Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Esam Yahya | -- | 5278 | 2023-05-29 10:17:46 | | | |
| 2 | Sirius Huang | Meta information modification | 5278 | 2023-05-30 03:21:35 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kciuk, M.; Yahya, E.B.; Mohamed Ibrahim Mohamed, M.; Rashid, S.; Iqbal, M.O.; Kontek, R.; Abdulsamad, M.A.; Allaq, A.A. Molecular Mechanisms of Cancer Immunotherapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/44954 (accessed on 09 August 2026).
Kciuk M, Yahya EB, Mohamed Ibrahim Mohamed M, Rashid S, Iqbal MO, Kontek R, et al. Molecular Mechanisms of Cancer Immunotherapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/44954. Accessed August 09, 2026.
Kciuk, Mateusz, Esam Bashir Yahya, Montaha Mohamed Ibrahim Mohamed, Summya Rashid, Muhammad Omer Iqbal, Renata Kontek, Muhanad A. Abdulsamad, Abdulmutalib A. Allaq. "Molecular Mechanisms of Cancer Immunotherapy" Encyclopedia, https://encyclopedia.pub/entry/44954 (accessed August 09, 2026).
Kciuk, M., Yahya, E.B., Mohamed Ibrahim Mohamed, M., Rashid, S., Iqbal, M.O., Kontek, R., Abdulsamad, M.A., & Allaq, A.A. (2023, May 29). Molecular Mechanisms of Cancer Immunotherapy. In Encyclopedia. https://encyclopedia.pub/entry/44954
Kciuk, Mateusz, et al. "Molecular Mechanisms of Cancer Immunotherapy." Encyclopedia. Web. 29 May, 2023.
Copy Citation
The cancer-immunity cycle is characterized by various stimulatory and inhibitory factors, which together regulate the immune response and halt the extreme response that may lead to autoimmune disease. Immunotherapy of cancer has rejuvenated the field of tumor immunology and revolutionized treatment options.
cancer immunotherapy
antitumor response
cytokines
immune checkpoints
1. Introduction
The human immune system comprises various defense mechanisms against all potential threats including arising cancer cells. Immune surveillance continuously recognizes and eliminates any transformed tumor cells through numerous mechanisms [1]. Cancer immunotherapy has been described as the fourth pillar of therapy against the tumor, which may surpass the effectiveness of conventional therapies such as surgery, radiotherapy, and chemotherapy [2]. Particularly, it has been listed by Science in 2013 among the top ten annual significantly valuable scientific breakthroughs [3]. Zhang et al. [4] classified the mechanisms of cancer immune therapies into five major groups (Figure 1)—the regulation of immune checkpoints, oncolytic virus therapies, cancer vaccines, cytokine therapies, and adoptive cell transfer.
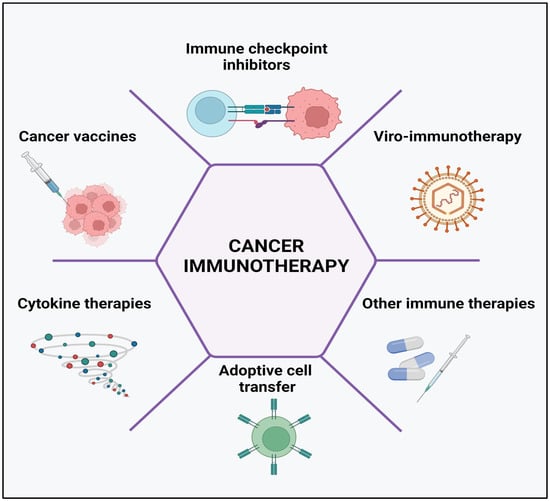
Figure 1. Cancer immunotherapy types include the use of immune-checkpoint inhibitors, cancer vaccines, cytokines, viruses, and adoptive cell transfer. Created with BioRender.com, accessed on 23 April 2023.
2. Regulation of Immune Checkpoints
Cancer therapy by regulation of immune checkpoints is an approach that aims to activate the immune system against cancer cells by targeting proteins that act as negative regulators of immune responses. Cancer cells can evade immune surveillance by expressing multiple immune-checkpoint proteins, such as programmed cell death ligand 1 (PD-L1), CD80/86, and Gal9, which inhibit T-cell activation and proliferation [5][6]. Therapies that target these immune-checkpoint proteins, such as immune-checkpoint inhibitors, have shown remarkable success in the treatment of various types of cancer [7][8][9]. These drugs block the interaction between immune-checkpoint proteins and their ligands, thereby releasing the inhibition of T-cell function.
PD-1 is an immunoglobulin (Ig) class protein that is expressed in T, B, myeloid, and NK cells. It has been demonstrated that PD-1 has two primary interacting partners: PD-L1 and PD-L2. PD-L1 is expressed by the APCs, hematopoietic and non-hematopoietic cells, whereas PD-L2 is expressed by activated macrophages and DCs. When PD-1 interacts with its ligands on other T cells, T-cell activation is hindered. The efficacy of TCR signaling is correlated with PD-1 expression in T cells. Its normal physiological function is to prevent the overactivation of T cells in the absence of the antigen. However, chronic viral infection and malignancy can trigger a constitutive PD-1/PD-L1 cell surface expression, which in turn inhibits the immune response and impairs T-cell function. This topic was reviewed by other authors [10][11][12][13][14] and the promises and limitations of the PD-1/PD-L1 blockage were recently described and will not be discussed here in detail [15].
CTLA-4 is a protein receptor found on the surface of T cells (CD4+ and CD8+). It is essential for immune response control, particularly in the context of T-cell activation and tolerance. CTLA-4 has a similar structure to CD28, another protein receptor produced on T cells that plays an important role in T-cell activation. CTLA-4, on the other hand, offers an inhibitory signal that reduces T-cell activation and proliferation, whilst CD28 gives a co-stimulatory signal that increases T-cell activation. CTLA-4 competes with CD28 for binding to APCs co-stimulatory molecules CD80 (B7-1) and CD86 (B7-2). When CD28 attaches to these molecules, it provides a stimulating signal, whereas CTLA-4 delivers a negative signal, limiting T-cell activation and proliferation [16][17][18].
Despite both of these molecules contribute to the inhibition of anticancer immunity, the timing of down-regulation, the underlying signaling mechanisms, and the anatomic locations of immunological suppression by these two immune checkpoints are different. PD-1 is more widely expressed on activated T cells, B cells, and myeloid cells than CTLA-4, which is only found on T cells. While CTLA-4 is active during the T-cell activation’s priming phase, PD-1 is active during the T-cell activation’s effector phase, largely within peripheral tissues. These characteristics suggest that anti-CTLA-4 and anti-PD-1 treatments may have cumulative and synergistic benefits in the treatment of the disease [19][20][21]. PD-1 inhibitors, such as pembrolizumab and nivolumab, have been approved for the treatment of melanoma, non-small-cell lung cancer, bladder cancer, and other types of cancer. CTLA-4 inhibitors, such as ipilimumab, have been approved for the treatment of melanoma, and combinations of PD-1 and CTLA-4 inhibitors are being investigated for the treatment of several types of cancer. Other immune-checkpoint inhibitors, such as those targeting T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), are currently under investigation in clinical trials [22][23].
