [62].
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653.
- Alfarouk, K.O.; Stock, C.M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15, 71.
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245.
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73.
- Wolinsky, J.B.; Colson, Y.L.; Grinstaff, M.W. Local drug delivery strategies for cancer treatment: Gels, nanoparticles, polymeric films, rods, and wafers. J. Control Release 2012, 159, 14–26.
- Fahr, A.; Liu, X. Drug delivery strategies for poorly water-soluble drugs. Expert Opin. Drug Deliv. 2007, 4, 403–416.
- Yolles, S.; Leafe, T.D.; Meyer, F.J. Timed—Release depot for anticancer agents. J. Pharm. Sci. 1975, 64, 115–116.
- Orive, G.; Hernández, R.M.; Gascón, A.R.; Pedraz, J.L. Micro and nano drug delivery systems in cancer therapy. Cancer Ther. 2005, 3, 131–138.
- Hristova, S.H.; Zhivkov, A.M. Montmorillonite colloid plates with adsorbed cytochrome c: In vitro cytotoxic effect on colon cancer cell culture. Cancer Nanotechnol. 2021, 12, 23.
- Hristova, S.H.; Zhivkov, A.M. Protein–Mineral Composite Particles with Logarithmic Dependence of Anticancer Cytotoxicity on Concentration of Montmorillonite Nanoplates with Adsorbed Cytochrome c. Pharmaceutics 2023, 15, 386.
- Benkhaya, S.; M'rabet, S.; El Harfi, A. A review on classifications, recent synthesis and applications of textile dyes. Inorg. Chem. Commun. 2020, 115, 107891.
- Zhao, H.; Javed, B.; Tian, F.; Liu, K. Hydrogel on a smart nanomaterial interface to carry theraupetics for digitalized glioma threatment. Gels 2022, 8, 664.
- Jiang, Y.; Krishnan, N.; Heo, J.; Fang, R.H.; Zhang, L. Nanoparticle–hydrogel superstructures for biomedical applications. J. Control Release 2020, 324, 505–521.
- Blanco, E.; Kessinger, C.W.; Sumer, B.D.; Gao, J. Multifunctional micellar nanomedicine for cancer therapy. Exp. Biol. Med. 2009, 234, 123–131.
- Cattel, L.; Ceruti, M.; Dosio, F. From conventional to stealth liposomes a new frontier in cancer chemotherapy. Tumori J. 2003, 89, 237–249.
- Gu, D.; O’Connor, A.J.; Qiao, G.G.H.; Ladewig, K. Hydrogels with smart systems for delivery of hydrophobic drugs. Expert Opin. Drug Deliv. 2017, 14, 879–895.
- Nguyen, M.K.; Lee, D.S. Injectable biodegradable hydrogels. Macromol. Biosci. 2010, 10, 563–579.
- Du, W.; Zong, Q.; Guo, R.; Ling, G.; Zhang, P. Injectable nanocomposite hydrogels for cancer therapy. Macromol. Biosci. 2021, 21, e2100186.
- Kelland, L.R.; Clarke, S.J.; McKeage, M.J. Advances in platinum complex cancer chemotherapy. Platin. Met. Rev. 1992, 36, 178–184.
- Elstad, N.L.; Fowers, K.D. OncoGel (ReGel/paclitaxel)—Clinical applications for a novel paclitaxel delivery system. Adv. Drug Deliv. Rev. 2009, 61, 785–794.
- Bajaj, G.; Kim, M.R.; Mohammed, S.I.; Yeo, Y. Hyaluronic acid-based hydrogel for regional delivery of paclitaxel to intraperitoneal tumors. J. Control Release 2012, 158, 386–392.
- Utech, S.; Boccaccini, A.R. A review of hydrogel-based composites for biomedical applications: Enhancement of hydrogel properties by addition of rigid inorganic fillers. J. Mater. Sci. 2016, 51, 271–310.
- Favre, E.; Leonard, M.; Laurent, A.; Dellacherie, E. Diffusion of polyethyleneglycols in calcium alginate hydrogels. Colloids Surf. A Physicochem. Eng. Asp. 2001, 194, 197–206.
