Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Manoj Kumar | -- | 3550 | 2023-05-31 08:52:17 | | | |
| 2 | Rita Xu | Meta information modification | 3550 | 2023-05-31 08:58:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Johnson, V.; Vasu, S.; Kumar, U.S.; Kumar, M. Extracellular Vesicles Surfaces for Cancer Immunotherapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/45038 (accessed on 26 July 2026).
Johnson V, Vasu S, Kumar US, Kumar M. Extracellular Vesicles Surfaces for Cancer Immunotherapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/45038. Accessed July 26, 2026.
Johnson, Vinith, Sunil Vasu, Uday S. Kumar, Manoj Kumar. "Extracellular Vesicles Surfaces for Cancer Immunotherapy" Encyclopedia, https://encyclopedia.pub/entry/45038 (accessed July 26, 2026).
Johnson, V., Vasu, S., Kumar, U.S., & Kumar, M. (2023, May 31). Extracellular Vesicles Surfaces for Cancer Immunotherapy. In Encyclopedia. https://encyclopedia.pub/entry/45038
Johnson, Vinith, et al. "Extracellular Vesicles Surfaces for Cancer Immunotherapy." Encyclopedia. Web. 31 May, 2023.
Copy Citation
Extracellular vesicles are small membranous particles secreted by cells. Extracellular vesicles facilitate the transportation of biomolecules, such as protein, RNA, and DNA fragments, to communicate with neighboring and distant cells. Cancer cells use extracellular vesicles to hijack the immune system and induce cancer-promoting signals. Modifying extracellular vesicles using surface engineering tools allows the addition of biomolecules for targeted delivery, thus modulating the hijacked tumor immune microenvironment to improve therapeutic efficacy.
extracellular vesicles
exosomes
microvesicles
apoptotic bodies
surface engineering
1. Introduction
Extracellular vesicles (EVs) are small membrane-bound particles that mediate cell-to-cell communication. All cells secrete EVs generated via intrinsic cellular biogenic pathways as the cargo of signaling molecules typically involved in cell regeneration, differentiation, and proliferation [1]. As small lipid bilayer particles, EVs carry surface proteins, lipids, cytokines, glycans, and other encapsulated molecules [2]. Apart from bioactive molecules, EVs can also encapsulate various cellular organelles, including the transfer of functional mitochondria to other cells, and promote cell survival and tissue regeneration [3]. The composition of each EV subpopulation differs based on their formation mechanisms. These EVs are broadly categorized into exosomes, microvesicles, and apoptotic bodies, ranging between 30–5000 nanometers (nm) [4]. Exosome biosynthesis involves an endosomal sorting complex required for transport (ESCRT), which guides intraluminal vesicle transport within late endosomes to the plasma membrane. The matured endosomal membrane fuses with the plasma membrane and causes the exocytosis of exosomes in the size range of 30–150 nm in diameter [5]. Unlike exosome formation, microvesicle formation does not require exocytosis. Microvesicles are solely derived from the budding of plasma membranes under external stimuli or stress factors. The membrane composition of microvesicles closely represents that of the plasma membrane of parent cells [6]. On the contrary, apoptotic bodies manifest from membrane blebbing, which involves the formation of small protrusions or blebs of the plasma membrane [7]. The membrane and luminal compositions of apoptotic bodies have distinct characteristics. Apoptotic bodies are enriched with phosphatidylserine (PS) phospholipid on the outer leaflet as opposed to their presence in the inner leaflet in the plasma membrane of a healthy cell [8]. PS on the outer leaflet of apoptotic bodies serves as an “eat me” signal and facilitates recognition by the macrophages for disposal.
The cellular uptake mechanism of EVs depends on the recipient cells’ membrane receptor components. The surface proteins and receptors facilitate the initial adhesion of exosomes to recipient membranes [9]. Upon adhesion, the membrane-associated exosomes are usually endocytosed via phagocytosis, micropinocytosis, or clathrin/caveolae-mediated endocytosis, trigger the transduction of intracellular signaling pathways or integrate with cell membrane-transferring protein for cell internalization. Microvesicles can also internalize via surface receptors or ligands, including integrins, ligands such as tetraspanins (CD63, CD9, or CD81), and heat-shock proteins. These receptors are core protein signatures of EVs and play a prominent role in cellular uptake [10]. Since microvesicles exhibit a broad range of size distribution (50 nm to 5 µm), their uptake mechanism is also determined, to a certain extent, by their sheer size. The membrane of apoptotic bodies carries a significant extent of tumor-associated macrophage (TAM) receptors, phospholipids, integrins, and scavenger receptors that enable the specific recognition of apoptotic bodies by immune cells such as macrophages and dendritic cells. Their internalization by cells usually culminates in lysosomal degradation [11].
Tumors deploy EVs to alter stromal cell behavior and promote metastasis. EVs are increasingly shown to be involved in the invasive–metastatic cascade. Tumors secrete a heterogenous population of EVs to shape the tumor microenvironment (TME) and change its malignant behavior in response to immune surveillance or therapies [12]. EVs could play the role of a critical biomarker in the diagnosis and prognosis of cancer [13]. While the current understanding of spatial interactions between the host and tumor cells remains poor, EV-mediated signaling has provided critical insights into these interactions. Malignant cells secrete EVs to promote angiogenesis and modulate the immune system to support cancer progression [14][15][16][17]. EVs isolated from cancer patients have been associated with metastasis or relapse [13]. Tumors orchestrate stromal cells via EV-mediated systemic reprogramming and support pre-metastatic niche formation and subsequent metastasis [12][18]. This allows cancers to differentially exhibit the temporal course of proliferation and the metastatic progression of distant organs.