TIM-3 is another cell surface immune-checkpoint molecule expressed in immunological cells such as T cells, Tregs, DCs, B cells, macrophages, NK cells, and mast cells. TIM-3 binds to four different ligands: galectin-9 (Gal-9), CEACAM-1 (carcinoembryonic antigen cell adhesion molecule 1), HMGB1 (high-mobility group protein B1), and PS (phosphatidylserine). TIM-3 mediates T-cell exhaustion, which suppresses antitumor immunity. The selective blockage of TIM-3 enhances anticancer immunity through increased IFN-γ production by T cells. TIM-3 inhibitors have been studied in animal models and clinical trials [24][25][26][27]. The blockade of a single checkpoint in many cases may not achieve the desired therapeutic effect [28][29].
TIGIT (T-cell immunoreceptor with Ig and ITIM domains) is a protein that is expressed on various types of immune cells, including T cells and NK cells. TIGIT is up-regulated on activated T cells, particularly on Tregs, follicular helper T cells (Tfh), and memory T cells [30]. TIGIT is a co-inhibitory receptor that interacts with its ligands, CD155 (poliovirus receptor) and CD112 (nectin-2), which are expressed on APCs and some tumor cells. The binding of TIGIT to its ligands leads to the inhibition of T-cell activation and proliferation, as well as the suppression of cytokine production. Therefore, TIGIT plays an important role in the regulation of the immune response and maintenance of immune homeostasis. Targeting TIGIT has emerged as a potential immunotherapy strategy for cancer and autoimmune diseases [31]. On the other hand, CD96 (also known as TACTILE) is a protein that is primarily expressed on activated T cells, including CD8+ T cells, Tregs, NKT cells, and some subsets of memory T cells. CD96 is also expressed in some tumor cells [32]. CD96 interacts with its ligand, CD155, which is widely expressed in various cell types, including T cells, DCs, tumor cells, and many other cell types. CD96 has been shown to play a role in T-cell activation and effector functions, as well as in the regulation of immune tolerance and autoimmunity. CD226 (also known as DNAM-1) and CD112 (nectin-2) are also cell surface receptors that are expressed on various immune cells, including T cells, NK cells, and DCs, as well as on many tumor cells. CD226 interacts with its ligands, CD155 and CD112, and plays a role in the activation and cytotoxicity of NK cells and CD8+ T cells [33][34]. TIGIT and CD96 are more likely to bind and suppress T-cell activation than CD226, which has a lower affinity for CD155. TIGIT binding to CD155 on the tumor cells leads to the up-regulation of IL-10 and down-regulation of IL-12 [32].
Vascular endothelial growth factor receptor-1 (VEGFR-1) is a crucial receptor that plays a major role in tumor progression as well as in the resistance to treatment methods based on immune-checkpoint inhibitors [35]. Activation of VEGFR is involved in myeloid progenitors mobilization from the bone marrow which later infiltrates the tumor. Lacal et al. [36] recently developed an anti-VEGFR monoclonal antibody (anti-VEGFR-1 mAb) able to inhibit the growth and proliferation of melanoma in preclinical in vivo models. In addition, the reduction in macrophage progenitor mobilization and tumor cell infiltration by myeloid cells was observed. The same authors indicated significant up-regulation in the expression of VEGFR-1 in human-activated macrophages 2 (M2) compared to activated macrophage 1 (M1) cells. The exposure to the designed monoclonal antibody (D16F7 mAb) decreases the chemotaxis of macrophages in vitro. The results of in vivo treatment in melanoma-bearing mice resulted in significant inhibition of tumor growth due to the alterations in the tumor microenvironment, which lead to a decrease in melanoma infiltration by M2 as well as PD-1+ and FoxP3+ cells. These modifications resulted in an increase in M1/M2 and CD8+/FoxP3+ cell ratios in favor of antitumor immune response [36]. Studies by Chauvin et al. [37] revealed the overexpression of PD-1 and TIGIT on most tumor-infiltrating CD8+ T cells and tumor antigen-specific circulating CD8+ T cells. Combined blockade of both checkpoints (TIGIT and PD-1) has shown significant enhancement in the antitumor function of cytotoxic CD8+ T cells compared to blocking PD-1 alone [38], this dual blockade seems to depend on CD226 signal transduction [39]. The combination of specific checkpoint blockade can be used to improve immune response and constitutes a promising direction for cancer immunotherapy.
Immune-checkpoint inhibition can be used to treat various malignancies including melanoma, lung cancer, bladder cancer, kidney cancer, and Hodgkin’s lymphoma. Unlike standard cancer therapies such as chemotherapy, immune-checkpoint inhibition can result in long-term remission and even tumor eradication in some individuals. Because they exclusively target the immune system rather than all rapidly dividing cells in the body, immune-checkpoint inhibitors have fewer side effects than standard chemotherapy. Despite increased safety profile, they can still induce substantial consequences such as lung, liver, and other organ inflammation. Additionally, the inhibition success rate depends on the type of cancer and other factors such as the patient’s immune system status. Additionally, certain cancer cells may acquire resistance to immune-checkpoint inhibitors, resulting in therapy failure. Because immune-checkpoint inhibitors are a relatively new type of cancer treatment, long-term data on their efficacy and potential side effects is also lacking. This topic was previously described by other authors [40][41][42][43].
3. Viro-Immunotherapy
Viruses have been widely investigated for being the ideal therapeutic option for various types of cancers in combination with immune-checkpoint inhibitors and many other types of immunotherapeutic approaches [44][45]. Oncolytic virotherapy focuses on the development of highly specific genetically engineered viruses for targeting only cancer cells. Following the recognition of cancer cells expressing the particular molecules, these viruses infect and kill cancer cells through their lysis. Owing to their ability to accommodate gene insertions, oncolytic viruses have been armed with transgenes to produce immune-stimulatory signals to improve antitumor immune response [46][47]. Numerous clinical and preclinical studies on the use of different oncolytic viruses in cancer immunotherapy have been conducted and reported promising antitumor potential. Table 1 presents the most commonly used oncolytic viruses for viro-immunotherapy.