- Bock, N.; Dargaville, T.R.; Woodruff, M.A. Electrospraying of polymers with therapeutic molecules: State of the art. Prog. Polym. Sci. 2012, 37, 1510–1551.
- Ma, G.; Lin, W.; Yuan, Z.; Wu, J.; Qian, H.; Xu, L.; Chen, S. Development of ionic strength/pH/enzyme triple-responsive zwitterionic hydrogel of the mixed l-glutamic acid and l-lysine polypeptide for site-specific drug delivery. J. Mater. Chem. B 2017, 5, 935–943.
- Huang, H.; Pierstorff, E.; Osawa, E.; Ho, D. Active nanodiamond hydrogels for chemotherapeutic delivery. Nano Lett. 2007, 7, 3305–3314.
- Lundqvist, M.; Stigler, J.; Elia, G.; Lynch, I.; Cedervall, T.; Dawson, K.A. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc. Natl. Acad. Sci. USA 2008, 105, 14265–14270.
- Rahman, M.; Laurent, S.; Tawil, N.; Yahia, L.H.; Mahmoudi, M.; Rahman, M.; Mahmoudi, M. Nanoparticle and protein corona. Protein-Nanopart. Interact. Bio-Nano Interface 2013, 15, 21–44.
- Monopoli, M.P.; Walczyk, D.; Campbell, A.; Elia, G.; Lynch, I.; Baldelli Bombelli, F.; Dawson, K.A. Physical−chemical aspects of protein corona: Relevance to in vitro and in vivo biological impacts of nanoparticles. J. Am. Chem. Soc. 2011, 133, 2525–2534.
- Hajipour, M.J.; Laurent, S.; Aghaie, A.; Rezaee, F.; Mahmoudi, M. Personalized protein coronas: A “key” factor at the nanobiointerface. Biomater. Sci. 2014, 2, 1210–1221.
- Corbo, C.; Molinaro, R.; Tabatabaei, M.; Farokhzad, O.C.; Mahmoudi, M. Personalized protein corona on nanoparticles and its clinical implications. Biomater. Sci. 2017, 5, 378–387.
- Hajipour, M.J.; Raheb, J.; Akhavan, O.; Arjmand, S.; Mashinchian, O.; Rahman, M.; Mahmoudi, M. Personalized disease-specific protein corona influences the therapeutic impact of graphene oxide. Nanoscale 2015, 7, 8978–8994.
- Wichterle, O.; Lím, D. Hydrophilic Gels for Biological Use. Nature 1960, 185, 117–118.
- Kashyap, N.; Kumar, N.; Kumar, M.N.V.R. Hydrogels for Pharmaceutical and Biomedical Applications. Crit. Rev. Ther. Drug Carr. Syst. 2005, 22, 107–150.
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007.
- Larrañeta, E.; Stewart, S.; Ervine, M.; Al-Kasasbeh, R.; Donnelly, R.F. Hydrogels for Hydrophobic Drug Delivery. Classification, Synthesis and Applications. J. Funct. Biomater. 2018, 9, 13.
- Rafikov, S.R.; Budtov, V.P.; Monakov, Y.B. Introduction in the Physical Chemistry of Polymers; Nauka: Moscow, Russia, 1978.
- Tenford, C. Physical Chemistry of Polymers; Himiya: Moscow, Russia, 1965.
- Ta, H.T.; Dass, C.R.; Dunstan, D.E. Injectable chitosan hydrogels for localised cancer therapy. J. Control Release 2008, 126, 205–216.
- Klouda, L.; Mikos, A.G. Thermosensitive hydrogels in biomedical applications. Eur. J. Pharm. Biopharm. 2008, 68, 34–45.
- Norouzi, M.; Nazari, B.; Miller, D.W. Injectable hydrogel-based drug delivery systems for local cancer therapy. Drug Discov. Today 2016, 21, 1835–1849.
- Graham, S.; Marina, P.F.; Blencowe, A. Thermoresponsive polysaccarides and their thermoriversible physical hydrogel networks. Carbohydr. Polym. 2019, 207, 143–159.
- Kojima, H. Studies on the gel transition and aqueous solutions of thermosensitive polymers. Polym. J. 2018, 50, 411–418.