2. Surface Engineering of EVs
EVs have a unique origin and molecular composition that renders them highly stable in the bloodstream; thus, they have potential in cancer immunotherapy [19]. However, their efficacy is hampered by ineffective tumor targeting and many surface modification strategies have been implemented to improve tumor targeting (Figure 1) [20]. These strategies enhanced targeting specificity and therapy by introducing new functionalities via a targeted ligand and therapeutic molecules [19]. Precise surface engineering of EVs can be achieved via genetic engineering tools controlling cellular biosynthesis pathways. The two primary tumor immunomodulatory approaches used in this strategy are the modification of EV surfaces to express tumor-specific antigens or immune checkpoint inhibitors to recruit the immune cells [21]. Unlike the genetic engineering approaches, chemical conjugation involves attaching specific molecules or ligands to the surface of EVs using chemical reactions. These molecules could be antibodies, peptides, or other targeting ligands that selectively bind to cancer cells or immune cell receptors to enhance therapeutic efficacy [22]. Lipid insertion is another promising approach for engineering EV surfaces for cancer immunotherapy [23]. Unlike the targeted antibodies and peptides involved, lipid-insertion molecules are generally immunostimulatory, play a role in the stability and longevity of vesicles, and, at times, act as therapeutic agents [24]. The unique advantage of lipid insertion over chemical conjugation is that it allows the precise control of the density and orientation of the inserted molecules on the EV surfaces, which can optimize their targeting and therapeutic efficacy. The preferential interaction of lipids with immune cells acts as a double-edged sword; it comes at the cost of possible toxicity and acute immunogenicity if the lipid insertions are not well-optimized [25].
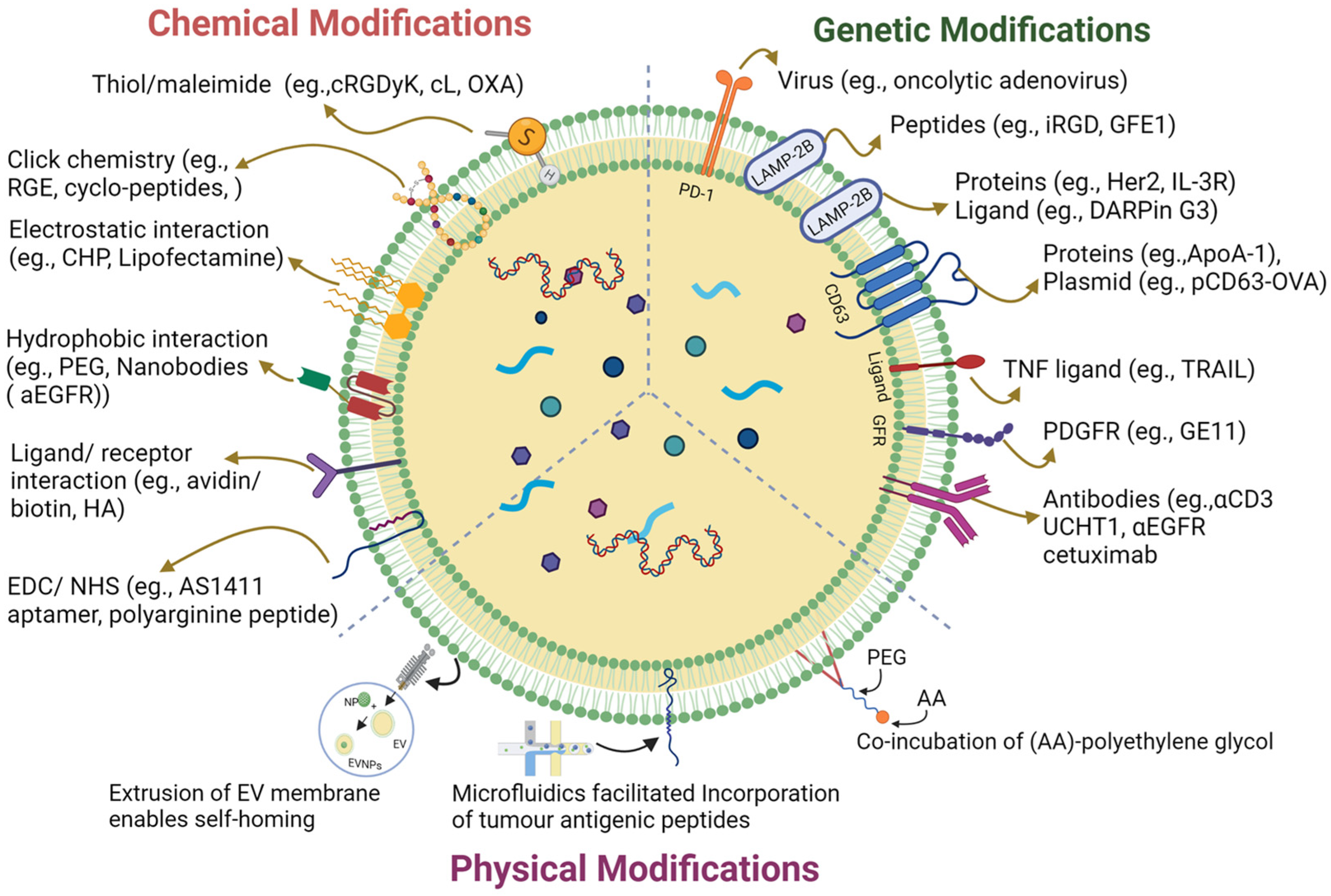
Figure 1. A graphical illustration of extracellular vesicle surface engineering strategies.
3. Genetic Engineering of EV Surfaces for Cancer Immunotherapy
As a transport cargo, the intrinsic property that allows EVs to bypass immune surveillance favors EV fabrication. Cell genetic engineering allows the obtention of desired EV functions to target specific cell types [26]. A good example is using EVs for the controlled activation of immune cells, such as dendritic cells, T-cells, and natural killer cells, in the immunomodulation of TME. Engineered EVs shed from parent cells could improve the specific target recognition efficiency, targeting ability, and anti-tumor efficacy [27][28][29]. Thus, EV surface modification provides a promising clinical application [30]. Typically, the genetic modification of the EV surface is achieved via transfection and activation through protein expression, via tagging molecules on the surface protein and DNA or via RNA delivery to TME [31]. The surface-engineered exosomes with aptamer-based DNA nano assemblies have benefited theragnostic applications [32]. For example, EVs were engineered to express Toll-like receptor (TLR) agonists or co-stimulatory molecules as immunostimulatory proteins [33]. These modifications enhance EVs’ ability to induce a robust anti-tumor response via immune activation in another study [34]. The study demonstrated EV-based activation of dendritic cells and a robust anti-tumor immune response being induced in mice. Thus, using EVs as a delivery system molecule holds great promise for effective cancer immunotherapies.