Table 1. Important and commonly used oncolytic viruses in cancer immunotherapy.
| Virus (Family) | Nucleic Acid Type (Group) | Study Remarks | Ref |
|---|---|---|---|
| Reovirus (Reoviridae) | dsRNA (G III) | Viruses exhibited a safe and tolerable toxicity profile in early clinical trials, with minimal viral shedding, replication, localization, and cytotoxic effects. | [48] |
| Influenza A Virus (Orthomyxoviridae) |
ssRNA-ve (G V) | Engineered viruses exhibited tumor-ablative potential and do not replicate in normal cell lines. | [49] |
| Herpes Simplex Virus1 (Herpesviridae) | dsDNA (G I) | A virus exhibited high antitumor efficacy, has a high safety margin, is well tolerated and its use is associated with a low frequency of adverse effects in most of the treated patients. | [50] |
| Adenovirus (Adenoviridae) | dsDNA (G I) | Clinical data revealed that the virus was tolerable and effective even when combined with other therapeutic approaches. | [51] |
| Measles virus (Paramyxoviridae) | ssRNA-ve (G V) | The virus replicated selectively in the tumor and significantly suppressed its development. It was completely safe and did not result in any measles-like symptoms. | [52] |
| Vaccinia virus (Poxviridae) | dsDNA (G I) | The virus replicated rapidly in tumor cells with significant antitumor effects, but its cytotoxicity varied based on cell lines. | [53] |
| Pseudorabies (Herpesviridae) | dsDNA (G I) | Selective replication of viruses has been observed in many kinds of cancers, with tumor growth being significantly slowed by more than 50%. | [54] |
Vesicular stomatitis viruses (VSV) have been reported to have several beneficial properties that favor their use for immunotherapy, including inherent tumor specificity, rapid replication kinetics, and the capacity to elicit a wide variety of immune responses [55]. Melzer et al. [56] summarized the strategies of this novel platform for the enhancement of the immune-stimulating potential. Modification of the virus’s endogenous genes to stimulate more interferon induction and virus-mediated expression of immune-stimulatory cytokines are two strategies that have been widely used with VSVs-based viro-immunotherapy or employed with other viruses to boost antitumor immune responses [57][58]. Vaccination approaches based on viruses have also been used to stimulate and even improve adaptive immune responses against particular tumor antigens [59].
In comparison to chemotherapies, oncolytic viro-immunotherapy has many advantages [60][61] including (a) selective targeting of cancer cells. Cancer cells can be specifically targeted and destroyed with viro-immunotherapy while healthy cells are unharmed. This is because the viruses have been engineered to specifically target cancer cells, reducing the likelihood of unintended effects. (b) Induction of immunogenic cell death: Immunogenic cell death triggered by oncolytic viruses primes the immune system to target and destroy cancer cells. As a result, the immune system has an increased likelihood of killing cancer cells even if they have metastasized. (c) Synergistic effect with other therapies: Combining viro-immunotherapy with standard cancer therapies such as chemotherapy and radiation has been shown to improve patient outcomes. Some clinical and preclinical research indicates that combination therapy is more effective than either medication alone. (d) Minimal systemic toxicity: As the viruses used in viro-immunotherapy are engineered to specifically target and destroy cancer cells, they are found to have low systemic toxicity. This lessens the likelihood of side effects that are usually associated with conventional anticancer therapies. (e) Potential to overcome drug resistance: Drug resistance is a serious problem impacting the usefulness of cancer therapies. Oncolytic viruses can specifically target and kill cancer cells that have developed resistance to other therapies. (f) Ability to target cancer stem cells: Cancer stem cells are a subpopulation of cancer cells that are believed to be responsible for tumor initiation, upkeep, and resurgence. Viro-immunotherapy may be able to target these cells. Treatment outcomes may improve and last longer if cancer stem cells are targeted [47][62][63][64]. However, their use can be also associated with some limitations such as (1) Limited efficacy in some types of cancer: The presence of a protective stromal barrier that blocks the virus from accessing the cancer cells has restricted the effectiveness of viro-immunotherapy in certain kinds of cancer. This may reduce the efficacy of viro-immunotherapy in some conditions. (2) Potential for immune-related adverse events: Viro-immunotherapy raises the risk of immunological-related side effects because it stimulates the immune system. Symptoms including the flu, inflammation, and autoimmune disorders are all potential side effects. (3) Pre-existing immunity to the virus: The therapeutic efficacy of the oncolytic virus may be reduced in patients who already have developed an immune response to the virus. This may occur as a result of previous contact with viruses or a tendency to mount an immune response against the virus as a result of a person’s genetic makeup. (4) Limited availability of clinical trials: Since viro-immunotherapy is still in its experimental stages, there are not many clinical trials available, that determine its safety and efficacy. Additionally, due to the novelty of viro-immunotherapy patients’ accessibility to this treatment option may be limited. (5) Potential for viral shedding: Viruses used in viro-immunotherapy can spread to other people especially patients with compromised immune systems. (6) Cost: Because of its complexity and high expense, viro-immunotherapy may be out of reach for many individuals. As indicated by the previous authors [45][47][62][63][64].
4. Cancer Vaccines
Cancer-preventive vaccines are intended to thwart the progression of cancer by either focusing on cancer-specific antigens that are expressed on precancerous or early-stage cancer cells or on viruses that are known to cause cancer [65]. On the other hand, therapeutic cancer vaccines (Figure 2) are intended to cure tumors that have already developed by enhancing the immune system’s capacity to recognize and destroy cancer cells. In recent decades, tremendous effort has been devoted to the development of cancer vaccines. One strategy that showed promise as a potential therapeutic option is the use of cancer-specific antigens (CSAs). CSAs are proteins or peptides that are exclusive to cancer cells and are identified by the immune system as being of a foreign origin. Several CSAs have been investigated, and they are now being incorporated into cancer vaccines to elicit an immune response against cancer cells [66][67].