- Zhivkova, I.V.; Zhivkov, A.M.; Stoychev, D.S. Electrostatic behaviour of polyethylene oxide. Eur. Polym. J. 1998, 34, 531–538.
- Bekturov, E.A.; Bakaunova, Z.K. Synthetic Water-Soluble Polymers in Solutions; Nauka: Alma-Ata, Kazakh SSR, 1981.
- Xin, H.; Nafisy, S. Drug delivery based on stimuli-responsive injectable hydrogels for breast cancer therapy: A review. Gels 2022, 8, 45.
- Kwon, S.S.; Kong, B.J.; Park, S.N. Physicochemical properties of pH-sensitive hydrogels based on hydroxyethyl cellulose–hyaluronic acid and for applications as transdermal delivery systems for skin lesions. Eur. J. Pharm. Biopharm. 2015, 92, 146–154.
- Singh, N.K.; Lee, D.S. In situ gelling pH- and temperature-sensitive biodegradable block copolymer hydrogels for drug delivery. J. Control Release 2014, 193, 214–227.
- Nguyen, M.K.; Park, D.K.; Lee, D.S. Injectable poly(amidoamine)-poly(ethylene glycol)-poly(amidoamine) triblock copolymer hydrogel with dual sensitivities: pH and temperature. Biomacromolecules 2009, 10, 728–731.
- Patrickios, C.S.; Georgiou, T.K. Covalent amphiphilic polymer networks. Curr. Opin. Colloid Interface Sci. 2003, 8, 76–85.
- Rikkou-Kalourkoti, M.; Kitiri, E.N.; Patrickios, C.S.; Leontidis, E.; Constantinou, M.; Constantinides, G.; Zhang, X.; Papadakis, C.M. Double networks based on amphiphilic cross-linked star block copolymer first conetworks and randomly cross-linked hydrophilic second networks. Macromolecules 2016, 49, 1731–1742.
- Swarnalatha, S.; Gopi, R.; Ganesh Kumar, A.; Selvi, P.K.; Sekaran, G. A novel amphiphilic nano hydrogel using ketene based polyester with polyacrylamide for controlled drug delivery system. J. Mater. Sci. Mater. Med. 2008, 19, 3005–3014.
- Kamath, K.R.; Park, K. Biodegradable hydrogels in drug delivery. Adv. Drug Deliv. Rev. 1993, 11, 59–84.
- Gunatillake, P.A.; Adhikari, R.; Gadegaard, N. Biodegradable synthetic polymers for tissue engineering. Eur. Cell Mater. 2003, 5, 1–16.
- Hyun, H.; Kim, Y.H.; Song, I.B.; Lee, J.W.; Kim, M.S.; Khang, G.; Park, K.; Lee, H.B. In vitro and in vivo release of albumin using a biodegradable MPEG-PCL diblock copolymer as an in situ gel-forming carrier. Biomacromolecules 2007, 8, 1093–1100.
- Ranucci, E.; Spagnoli, G.; Ferruti, P.; Sgouras, D.; Duncan, R. Poly (amidoamine) s with potential as drug carriers: Degradation and cellular toxicity. J. Biomater. Sci. Polym. Ed. 1991, 2, 303–315.
- Zhong, S.P.; Campoccia, D.; Doherty, P.J.; Williams, R.L.; Benedetti, L.; Williams, D.F. Biodegradation of hyaluronic acid derivatives by hyaluronidase. Biomaterials 1994, 15, 359–365.
- Matica, A.; Menghiu, G.; Ostafe, V. Biodegradability of chitosan based products. New Front. Chem. 2017, 26, 75–86.
- Alimirzaei, F.; Vasheghani-Farahani, E.; Ghiaseddin, A.; Soleimani, M. pH-Sensitive Chitosan Hydrogel with Instant Gelation for Myocardial Regeneration. J. Tissue Sci. Eng. 2017, 8, 3.
- Pillai, O.; Panchagnula, R. Polymers in drug delivery. Curr. Opin. Chem. Biol. 2001, 5, 447–451.
More
Information
Subjects:
Chemistry, Physical
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
679
Revisions:
4 times
(View History)
Update Date:
31 Jul 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No