3.1. EVs Carrying Immune Receptor and Ligand Protein
Another EV surface modification strategy is attaching monoclonal antibody-derived chimeric antigen receptors (CARs) for specific binding on cancer cells [35]. CARs are synthetic proteins that can specifically recognize and bind to tumor cell-surface antigens [36]. Engineered EVs bind to specific receptors on tumor cells when engineered to express CARs, deliver immunomodulatory proteins to target cells, and induce an immune response against cancer cells. For example, Shi et al., 2020, engineered EVs to express a human epidermal growth factor receptor-2 (HER2)-specific CAR against HER2 protein overexpressed in breast cancer [37]. This study demonstrated CAR-EVs’ potential as novel cancer immunotherapy using in vitro and in vivo breast cancer models. Likewise, Fu et al., 2019, demonstrated that exosomes derived from CAR-T-cells have potent anti-tumor effects and low toxicity [38]. The exosomes produced by the engineered CAR-T-cells also expressed the same chimeric antigen receptor as the parent CAR-T cells did. The study found that these CAR-exosomes could target and kill cancer cells with a much lower toxicity profile than CAR-T-cell therapy [38]. Therefore, CAR-exosomes could be a promising alternative to CAR-T-cells in cancer immunotherapy. This approach has been reviewed and extensively discussed in a recent review article by Pagotto et al., 2023 [36]. EVs naturally contain membrane-associated immunoregulatory molecules, including the immune-checkpoint molecules such as programmed death ligand s1 (PD-L1), cytotoxic T lymphocyte antigen-4 (CTLA-4), and the apoptosis-inducing ligands FASL and TNF-related apoptosis-inducing ligand (TRAIL). These immune checkpoints enable EVs to interact with cognate ligands and receptors expressed by T-cells and natural killer (NK) cells in TMEs [39]. An alternative strategy with which to modify the surface of EVs for targeting specific immune cells or tumor cells is the incorporation of peptides, antibodies, or ligands [40]. This approach can also deliver therapeutic cargo, such as RNA and chemotherapy drugs, to specific cells.
The surface engineering of exosomes by the aptamer-based DNA nano assemblies has significantly benefited the theragnostic applications (Figure 1) [32]. TRAIL is a therapeutic agent which induces apoptosis via targeting the death receptors 4 and 5 on cancer cells [41][42]. The TRAIL can be loaded onto the cells via transfection [43][44]. TRAIL-containing exosomes were developed by transducing K562 leukemic cells with the TRAIL lentivirus expression vector [43]. The secreted exosomes exhibited the enhanced apoptosis of lymphoma and melanoma cells. Additionally, exosomes with TRAIL were created using engineered mesenchymal stem cells (MSCs), resulting in the apoptosis of various cancer cell lines, including breast, renal, lung, and mesothelioma [44].
One of the most widely used exosome surface proteins to display a targeting motif is LAMP-2B, a lysosome-associated membrane protein (LAMP) member. LAMP-2B is predominantly available on the lysosomes and endosomes, and a smaller fraction is expressed on the cell surface. The abundant expression of the LAMP-2B protein was reported on the dendritic cell-derived exosomes [45]. LAMP-2B, present on the surface of the exosomes, is a conventional site on which to fuse biomolecules for several functions. Indeed, the surface of the exosomes has a large N-terminal extra-membrane domain of LAMP-2B, which provides a golden ticket for researchers to connect biomolecules and therapeutic agents [46]. For instance, engineering to express LAMP2B on the surface of the mouse immature dendritic cells (imDCs) was achieved via the fusion of the αv-integrin-specific (iRGD) peptide (CRGDKGPDC), which reduces the immunogenicity and toxicity of the extraneous exosomes. imDC-derived EVs were naturally equipped with the iRGD peptide, improving the tumor-targeting capability [29]. These iRGD exosomes were used to specifically target αvβ3-harboring A549 tumors via delivering the KRAS siRNA in vivo, which resulted in tumor suppression via the knocking down of the KRAS gene [47].
Similarly, the N-terminal of LAMP2B fused with interleukin-3 receptor (IL-3Rα) improved exosome-targeting efficiency at treating chronic myeloid leukemia (CML) [48]. The IL-3Rα-exosomes, derived from CML cells with highly expressed IL-3Rα, were further loaded with breakpoint cluster region (BCR)-ABL siRNA and imatinib. These exosomes were highly accumulated at the tumor site, showed an intense anti-tumor effect and increased survival rate of xenografted mice. Another study used a similar strategy to fuse the N-terminus of LAMP2B with HER2 and efficiently targeted colon cancer [49]. The surface of the exosomes expressing HER2-LAMP2B fusion protein promoted tumor-specific uptake via epidermal growth factor receptor (EGFR)-mediated endocytosis. The incorporated HER2-LAMP2B proteins were expressed on exosomes with 5-fluorouracil (5-FU) and miRNA (target-HER2-LAMP2-GFP) via electroporation and incubation, respectively. These exosomes accumulated well on the tumor site and showed extensive suppression of colon cancer in BALB/c nude mice [49]. Likewise, a lentiviral construct containing LAMP-2B-DARPin the G3 chimeric gene was transduced in HEK293T cells to produce exosomes bearing DARPin G3 [50]. Exosomes were loaded with siRNA for targeted delivery to SKB3 tumor cells. These exosomes could specifically target the SKB3 cells and deliver siRNA to inhibit gene expression [50]. Another study transfected the HEK293T cells with a tLyp-1 (tumor-homing and -penetrating peptide CGNKRTR) LAMP2B plasmid construct. The derived exosomes from these cells were electroporated to be loaded with the synthesized siRNA [51]. These tLyp-1-siRNA exosomes showed enhanced delivery for lung cancer via selectively targeting neuropilin receptors (NRP1 and NRP2) expressed on the tumor tissues.
In addition to LAMP2B, the transmembrane protein platelet-derived growth factor receptor (PDGFR) is another commonly used membrane display. In a study, the PDGFR transmembrane region GE11 (YHWYGYTPQNVI) was genetically fused using a phage-display vector transfected in the 293T cells to produce the exosome with GE11 [52]. These exosomes showed low mitogenic activity and a high affinity for EGFR-overexpressing cancer cells; thus, they were demonstrated as part of a tailor-made delivery system for EGFR-targeted therapy. The GE11 exosomes were loaded with anti-tumor nucleic acid inhibitor miRNA let-7 and consistently inhibited mouse tumor growth [52]. Similarly, two antibody fragments (αCD3 UCHT1 and scFv fragments of αEGFR cetuximab) were genetically introduced on the exosome surface [53]. This work demonstrated how the cross-linking of EGFR-expressing breast cancer cells and T-cells effectively promoted anti-tumor immunity. Furthermore, a study combined immune checkpoint blockade and oncolytic virotherapy in single-particle nanovesicles with programmed cell death protein 1 (PD1) to create bioengineered cell membrane nanovesicles (PD1-BCMNs) [54]. The PD1-BCMN nanovesicles were harbored with oncolytic adenovirus (OA). PD1-BCMN nanovesicles specifically delivered the OA to immunologically cold tumor tissue, turning it into an immunologically hot tumor. This led to the presentation of more targets for enhanced delivery and showed a strong anti-tumor immune response via effectively activating the tumor-infiltrating T-cells [54].