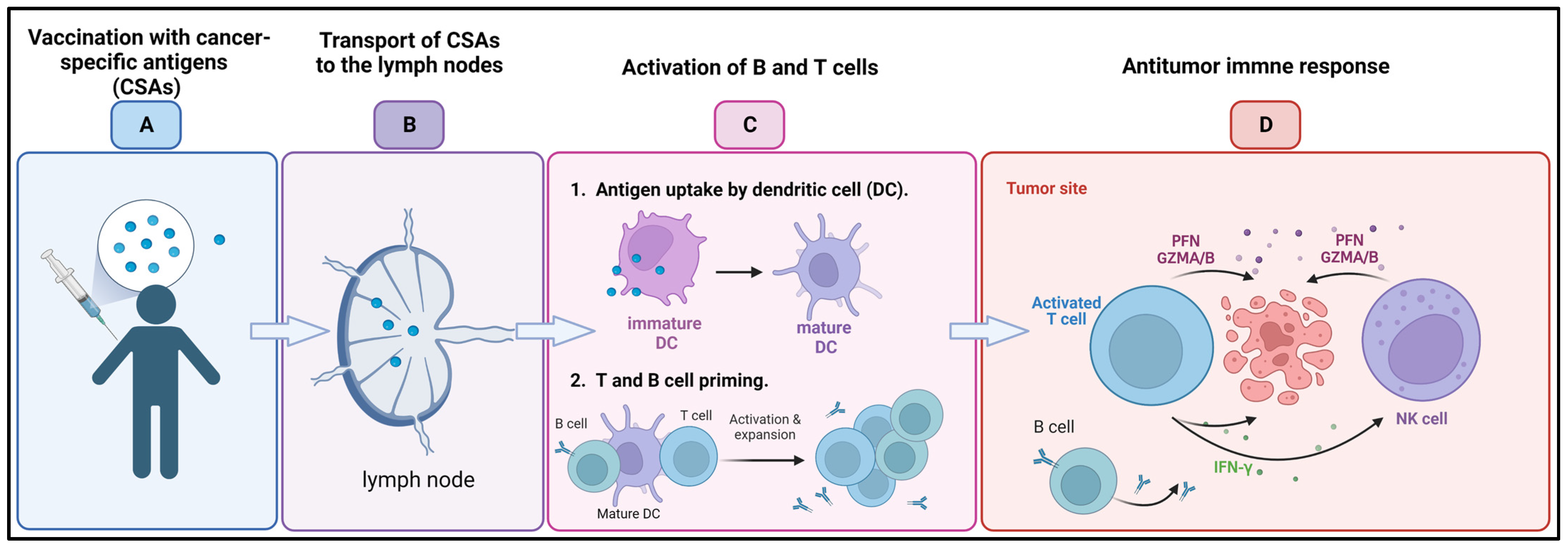
Figure 2. Cancer vaccine: (A) Following administration of cancer-specific antigens (CSAs) through vaccination (B) antigens are transported to the lymph nodes, where (C) they are uptaken by dendritic cells (DCs) and activate the B and T cells. (D) B cells produce tumor-targeted antibodies, while activated T cells and natural killer (NK) recruited to the tumor microenvironment release perforins (PFNs) and granzyme A/B (GZMA/B) and interferon γ (IFN-γ) leading to cancer cell death. Created with BioRender.com, accessed on 23 April 2023.
Exosomes are small vesicles that are connected to membranes and are produced by the majority of cells for intercellular exchange and regulation. These exosomes deliver materials or messages to the cells they are targeting for a variety of physiologically significant purposes. Exosomes that were derived from M1-polarized, proinflammatory macrophages were the subject of the research that was conducted by Cheng et al. The researchers were interested in the possibility of using M1 exosomes as an immunopotentiator for a cancer vaccine. After being injected subcutaneously, the M1 exosomes showed a tropism toward lymph nodes. They were predominantly uptaken by the local macrophages and DCs and triggered the secretion of a pool of Th1 cytokines. M1, but not M2, exosomes increased the activity of a lipid calcium phosphate (LCP) nanoparticle-encapsulated tyrosinase-related protein 2 (TRP2) vaccine and elicited a stronger antigen-specific cytotoxic T-cell response. In a melanoma growth inhibition study, M1 exosomes showed to be a more potent immunopotentiator than an adjuvant, such as CpG oligonucleotides (ODNs) when combined with LCP nanoparticle vaccine in a melanoma. The study demonstrated the potential of M1-polarized macrophage-derived exosomes as a vaccine adjuvant [68].
Despite the large number of vaccine strategies that are being developed regularly, the construction of safe and successful therapeutic cancer vaccines remains a challenge [66][69][70]. Advantages of their use include the prevention of cancer—vaccines against cancer-causing viruses including human papillomavirus (HPV) and hepatitis B virus (HBV) can be used to protect against the onset of cancer. Cancers such as cervical and liver cancer have been proven to decrease in frequency after vaccination against these viruses. Fewer side effects are another advantage due to cancer vaccines target only cancer cells, leaving healthy cells unharmed, making them safer than standard cancer therapies such as chemotherapy and radiation. On the other hand, the disadvantages include limited efficacy: not all individuals who receive a vaccine will experience a positive response. Cancer vaccines’ success rates may differ among patients with different cancer stages. High cost: cancer vaccines can be pricey, especially if they are customized for each patient, and may not be affordable for everyone. Additionally, development challenges: it can be difficult and time-consuming to develop a vaccine against cancer since accomplishing it requires finding the right target antigens, refining the vaccine’s formulation, and testing it in humans to make sure it is safe and effective.
5. Cytokine Therapies
Cytokines are chemical messengers secreted by immune and non-immune cells in reaction to cellular stresses such as bacterial infections, inflammation, and tumorigenesis to control cellular interactions [71]. The first use of cytokines in cancer treatment occurred in 1976, following the discovery of interleukin-2 (IL-2), initially known as a T-cell growth factor, which appeared to have the capacity to activate T cells and thus exert immune-stimulatory properties [71][72]. In vitro incubation of inactive lymphoid cells with recombinant IL-2 results in the production of lymphokine-activated killer (LAK) cells, which are cells that are capable to lyse tumor cells. The administration of LAKs and large doses of IL-2 in patients with various types of cancer have been reported to cause cancer regressions and significantly enhance antitumor response in preclinical studies and clinical trials [73][74].
More than half a century ago, type I interferons (IFNs), were identified for the first time as the factors responsible for virus inhibition. However, in the last ten years, researchers have only just started to understand the precise function that type I IFNs play in the body’s natural immune response to cancer. IFNs have been implicated in two primary functions in tumor control—induction of tumor cell senescence and stimulation of DC maturation to boost the T-cell cytotoxicity [75]. Cauwels et al. [76] developed a novel strategy for cancer treatment using Activity-on-Target cytokines, which are modified immune-cytokines that possess up to 1000-fold more potency for target cells. The authors reported that modified type I interferon designed to target Clec9A+ DCs was able to limit the progression of tumors and showed strong antitumor activity in breast carcinoma, murine melanoma, and lymphoma models, without toxic side effects. IFN activation of immune cells may inhibit tumor angiogenesis, whereas ILs promote the activation and growth of helper and cytotoxic T lymphocytes (CD4+ and CD8+ T cells) [77][78]. Granulocyte-macrophage colony-stimulating factor (GM-CSF) is another cytokine that, in reaction to stress, infections, and malignancies, stimulates the formation of myeloid cell subgroups including neutrophils, monocytes, macrophages, and DCs. GM-CSF has a global effect on the host immune surveillance system when pathologic conditions are present. When compared to other soluble immune mediators, it has been discovered that either an excess of or a deficiency in GM-CSF can encourage the aggressiveness of cancer. An insufficient amount of GM-CSF inhibits the proper production of innate immune cells, which in turn prevents the resulting induction of adaptive anticancer immune responses. On the other hand, too much GM-CSF can exhaust immune cells and encourage the growth of cancer [79][80]. GM-CSF has been also used to accelerate and augment granulocyte recovery after chemotherapy of cancers [81][82]. Cytokines such as INFγ or tumor necrosis factors (TNFs) are essential in the development of an immunogenic microenvironment. Anthracyclines as well as other treatments such as photodynamic therapy (PDT) exert their impacts on cancer cells in a way that stimulates the body’s immune system. This process, also known as immunogenic cell death (ICD), is distinguished by the release of membrane-bound and soluble factors that boost the function of immune cells [83] and can act as pro-inflammatory factors (TNFs, IL-1β, and IL-6) or can lead to increase in the expression of MHC class I on the surface of antigen-presenting cells. This leads to improved immune response via the differentiation promotion or the activation of both T cells and NK cells [84].