3.2. EV Signature Protein Fusion
Exosomes highly express signature proteins, such as CD9, CD63, and CD81, that can be fused with the targeting molecules [55]. In particular, Ran et al., 2020, fused myostatin propeptide with the second extracellular loop of CD63, which increased exosome serum stability and its delivery efficacy in MDX mice [56]. The exosome surface presents a high-density lipoprotein (HDL), ApoA-1, which binds to the scavenger receptor class B type 1 (SR-B1) abundantly located on hepatocellular carcinoma cells. Liang et al., 2018, genetically introduced Apo-A1 in 293T cells, subsequently inserting the extracellular loop of CD63 on the surface of exosomes [57]. The exosomes derived from the ApoA-1-overexpressing donor cells were primed with miR-26a via electroporation. These engineered exosomes selectively bound the HepG2 cells via SR-B1 and were captured via receptor-mediated endocytosis, leading to the release of miRNA in HepG2 cells, reducing cell migration and proliferation [57]. Another study fused CD63 with the ovalbumin antigen (OVA-Ag), transfecting encoding plasmid DNA into parent cells to produce OVA-carrying exosomes [58]. Vaccinating mice with these exosomes showed a strong Ag-specific CD8+ T-cell response repressing tumor growth [59]. Exosome secretion and uptake were visualized by fusing the extracellular loop of CD63 with the fluorescent protein pHluorin.
Similarly, functionally customized exosomes were made through genetic modification to accommodate actively integrated membrane proteins or soluble protein cargos (GIFTed-Exos) [60]. The remarkable properties of GIFTed-Exos that stimulate T-cells were obtained by genetically combining the glucocorticoid-induced tumor necrosis factor receptor family-related ligand (GITRL) with exosome-associated tetraspanin CD9 and transmembrane protein CD70. The feasibility of genetically linking the fluorescent protein mCherry to a membrane protein fusion facilitated the release of apoptin-inducing proteins, of apoptin, and of antioxidant enzymes through light-stimulated delivery. Therefore, a vast array of proteins can be delivered to the target cells through GIFTed-Exos [60]. Surface-displayed antigens on exosomes have been used as in anti-cancer vaccines. A study fused CD63 with OVA-Ag to produce OVA exosomes, improving DNA vaccine immunogenicity and preventing mouse tumor growth [58]. The PS-containing exosome membrane has localized C1C2 domains of lactadherin that can be used for anchoring recombinant proteins to promote the increase in cytokine levels for immunogenicity and therapeutic efficacy in TME. This approach successfully suppressed murine lymphoma via exosome-targeting tumor antigen vaccines in the in vivo model [61]. The exosomal protein vesicular stomatitis infection glycoproteins (VSVGs) are also available on exosome surfaces [62]. The VSVG contains a cytoplasmic region and a transmembrane region. Upon replacing the extracellular and cytoplasmic regions of VSVG with the other proteins (red fluorescent proteins (RFP), luciferase, or green fluorescent proteins (GFP)), a permissible enrichment of surface proteins was created without altering the transmembrane space and symbolic peptide. The surface presentation using VSVG fusion proteins on the exosome promoted the target cell uptake of engineered exosomes via the enhanced protein surface display [63].
3.3. Genetic Engineering Cancer Cell-Derived EVs
In a proof-of-concept study, genetic manipulation of exosomes was achieved via transfection of lung adenocarcinoma cells, SK-LU-1 cells. The isolated exosomes were shown to promote the transmission of TAMs to the M1 active profiles [64]. Likewise, human pancreatic cancer cell (Panc-1 cell)-derived exosomes co-transfected with HA-PEI/HA-PEG NP transmissive miR-155 and miR-125b2 plasmid DNA had synergistic roles in converting M2-like macrophages into M1-like ones [65]. Additionally, genetically engineered human epidermal growth factor and anti-HER2 antibodies have been used as targeting moieties for targeting MDA-MB-468 tumor xenografts [66]. In contrast, the biofunctionalized liposome-like nanovesicles (BLNs) synthesized via attaching the hEGF covalently to the artificial liposomes held outstanding targeting capabilities and high biological functionalities. In addition, doxorubicin (DOX)-conjugated BLNs exhibited significantly higher anti-tumor therapeutic outcomes than the liposome doxorubicin (Doxil), a clinically approved chemotherapy, did [66]. Given the ease of manufacturing and excellent targeting capabilities, BLNs are a promising alternative for immune liposomes and proteoliposomes. In another recent study, α-lactalbumin (α-LA)-engineered breast cancer cell-derived exosomes were loaded with immunogenic cell death (ICD) inducers Hiltonol (a Toll-like receptor agonist) and human neutrophil elastase (ELANE) to form an in situ DC vaccine (HELA-Exos) [67]. Exposure to Hiltonol and tumor antigens adequately stimulated the induction of immunogenic cell death (ICD) in cancer cells by HELA-Exo. This process activated type-one conventional DCs (cDC1s) locally and triggered a robust CD8+ T-cell response against tumor cells. Consequently, this immune response effectively suppressed poorly immunogenic triple-negative breast cancer (TNBC) in both a xenograft mouse model and patient-derived tumor organoids [67].
Similarly, the NIH 3T3 cell lines were engineered to express IL-15/IL-15R𝛼 on the cell membrane. Nanovesicles derived through extrusion overexpressing the IL-15/IL-15R𝛼 (IL-15/IL-15R𝛼-NVs) complex boosted the proliferation, activation, and survival of tumor-infiltrated T-cells via the trans presentation of IL-15 to T-cells and the eliminated tumor. These nanovesicles also promote the activation and proliferation of tumor-specific CD8+ T-cells and TRM cells, effectively inhibiting melanoma growth in mice and increasing the survival rate of mice. An IL-15/IL-15R𝛼-nanovesicle complex was used as cargo to carry the PD-1/PD-L1 inhibitor to a T-cell. The PD-1/PD-L1-inhibitor combined with the IL-15/IL-15R𝛼-nanovesicle complex significantly enhanced the anti-tumor responses [68]. Similarly, Shi et al., 2020, expressed an anti-CD3-anti-HER2 bispecific scFv antibody in Expi293 cells and derived a synthetic multivalent antibody-retargeted exosome (SMART-Exo) to control cellular immunity. The derived SMART-Exo targeted breast cancer-associated HER2 receptors and the T-cell CD3 (Figure 2a). By activating cytotoxic T-cells and redirecting towards attacking HER2-expressing breast cancer cells, the SMART-Exo exhibited specific anti-tumor activity and high potency [37].
CXCR4 is a chemokine receptor that regulates T-cell migration. In a study, MSCs were infected with the PGMLV-PA6-containing virus expressing CXCR4 protein and GFP [69]. The derived exosomes exhibited high CXCR4 expression as a targeted gene–drug delivery system. In addition, the surviving gene (si-survivin) was loaded via electrotransformation to target the tumor site and inhibit growth. RNAi was loaded in the gene–drug delivery system (CXCR4high Exo/si-Survivin) to target lung and gastric cancer. [69]. In another study, murine melanoma B16BL6 cells were transfected with a fusion of streptavidin–cadherin (SAV-LA) protein-encoding plasmid yielding SAV-LA-expressing exosomes (SAV-Exo). It was modified by combining biotinylated CpG (5′-C-phosphate-G-3′) DNA-producing CpG-DNA-modified exosomes (CpG-SAV-Exo). The CpG-SAV-Exo exhibited efficient delivery of the exosome with CpG DNA which promoted the activation of murine dendritic DC2.4 cells in culture and enhanced their the tumor antigen presentation capacity(Figure 2b) [70].
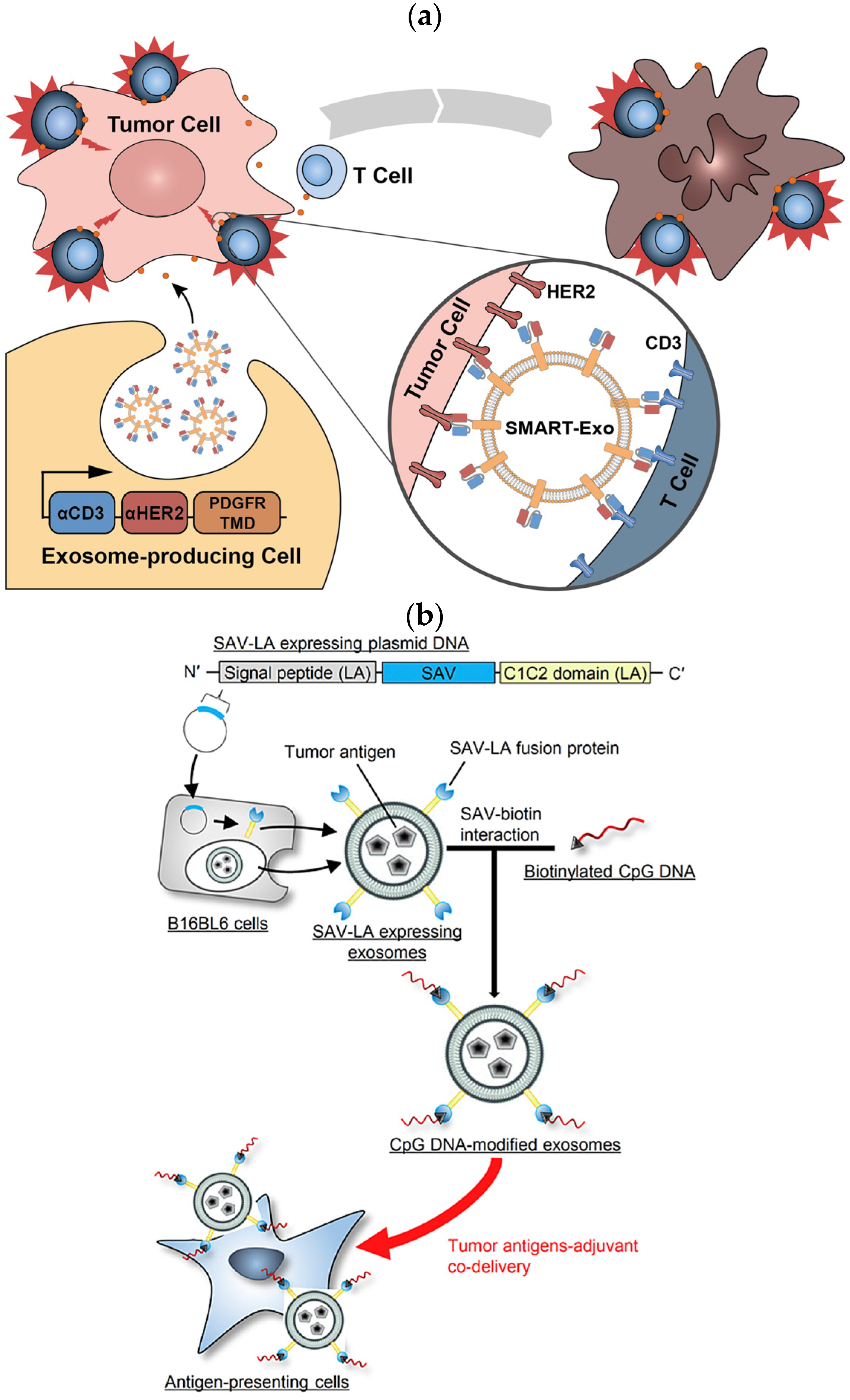
Figure 2. (a) Schematic representation of the surface-expressed SMART exosomes derived from the Anti-CD3-anti-HER2-engineered Expi293 cells exhibiting a bispecific scFv antibody, targeting breast cancer-associated HER2 receptors and the T-cell CD3. (b) Preparation of SAV-LA-expressing exosomes (SAV-exo). B16BL6 cells were transfected with plasmid DNA encoding streptavidin (SAV) fusion with lactahedrin (LA). SAV-exo were collected from the culture supernant of B16BL6 cells. Further, CpG-SAV-exo were prepared by mixing SAV-exo and biotinylated CpG DNA for enhanced tumor antigen presentation (red arrow).