Immunogenic cell death inducers trigger the release of damage-associated molecular patterns (DAMPs) such as high mobility group box protein 1 (HMGBP1), CRT, and ATP, as well as inflammatory cytokines from cancer cells. Both DAMPs and cytokines send activating signals for DCs and NK cells, which respond instantly by releasing effector cytokines [85]. Dying tumor cells treated with ICD inducers trigger the release of various cytokines, which modulate an effective immune response, such as IL-6 and IL-8, [86]. NK cells functionality appears to be increased by cytokines released from activated DCs, such as IL-12, as well as cytokines produced by other innate immune cells, such as IFN-α/β, leading to the secretion of IFN-γ and TNFα (Figure 3) [84].
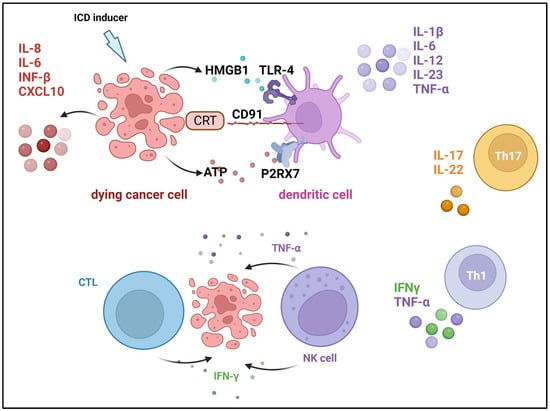
Figure 3. Cytokines that are involved in immunogenic cell death induction. Damage-associated molecular patterns (DAMPs) are released from dying cells during immunogenic cell death (ICD) and facilitate tumor antigen presentation and augment adaptive immunity. Cancer cells secrete calreticulin (CRT), which binds to the CD91 receptor on dendritic cells (DCs), and high-mobility group protein 1 (HMGB1), which is a ligand for the Toll-like receptor (TLR-4) on DCs. In addition, DSc’s P2X purinoceptor 7 (P2RX7) engages with ATP that cancer cells secrete. Created with BioRender.com, accessed on 23 April 2023. Modified from Showalter et al. [83].
While cytokine treatments have the potential to improve cancer treatment, they also pose considerable risks [87][88][89][90][91][92]. Below are the pros and negatives of their use:
-
Stimulation of the immune system: the immune system can be stimulated with cytokine treatments to better target and destroy cancer cells.
-
Reduced toxicity: compared to conventional treatments such as chemotherapy and radiation, cytokine therapies are safer and have fewer adverse effects.
-
Versatility: cytokine treatments are adaptable because they can be used to treat a wide variety of cancers.
-
May provide long-term benefits: some cytokine therapies have been demonstrated to produce lasting advantages in cancer patients, increasing the likelihood of a long-lasting response to treatment.
-
Can be used in combination with other treatments: combining cytokine therapies with standard cancer treatments such as chemotherapy, radiation therapy, and immunotherapy has been shown to improve patient outcomes.
-
May induce remission: some cytokine therapies, such as high-dose IL-2 and IFN-α, have been shown to induce complete or partial remission in some cancer patients.
-
Long-term immune memory: long-term immunological memory can be induced by some cytokine therapies, such as IL-2, meaning that the immune system will continue to detect and fight cancer cells even after treatment has ended.
-
Fewer treatment sessions: patients may find cytokine therapies more convenient because they often need fewer treatment sessions than treatments such as chemotherapy.
Although cytokine treatments have shown promise in the treatment of cancer, further study is needed to determine their full potential. It is important to overcome the potential limitations of this therapeutic approach:
-
Limited effectiveness: some patients may only have a partial response to cytokine treatments, and they may not work for all forms of cancer.
-
Side effects: side effects from cytokine therapies include fever, exhaustion, and muscle aches, but they are typically less severe than those from chemotherapy and radiation. Some cytokine therapies, such as high-dose IL-2, can cause damage to organs such as the kidneys and liver, which can be life-threatening.
-
Expensive: some patients may not be able to afford cytokine therapy due to their high cost.
-
Can cause autoimmune reactions: there are risks associated with cytokine treatments, including autoimmune reactions and tissue damage.
6. Adoptive Cell Transfer
Adoptive cell transfer (ACT) is a cancer treatment method that involves injecting immune cells, such as T cells, into a patient’s body, where they seek out and destroy cancer cells. Before being administered to a patient, T cells are usually modified or enhanced in the lab to strengthen their ability to recognize and target cancer cells. CAR T-cell therapy is a very successful form of ACT that involves engineering T cells to express chimeric antigen receptors (CARs) that can recognize and adhere to specific proteins on the surface of cancer cells (Figure 4) [93][94].
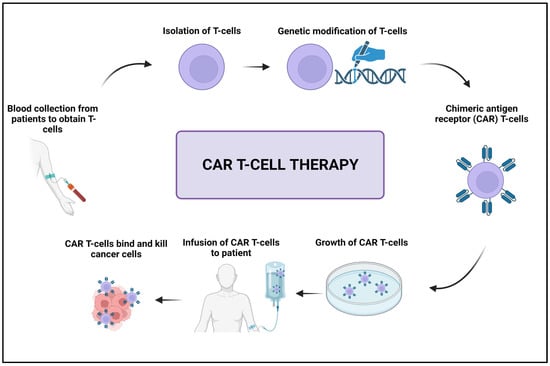
Figure 4. Schematic illustration of the CAR T-cell therapy. Created with BioRender.com, accessed on 23 April 2023.
Rosenberg et al. [73] revealed that the administration of modified autologous LAK cells in addition to recombination-derived IL-2 was effective in the treatment of patients with metastatic cancers. High response rates in patients with follicular lymphoma and diffuse large B-cell lymphoma have been reported with modified T cell-based therapy, using CAR able to target CD19 receptors on the surface of B-cell cancers [95]. Schuster et al. [96] used autologous modified T cells expressing a CD19-directed CAR to treat patients with follicular lymphoma or diffuse large B-cell lymphoma. The authors revealed that the engineered cells were effective in the treatment. High rates of durable disease reduction were observed, with significant recovery of B cells and immunoglobulins in some patients. Similarly, Wang et al. [97] conducted a trail on seven patients suffering from advanced diffuse large B cell lymphomas and concluded that anti-CD20 CAR T cells caused prolonged tumor regression. In a different study, Mohammed et al. [98] generated chimeric antigen receptor T cells directed against a specific antigen located on prostate stem cells and revealed specific tumor lysis. The authors generated an inverted receptor for the cytokine to protect the modified cells from the immunosuppressive cytokines, resulting in significant enhancement in antitumor activity.
Similarly to the previously discussed immunotherapeutic approaches CAR T-therapies possess significant advantages over conventional chemotherapy and radiotherapy. In contrast to conventional chemotherapy, which is harmful to normal cells as well as cancerous ones, CAR T-cell therapy is a targeted technique that can selectively destroy cancer cells while sparing healthy ones. This lowers the toxicity and adverse effects of CAR T-cell treatment. CAR T cells may also persist in the body for an extended time and continue to attack cancer cells, offering protection against the disease even after the cells have been eliminated. The elimination of cancer cells using CAR T-cell therapy is curative in many patients. However, similar to other novel immunotherapies, the CAR T-cell approach is an expensive treatment. This can be attributed to the sophisticated manufacturing procedure for CAR T cells that can cause treatment delays for some patients. Moreover, it is crucial to determine which patients could benefit the most form the treatment and those with increased risk of detrimental side effects such as cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) [99][100].
7. Other Immune Therapies
Lag-3 (Lymphocyte Activation Gene 3) is a protein receptor expressed on the surface of certain immune cells, including T cells, B cells, and natural killer (NK) cells. Lag-3 plays a role in regulating immune responses and is considered an immune-checkpoint protein [101]. In the context of immunotherapy, Lag-3 has gained attention as a potential target for cancer treatment. Immune-checkpoint inhibitors, such as antibodies targeting PD-1 (programmed cell death protein 1) and CTLA-4 (cytotoxic T-lymphocyte-associated protein 4), have shown success in unleashing the immune system against cancer cells. However, not all patients respond to these therapies, highlighting the need for additional targets [102]. Several clinical trials are underway to investigate the efficacy of Lag-3 inhibitors either as monotherapy or in combination with other immunotherapies. Preliminary data from these trials suggest that Lag-3 inhibitors may provide clinical benefit in certain types of cancers, such as melanoma, lung cancer, and renal cell carcinoma [103][104]. It is important to note that the field of Lag-3 and immunotherapy is still evolving, and more research is needed to fully understand its role and potential in cancer treatment. Clinical trials will continue to provide valuable insights into the efficacy and safety of Lag-3-targeted therapies, as well as their optimal use in different cancer types and patient populations.
Nanoparticles and biomaterials have been employed to enable programmed localization, improve pharmacokinetics, and co-deliver immune-modulatory substances that were not previously possible with direct administration of these compounds in solution [105]. In a recent investigation, Ji et al. [106] designed a biopolymer immune implant for post-surgical therapy of colorectal cancer and revealed that an immune implant was able to eradicate residual tumors post-surgery for more than 150 days. The gradual release of the loaded resiquimod (Toll-like receptor 7/8 (TLR7/TLR8) agonist) was achieved and the anti-CD134 (OX-40) antibody elicited immune memory and inhibited the growth of distal tumors when used in the implant [106]. In previous studies, the OX40 and OX40 ligand interaction was shown to promote the growth and division of T cells while attenuating the immunosuppression of Treg cells. The OX40 plays a crucial part in immunity, hence it is the subject of numerous clinical trials to assess its therapeutic effects in the treatment of cancer [107]. Immunological studies demonstrated that treating patients with a biopolymer immune implant has a two-phase effect: initially, a rise in the infiltration of NK cells and DCs is observed in the first few days, followed by an increase in the infiltration of T cells and the establishment of immune memory in the following weeks [106]. Peptide-based materials constitute another approach to cancer immunotherapies that have been used to solve several traditional treatment challenges and have demonstrated specific and considerable efficacy as cancer immunotherapies [108]. Peptide-based cancer vaccines and delivery systems have been the subject of extensive research. These vaccines and systems aim to replicate the functional domains of proteins with highly specialized immuno-regulatory capabilities [108][109]. Shen et al. [110] reported a mono-palmitoylated peptide able to induce an anticancer immune response without any addition of an adjuvant. The authors also developed a long peptide of a di-palmitic acid conjugated with TLR2 agonist and achieved significant improvement in antitumor immunity by diminishing the function of tumor-associated macrophages (TAMs). Using a different approach, Choi et al. [111] investigated different peptide antigens from cancer stem cells to compose DC vaccination for hepatocellular carcinoma and human breast cancer. By pulsing dendritic cells with CD44 and epithelial cell adhesion molecule-based peptides, the authors reported an effective stimulation and mature DC production in addition to enhanced T-cell stimulation and significant increase in the number of CTLs [111].
References
- Yahya, E.B.; Amirul, A.; HPS, A.K.; Olaiya, N.G.; Iqbal, M.O.; Jummaat, F.; AK, A.S.; Adnan, A. Insights into the Role of Biopolymer Aerogel Scaffolds in Tissue Engineering and Regenerative Medicine. Polymers 2021, 13, 1612.
- Shi, T.; Ma, Y.; Yu, L.; Jiang, J.; Shen, S.; Hou, Y.; Wang, T. Cancer immunotherapy: A focus on the regulation of immune checkpoints. Int. J. Mol. Sci. 2018, 19, 1389.
- Couzin-Frankel, J. Cancer immunotherapy. Science 2013, 342, 1432–1433.
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821.
- Witkowska, M.; Smolewski, P. Immune checkpoint inhibitors to treat malignant lymphomas. J. Immunol. Res. 2018, 2018, 1982423.
- Zhang, C.; Peng, Y.; Hublitz, P.; Zhang, H.; Dong, T. Genetic abrogation of immune checkpoints in antigen-specific cytotoxic T-lymphocyte as a potential alternative to blockade immunotherapy. Sci. Rep. 2018, 8, 5549.
- Akhbariyoon, H.; Azizpour, Y.; Esfahani, M.F.; Firoozabad, M.S.M.; Rad, M.R.; Esfahani, K.S.; Khoshavi, N.; Karimi, N.; Shirinisaz, A.; Abedi, F. Immune checkpoint inhibition for the treatment of cancers: An update and critical review of ongoing clinical trials. Clin. Immunol. 2021, 232, 108873.
- Gadducci, A.; Guerrieri, M.E. Immune checkpoint inhibitors in gynecological cancers: Update of literature and perspectives of clinical research. Anticancer Res. 2017, 37, 5955–5965.
- Kudo, M. Immune checkpoint inhibition in hepatocellular carcinoma: Basics and ongoing clinical trials. Oncology 2017, 92, 50–62.
- Munari, E.; Mariotti, F.R.; Quatrini, L.; Bertoglio, P.; Tumino, N.; Vacca, P.; Eccher, A.; Ciompi, F.; Brunelli, M.; Martignoni, G. PD-1/PD-L1 in cancer: Pathophysiological, diagnostic and therapeutic aspects. Int. J. Mol. Sci. 2021, 22, 5123.
- Chang, E.; Pelosof, L.; Lemery, S.; Gong, Y.; Goldberg, K.B.; Farrell, A.T.; Keegan, P.; Veeraraghavan, J.; Wei, G.; Blumenthal, G.M. Systematic review of PD-1/PD-L1 inhibitors in oncology: From personalized medicine to public health. Oncol. 2021, 26, e1786–e1799.