In another study, MSCs were transfected with the pEGFP-C1-GFE1-LAMP2B plasmid. The functional exosome (GExoI) was decorated with pulmonary targeting peptide GFE1 on the membrane surface. These exosomes were loaded with the PI3Kγ inhibitor (IPI549) to suppress melanoma lung metastasis. In a postoperative mouse model, the accumulation of intravenously injected GExoI in the lungs released IPI549 to block G-MDSC recruitment by interfering with CXCLs/CXCR2/PI3Kγ signaling. The increased percentages of CD4+ and CD8+ T-cells in the lungs inhibited metastasis and immunostimulation of the TME [71]. In another study, non-small cell lung cancer (NSCLC) cell lines were overexpressed with PD-1. The biomimetic nanovesicles derived from the NSCLC cell lines exhibited PD-1 (P-NV) and efficiently targeted the NSCLC cells. Further loading P-NVs with doxorubicin (DOX) and 2-deoxy-D-glucose (2-DG) efficiently shrank autochthonous and allografted lung cancers in a mouse model. This P-NV-loaded DOX effectively caused cytotoxicity and activated the anti-tumor immune function of infiltrating T-cells [72]. In another study, U937 monoblastic cells were engineered with the anti-PSMA peptide (WQPDTAHHWAT) to generate exosome mimetics (Ems). These EMs presented an anti-PSMA peptide that targeted advanced prostate cancer (PC). In addition to anti-PSMA peptides, these nanosized EMs displayed monocyte proteins and exosomal markers on the surface. The cellular internalization of the anti-PSMA-EMs was increased in PSMA-positive PC cell lines (LNCaP and C4-2B) and enhanced tumor targeting in solid C4-2B tumors [73].
References
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell Mol. Neurobiol. 2016, 36, 301–312.
- Buzas, E.I. The roles of extracellular vesicles in the immune system. Nat. Rev. Immunol. 2023, 23, 236–250.
- Gomzikova, M.O.; James, V.; Rizvanov, A.A. Mitochondria Donation by Mesenchymal Stem Cells: Current Understanding and Mitochondria Transplantation Strategies. Front. Cell Dev. Biol. 2021, 9, 653322.
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066.
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977.
- Lv, Y.; Tan, J.; Miao, Y.; Zhang, Q. The role of microvesicles and its active molecules in regulating cellular biology. J. Cell Mol. Med. 2019, 23, 7894–7904.
- Coleman, M.L.; Sahai, E.A.; Yeo, M.; Bosch, M.; Dewar, A.; Olson, M.F. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 2001, 3, 339–345.
- Marino, G.; Kroemer, G. Mechanisms of apoptotic phosphatidylserine exposure. Cell Res. 2013, 23, 1247–1248.
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641.
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47.
- Fadok, V.A.; Bratton, D.L.; Henson, P.M. Phagocyte receptors for apoptotic cells: Recognition, uptake, and consequences. J. Clin. Investig. 2001, 108, 957–962.
- Forder, A.; Hsing, C.Y.; Trejo Vazquez, J.; Garnis, C. Emerging Role of Extracellular Vesicles and Cellular Communication in Metastasis. Cells 2021, 10, 3429.
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials—An ISEV position paper. J. Extracell. Vesicles 2015, 4, 30087.
- Ciardiello, C.; Cavallini, L.; Spinelli, C.; Yang, J.; Reis-Sobreiro, M.; de Candia, P.; Minciacchi, V.R.; Di Vizio, D. Focus on Extracellular Vesicles: New Frontiers of Cell-to-Cell Communication in Cancer. Int. J. Mol. Sci. 2016, 17, 175.
- Peinado, H.; Lavotshkin, S.; Lyden, D. The secreted factors responsible for pre-metastatic niche formation: Old sayings and new thoughts. Semin. Cancer Biol. 2011, 21, 139–146.
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495.
- van der Pol, E.; Böing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705.
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012, 151, 1542–1556.
- Zhang, F.; Guo, J.; Zhang, Z.; Duan, M.; Wang, G.; Qian, Y.; Zhao, H.; Yang, Z.; Jiang, X. Application of engineered extracellular vesicles for targeted tumor therapy. J. Biomed. Sci. 2022, 29, 14.
- Ma, Y.; Dong, S.; Li, X.; Kim, B.Y.S.; Yang, Z.; Jiang, W. Extracellular Vesicles: An Emerging Nanoplatform for Cancer Therapy. Front. Oncol. 2020, 10, 606906.
- Mortezaee, K.; Majidpoor, J. Extracellular vesicle-based checkpoint regulation and immune state in cancer. Med. Oncol. 2022, 39, 225.
- Zhang, M.; Hu, S.; Liu, L.; Dang, P.; Liu, Y.; Sun, Z.; Qiao, B.; Wang, C. Engineered exosomes from different sources for cancer-targeted therapy. Signal Transduct. Target. Ther. 2023, 8, 124.
- Zhang, X.; Cui, H.; Zhang, W.; Li, Z.; Gao, J. Engineered tumor cell-derived vaccines against cancer: The art of combating poison with poison. Bioact. Mater. 2023, 22, 491–517.
- Khawar, M.B.; Abbasi, M.H.; Siddique, Z.; Arif, A.; Sheikh, N. An Update on Novel Therapeutic Warfronts of Extracellular Vesicles (EVs) in Cancer Treatment: Where We Are Standing Right Now and Where to Go in the Future. Oxid. Med. Cell Longev. 2019, 2019, 9702562.
- Fobian, S.F.; Cheng, Z.; Ten Hagen, T.L.M. Smart Lipid-Based Nanosystems for Therapeutic Immune Induction against Cancers: Perspectives and Outlooks. Pharmaceutics 2021, 14, 26.
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183.
- Sedgwick, A.E.; D’Souza-Schorey, C. The biology of extracellular microvesicles. Traffic 2018, 19, 319–327.
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763.
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390.
- Sun, Y.; Sun, F.; Xu, W.; Qian, H. Engineered Extracellular Vesicles as a Targeted Delivery Platform for Precision Therapy. Tissue Eng. Regen. Med. 2023, 20, 157–175.
- Zhang, Z.; Dombroski, J.A.; King, M.R. Engineering of Exosomes to Target Cancer Metastasis. Cell Mol. Bioeng. 2020, 13, 1–16.
- Wan, S.; Zhang, L.; Wang, S.; Liu, Y.; Wu, C.; Cui, C.; Sun, H.; Shi, M.; Jiang, Y.; Li, L.; et al. Molecular Recognition-Based DNA Nanoassemblies on the Surfaces of Nanosized Exosomes. J. Am. Chem. Soc. 2017, 139, 5289–5292.
- Wen, C.; Seeger, R.C.; Fabbri, M.; Wang, L.; Wayne, A.S.; Jong, A.Y. Biological roles and potential applications of immune cell-derived extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1400370.
- Lee, J.H.; Song, J.; Kim, I.G.; You, G.; Kim, H.; Ahn, J.H.; Mok, H. Exosome-mediated delivery of transforming growth factor-beta receptor 1 kinase inhibitors and toll-like receptor 7/8 agonists for combination therapy of tumors. Acta Biomater. 2022, 141, 354–363.
- Murphy, D.E.; de Jong, O.G.; Brouwer, M.; Wood, M.J.; Lavieu, G.; Schiffelers, R.M.; Vader, P. Extracellular vesicle-based therapeutics: Natural versus engineered targeting and trafficking. Exp. Mol. Med. 2019, 51, 1–12.
- Pagotto, S.; Simeone, P.; Brocco, D.; Catitti, G.; De Bellis, D.; Vespa, S.; Di Pietro, N.; Marinelli, L.; Di Stefano, A.; Veschi, S.; et al. CAR-T-Derived Extracellular Vesicles: A Promising Development of CAR-T Anti-Tumor Therapy. Cancers 2023, 15, 1052.
- Shi, X.; Cheng, Q.; Hou, T.; Han, M.; Smbatyan, G.; Lang, J.E.; Epstein, A.L.; Lenz, H.J.; Zhang, Y. Genetically Engineered Cell-Derived Nanoparticles for Targeted Breast Cancer Immunotherapy. Mol. Ther. 2020, 28, 536–547.
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F.; et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat. Commun. 2019, 10, 4355.
- Yin, Z.; Fan, J.; Xu, J.; Wu, F.; Li, Y.; Zhou, M.; Liao, T.; Duan, L.; Wang, S.; Geng, W.; et al. Immunoregulatory Roles of Extracellular Vesicles and Associated Therapeutic Applications in Lung Cancer. Front. Immunol. 2020, 11, 2024.
- Pham, T.C.; Jayasinghe, M.K.; Pham, T.T.; Yang, Y.; Wei, L.; Usman, W.M.; Chen, H.; Pirisinu, M.; Gong, J.; Kim, S.; et al. Covalent conjugation of extracellular vesicles with peptides and nanobodies for targeted therapeutic delivery. J. Extracell. Vesicles 2021, 10, e12057.
- Mitchell, M.J.; Wayne, E.; Rana, K.; Schaffer, C.B.; King, M.R. TRAIL-coated leukocytes that kill cancer cells in the circulation. Proc. Natl. Acad. Sci. USA 2014, 111, 930–935.
- Wayne, E.C.; Chandrasekaran, S.; Mitchell, M.J.; Chan, M.F.; Lee, R.E.; Schaffer, C.B.; King, M.R. TRAIL-coated leukocytes that prevent the bloodborne metastasis of prostate cancer. J. Control. Release 2016, 223, 215–223.
- Rivoltini, L.; Chiodoni, C.; Squarcina, P.; Tortoreto, M.; Villa, A.; Vergani, B.; Burdek, M.; Botti, L.; Arioli, I.; Cova, A.; et al. TNF-Related Apoptosis-Inducing Ligand (TRAIL)-Armed Exosomes Deliver Proapoptotic Signals to Tumor Site. Clin. Cancer Res. 2016, 22, 3499–3512.
- Yuan, Z.; Kolluri, K.K.; Gowers, K.H.; Janes, S.M. TRAIL delivery by MSC-derived extracellular vesicles is an effective anticancer therapy. J. Extracell. Vesicles 2017, 6, 1265291.
- Liang, Y.; Duan, L.; Lu, J.; Xia, J. Engineering exosomes for targeted drug delivery. Theranostics 2021, 11, 3183–3195.
- Terasawa, K.; Tomabechi, Y.; Ikeda, M.; Ehara, H.; Kukimoto-Niino, M.; Wakiyama, M.; Podyma-Inoue, K.A.; Rajapakshe, A.R.; Watabe, T.; Shirouzu, M.; et al. Lysosome-associated membrane proteins-1 and -2 (LAMP-1 and LAMP-2) assemble via distinct modes. Biochem. Biophys. Res. Commun. 2016, 479, 489–495.
- Zhou, Y.; Yuan, Y.; Liu, M.; Hu, X.; Quan, Y.; Chen, X. Tumor-specific delivery of KRAS siRNA with iRGD-exosomes efficiently inhibits tumor growth. ExRNA 2019, 1, 28.
- Bellavia, D.; Raimondo, S.; Calabrese, G.; Forte, S.; Cristaldi, M.; Patinella, A.; Memeo, L.; Manno, M.; Raccosta, S.; Diana, P.; et al. Interleukin 3- receptor targeted exosomes inhibit in vitro and in vivo Chronic Myelogenous Leukemia cell growth. Theranostics 2017, 7, 1333–1345.
- Liang, G.; Zhu, Y.; Ali, D.J.; Tian, T.; Xu, H.; Si, K.; Sun, B.; Chen, B.; Xiao, Z. Engineered exosomes for targeted co-delivery of miR-21 inhibitor and chemotherapeutics to reverse drug resistance in colon cancer. J. Nanobiotechnol. 2020, 18, 10.
- Limoni, S.K.; Moghadam, M.F.; Moazzeni, S.M.; Gomari, H.; Salimi, F. Engineered Exosomes for Targeted Transfer of siRNA to HER2 Positive Breast Cancer Cells. Appl. Biochem. Biotechnol. 2019, 187, 352–364.
- Bai, J.; Duan, J.; Liu, R.; Du, Y.; Luo, Q.; Cui, Y.; Su, Z.; Xu, J.; Xie, Y.; Lu, W. Engineered targeting tLyp-1 exosomes as gene therapy vectors for efficient delivery of siRNA into lung cancer cells. Asian J. Pharm. Sci. 2020, 15, 461–471.
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191.
- Cheng, Q.; Shi, X.; Han, M.; Smbatyan, G.; Lenz, H.J.; Zhang, Y. Reprogramming Exosomes as Nanoscale Controllers of Cellular Immunity. J. Am. Chem. Soc. 2018, 140, 16413–16417.