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92.
- Liu, J.; Chen, Z.; Li, Y.; Zhao, W.; Wu, J.; Zhang, Z. PD-1/PD-L1 checkpoint inhibitors in tumor immunotherapy. Front. Pharmacol. 2021, 12, 731798.
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8.
- Kciuk, M.; Kołat, D.; Kałuzińska-Kołat, Ż.; Gawrysiak, M.; Drozda, R.; Celik, I.; Kontek, R. PD-1/PD-L1 and DNA Damage Response in Cancer. Cells 2023, 12, 530.
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood J. Am. Soc. Hematol. 2018, 131, 58–67.
- Sobhani, N.; Tardiel-Cyril, D.R.; Davtyan, A.; Generali, D.; Roudi, R.; Li, Y. CTLA-4 in regulatory T cells for cancer immunotherapy. Cancers 2021, 13, 1440.
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367.
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 2016, 39, 98.
- De Silva, P.; Aiello, M.; Gu-Trantien, C.; Migliori, E.; Willard-Gallo, K.; Solinas, C. Targeting CTLA-4 in cancer: Is it the ideal companion for PD-1 blockade immunotherapy combinations? Int. J. Cancer 2021, 149, 31–41.
- Wojtukiewicz, M.Z.; Rek, M.M.; Karpowicz, K.; Górska, M.; Polityńska, B.; Wojtukiewicz, A.M.; Moniuszko, M.; Radziwon, P.; Tucker, S.C.; Honn, K.V. Inhibitors of immune checkpoints—PD-1, PD-L1, CTLA-4—New opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev. 2021, 40, 949–982.
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 1–12.
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: Mechanism, combinations, and clinical outcome. Front. Pharmacol. 2017, 8, 561.
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. OncoTargets Ther. 2018, 7005–7009.
- Saleh, R.; Toor, S.M.; Elkord, E. Targeting TIM-3 in solid tumors: Innovations in the preclinical and translational realm and therapeutic potential. Expert Opin. Ther. Targets 2020, 24, 1251–1262.
- Rezaei, M.; Tan, J.; Zeng, C.; Li, Y.; Ganjalikhani-Hakemi, M. TIM-3 in leukemia; immune response and beyond. Front. Oncol. 2021, 11, 753677.
- Gomes de Morais, A.L.; Cerdá, S.; de Miguel, M. New checkpoint inhibitors on the road: Targeting TIM-3 in solid tumors. Curr. Oncol. Rep. 2022, 24, 651–658.
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell 2014, 26, 923–937.
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; Van den Eertwegh, A.J.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712.
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957.
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863.
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD 96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120.
- Meyer, D.; Seth, S.; Albrecht, J.; Maier, M.K.; du Pasquier, L.; Ravens, I.; Dreyer, L.; Burger, R.; Gramatzki, M.; Schwinzer, R. CD96 interaction with CD155 via its first Ig-like domain is modulated by alternative splicing or mutations in distal Ig-like domains. J. Biol. Chem. 2009, 284, 2235–2244.
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119.
- Lisi, L.; Pia Ciotti, G.M.; Chiavari, M.; Ruffini, F.; Lacal, P.M.; Graziani, G.; Navarra, P. Vascular endothelial growth factor receptor 1 in glioblastoma-associated microglia/macrophages. Oncol. Rep. 2020, 43, 2083–2092.
- Lacal, P.M.; Atzori, M.G.; Ruffini, F.; Scimeca, M.; Bonanno, E.; Cicconi, R.; Mattei, M.; Bernardini, R.; D’Atri, S.; Tentori, L. Targeting the vascular endothelial growth factor receptor-1 by the monoclonal antibody D16F7 to increase the activity of immune checkpoint inhibitors against cutaneous melanoma. Pharmacol. Res. 2020, 159, 104957.
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.-h.T.; Maurer, M.; Korman, A.J. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058.
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 2018, 7, e1466769.
- Yadav, M.; Green, C.; Ma, C.; Robert, A.; Glibicky, A.; Nakamura, R.; Sumiyoshi, T.; Meng, R.; Chu, Y.-W.; Wu, J. Tigit, CD226 and PD-L1/PD-1 are highly expressed by marrow-infiltrating T cells in patients with multiple myeloma. Blood 2016, 128, 2102.
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801.
- Zhang, Y.; Chikata, T.; Kuse, N.; Murakoshi, H.; Gatanaga, H.; Oka, S.; Takiguchi, M. Immunological Control of HIV-1 Disease Progression by Rare Protective HLA Allele. J. Virol. 2022, 96, e01248-01222.
- Naimi, A.; Mohammed, R.N.; Raji, A.; Chupradit, S.; Yumashev, A.V.; Suksatan, W.; Shalaby, M.N.; Thangavelu, L.; Kamrava, S.; Shomali, N. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun. Signal. 2022, 20, 1–31.
- Sanmamed, M.; Chester, C.; Melero, I.; Kohrt, H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann. Oncol. 2016, 27, 1190–1198.
- Dyer, A.; Frost, S.; Fisher, K.; Seymour, L. The role of cancer metabolism in defining the success of oncolytic viro-immunotherapy. Cytokine Growth Factor Rev. 2020, 56, 115–123.
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567.
- Choi, K.-J.; Zhang, S.-N.; Choi, I.-K.; Kim, J.-S.; Yun, C.-O. Strengthening of antitumor immune memory and prevention of thymic atrophy mediated by adenovirus expressing IL-12 and GM-CSF. Gene Ther. 2012, 19, 711–723.
- Chiocca, E.A.; Rabkin, S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300.
- Gong, J.; Sachdev, E.; Mita, A.C.; Mita, M.M. Clinical development of reovirus for cancer therapy: An oncolytic virus with immune-mediated antitumor activity. World J. Methodol. 2016, 6, 25.
- Bergmann, M.; Romirer, I.; Sachet, M.; Fleischhacker, R.; García-Sastre, A.; Palese, P.; Wolff, K.; Pehamberger, H.; Jakesz, R.; Muster, T. A genetically engineered influenza A virus with ras-dependent oncolytic properties. Cancer Res. 2001, 61, 8188–8193.
- Eissa, I.R.; Naoe, Y.; Bustos-Villalobos, I.; Ichinose, T.; Tanaka, M.; Zhiwen, W.; Mukoyama, N.; Morimoto, T.; Miyajima, N.; Hitoki, H. Genomic signature of the natural oncolytic herpes simplex virus HF10 and its therapeutic role in preclinical and clinical trials. Front. Oncol. 2017, 7, 149.
- Yu, W.; Fang, H. Clinical trials with oncolytic adenovirus in China. Curr. Cancer Drug Targets 2007, 7, 141–148.
- Fujiyuki, T.; Amagai, Y.; Shoji, K.; Kuraishi, T.; Sugai, A.; Awano, M.; Sato, H.; Hattori, S.; Yoneda, M.; Kai, C. Recombinant SLAMblind Measles Virus Is a Promising Candidate for Nectin-4-Positive Triple Negative Breast Cancer Therapy. Mol. Ther. Oncolytics 2020, 19, 127–135.