- Lv, P.; Chen, X.; Fu, S.; Ren, E.; Liu, C.; Liu, X.; Jiang, L.; Zeng, Y.; Wang, X.; Liu, G. Surface engineering of oncolytic adenovirus for a combination of immune checkpoint blockade and virotherapy. Biomater. Sci. 2021, 9, 7392–7401.
- Akbari, A.; Nazari-Khanamiri, F.; Ahmadi, M.; Shoaran, M.; Rezaie, J. Engineered Exosomes for Tumor-Targeted Drug Delivery: A Focus on Genetic and Chemical Functionalization. Pharmaceutics 2022, 15, 66.
- Ran, N.; Gao, X.; Dong, X.; Li, J.; Lin, C.; Geng, M.; Yin, H. Effects of exosome-mediated delivery of myostatin propeptide on functional recovery of mdx mice. Biomaterials 2020, 236, 119826.
- Liang, G.; Kan, S.; Zhu, Y.; Feng, S.; Feng, W.; Gao, S. Engineered exosome-mediated delivery of functionally active miR-26a and its enhanced suppression effect in HepG2 cells. Int. J. Nanomed. 2018, 13, 585–599.
- Kanuma, T.; Yamamoto, T.; Kobiyama, K.; Moriishi, E.; Masuta, Y.; Kusakabe, T.; Ozasa, K.; Kuroda, E.; Jounai, N.; Ishii, K.J. CD63-Mediated Antigen Delivery into Extracellular Vesicles via DNA Vaccination Results in Robust CD8(+) T Cell Responses. J. Immunol. 2017, 198, 4707–4715.
- Sung, B.H.; von Lersner, A.; Guerrero, J.; Krystofiak, E.S.; Inman, D.; Pelletier, R.; Zijlstra, A.; Ponik, S.M.; Weaver, A.M. A live cell reporter of exosome secretion and uptake reveals pathfinding behavior of migrating cells. Nat. Commun. 2020, 11, 2092.
- Cheng, Q.; Dai, Z.; Shi, X.; Duan, X.; Wang, Y.; Hou, T.; Zhang, Y. Expanding the toolbox of exosome-based modulators of cell functions. Biomaterials 2021, 277, 121129.
- Rountree, R.B.; Mandl, S.J.; Nachtwey, J.M.; Dalpozzo, K.; Do, L.; Lombardo, J.R.; Schoonmaker, P.L.; Brinkmann, K.; Dirmeier, U.; Laus, R.; et al. Exosome targeting of tumor antigens expressed by cancer vaccines can improve antigen immunogenicity and therapeutic efficacy. Cancer Res. 2011, 71, 5235–5244.
- Mentkowski, K.I.; Snitzer, J.D.; Rusnak, S.; Lang, J.K. Therapeutic Potential of Engineered Extracellular Vesicles. AAPS J. 2018, 20, 50.
- Meyer, C.; Losacco, J.; Stickney, Z.; Li, L.; Marriott, G.; Lu, B. Pseudotyping exosomes for enhanced protein delivery in mammalian cells. Int. J. Nanomed. 2017, 12, 3153–3170.
- Trivedi, M.; Talekar, M.; Shah, P.; Ouyang, Q.; Amiji, M. Modification of tumor cell exosome content by transfection with wt-p53 and microRNA-125b expressing plasmid DNA and its effect on macrophage polarization. Oncogenesis 2016, 5, e250.
- Su, M.J.; Aldawsari, H.; Amiji, M. Pancreatic Cancer Cell Exosome-Mediated Macrophage Reprogramming and the Role of MicroRNAs 155 and 125b2 Transfection using Nanoparticle Delivery Systems. Sci. Rep. 2016, 6, 30110.
- Zhang, P.; Zhang, L.; Qin, Z.; Hua, S.; Guo, Z.; Chu, C.; Lin, H.; Zhang, Y.; Li, W.; Zhang, X.; et al. Genetically Engineered Liposome-like Nanovesicles as Active Targeted Transport Platform. Adv. Mater. 2018, 30, 1705350.
- Huang, L.; Rong, Y.; Tang, X.; Yi, K.; Qi, P.; Hou, J.; Liu, W.; He, Y.; Gao, X.; Yuan, C.; et al. Engineered exosomes as an in situ DC-primed vaccine to boost antitumor immunity in breast cancer. Mol. Cancer 2022, 21, 45.
- Fang, W.; Li, L.; Lin, Z.; Zhang, Y.; Jing, Z.; Li, Y.; Zhang, Z.; Hou, L.; Liang, X.; Zhang, X.; et al. Engineered IL-15/IL-15Rα-expressing cellular vesicles promote T cell anti-tumor immunity. Extracell. Vesicle 2023, 2, 100021.
- Xu, S.; Liu, B.; Fan, J.; Xue, C.; Lu, Y.; Li, C.; Cui, D. Engineered mesenchymal stem cell-derived exosomes with high CXCR4 levels for targeted siRNA gene therapy against cancer. Nanoscale 2022, 14, 4098–4113.
- Morishita, M.; Takahashi, Y.; Matsumoto, A.; Nishikawa, M.; Takakura, Y. Exosome-based tumor antigens-adjuvant co-delivery utilizing genetically engineered tumor cell-derived exosomes with immunostimulatory CpG DNA. Biomaterials 2016, 111, 55–65.
- Han, X.; Bi, L.; Wu, Y.; Yan, J.; Wu, X.; Zheng, R.; Sun, Y.; Zhang, H.; Wang, Z.; Wang, Y.; et al. Genetically engineered exosomes for targetedly preventing premetastatic niche formation and suppressing postoperative melanoma lung metastasis. Nano Today 2022, 46, 101597.
- Li, B.; Yang, T.; Liu, J.; Yu, X.; Li, X.; Qin, F.; Zheng, J.; Liang, J.; Zeng, Y.; Zhou, Z.; et al. Genetically engineered PD-1 displaying nanovesicles for synergistic checkpoint blockades and chemo-metabolic therapy against non-small cell lung cancer. Acta Biomater. 2023, 161, 184–200.
- Severic, M.; Ma, G.; Pereira, S.G.T.; Ruiz, A.; Cheung, C.C.L.; Al-Jamal, W.T. Genetically-engineered anti-PSMA exosome mimetics targeting advanced prostate cancer in vitro and in vivo. J. Control. Release 2021, 330, 101–110.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
929
Revisions:
2 times
(View History)
Update Date:
31 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No