- Deng, L.; Fan, J.; Ding, Y.; Zhang, J.; Zhou, B.; Zhang, Y.; Huang, B.; Hu, Z. Oncolytic cancer therapy with a vaccinia virus strain. Oncol. Rep. 2019, 41, 686–692.
- Shiau, A.-L.; Lin, Y.-P.; Shieh, G.-S.; Su, C.-H.; Wu, W.-L.; Tsai, Y.-S.; Cheng, C.-W.; Lai, M.-D.; Wu, C.-L. Development of a conditionally replicating pseudorabies virus for HER-2/neu-overexpressing bladder cancer therapy. Mol. Ther. 2007, 15, 131–138.
- Smedberg, J.R.; Westcott, M.M.; Ahmed, M.; Lyles, D.S. Signaling pathways in murine dendritic cells that regulate the response to vesicular stomatitis virus vectors that express flagellin. J. Virol. 2014, 88, 777–785.
- Melzer, M.K.; Lopez-Martinez, A.; Altomonte, J. Oncolytic vesicular stomatitis virus as a viro-immunotherapy: Defeating cancer with a “hammer” and “anvil”. Biomedicines 2017, 5, 8.
- Pikor, L.A.; Bell, J.C.; Diallo, J.-S. Oncolytic viruses: Exploiting cancer’s deal with the devil. Trends Cancer 2015, 1, 266–277.
- Atherton, M.J.; Evgin, L.; Keller, B.A.; Shenouda, M.M.; Stephenson, K.B.; Vile, R.G.; Bell, J.C.; Evans, D.H.; Lichty, B.D. Infectious Optimism following the 10th International Oncolytic Virus Meeting. Mol. Ther. Oncolytics 2017, 7, 12–16.
- Sultan, H.; Fesenkova, V.I.; Addis, D.; Fan, A.E.; Kumai, T.; Wu, J.; Salazar, A.M.; Celis, E. Designing therapeutic cancer vaccines by mimicking viral infections. Cancer Immunol. Immunother. 2017, 66, 203–213.
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379.
- Davola, M.E.; Mossman, K.L. Oncolytic viruses: How “lytic” must they be for therapeutic efficacy? Oncoimmunology 2019, 8, e1581528.
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788.
- Breitbach, C.J.; Burke, J.; Jonker, D.; Stephenson, J.; Haas, A.R.; Chow, L.Q.; Nieva, J.; Hwang, T.-H.; Moon, A.; Patt, R. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 2011, 477, 99–102.
- Holmes, M.; Scott, G.B.; Heaton, S.; Barr, T.; Askar, B.; Müller, L.M.; Jennings, V.A.; Ralph, C.; Burton, C.; Melcher, A. Efficacy of Coxsackievirus A21 against drug-resistant neoplastic B cells. Mol. Ther. Oncolytics 2023, 29, 17–29.
- Enokida, T.; Moreira, A.; Bhardwaj, N. Vaccines for immunoprevention of cancer. J. Clin. Investig. 2021, 131, e146956.
- Grimmett, E.; Al-Share, B.; Alkassab, M.B.; Zhou, R.W.; Desai, A.; Rahim, M.M.A.; Woldie, I. Cancer vaccines: Past, present and future; a review article. Discov. Oncol. 2022, 13, 31.
- Hargadon, K.M. Tumor microenvironmental influences on dendritic cell and T cell function: A focus on clinically relevant immunologic and metabolic checkpoints. Clin. Transl. Med. 2020, 10, 374–411.
- Cheng, L.; Wang, Y.; Huang, L. Exosomes from M1-polarized macrophages potentiate the cancer vaccine by creating a pro-inflammatory microenvironment in the lymph node. Mol. Ther. 2017, 25, 1665–1675.
- Emens, L.A. Cancer vaccines: On the threshold of success. Expert Opin. Emerg. Drugs 2008, 13, 295–308.
- Donninger, H.; Li, C.; Eaton, J.W.; Yaddanapudi, K. Cancer vaccines: Promising therapeutics or an unattainable dream. Vaccines 2021, 9, 668.
- Waldmann, T.A. Cytokines in cancer immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10, a028472.
- Yron, I.; Wood, T.; Spiess, P.; Rosenberg, S. In vitro growth of murine T cells. V. The isolation and growth of lymphoid cells infiltrating syngeneic solid tumors. J. Immunol. 1980, 125, 238–245.
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Leitman, S.; Chang, A.E.; Ettinghausen, S.E.; Matory, Y.L.; Skibber, J.M.; Shiloni, E.; Vetto, J.T. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N. Engl. J. Med. 1985, 313, 1485–1492.
- Rosenberg, S.A.; Mulé, J.J.; Spiess, P.J.; Reichert, C.M.; Schwarz, S.L. Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high-dose recombinant interleukin 2. J. Exp. Med. 1985, 161, 1169–1188.
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414.
- Cauwels, A.; Van Lint, S.; Paul, F.; Garcin, G.; De Koker, S.; Van Parys, A.; Wueest, T.; Gerlo, S.; Van der Heyden, J.; Bordat, Y. Delivering type I interferon to dendritic cells empowers tumor eradication and immune combination treatments. Cancer Res. 2018, 78, 463–474.
- Enomoto, H.; Tao, L.; Eguchi, R.; Sato, A.; Honda, M.; Kaneko, S.; Iwata, Y.; Nishikawa, H.; Imanishi, H.; Iijima, H. The in vivo antitumor effects of type I-interferon against hepatocellular carcinoma: The suppression of tumor cell growth and angiogenesis. Sci. Rep. 2017, 7, 12189.
- Cox, M.A.; Harrington, L.E.; Zajac, A.J. Cytokines and the inception of CD8 T cell responses. Trends Immunol. 2011, 32, 180–186.
- Yan, W.-L.; Shen, K.-Y.; Tien, C.-Y.; Chen, Y.-A.; Liu, S.-J. Recent progress in GM-CSF-based cancer immunotherapy. Immunotherapy 2017, 9, 347–360.
- Kumar, A.; Khani, A.T.; Ortiz, A.S.; Swaminathan, S. GM-CSF: A double-edged sword in cancer immunotherapy. Front. Immunol. 2022, 13, 901277.
- Bohlius, J.; Reiser, M.; Schwarzer, G.; Engert, A. Impact of granulocyte colony-stimulating factor (CSF) and granulocyte–macrophage CSF in patients with malignant lymphoma: A systematic review. Br. J. Haematol. 2003, 122, 413–423.
- Chen, Y.; Zhao, Z.; Chen, Y.; Lv, Z.; Ding, X.; Wang, R.; Xiao, H.; Hou, C.; Shen, B.; Feng, J. An epithelial-to-mesenchymal transition-inducing potential of granulocyte macrophage colony-stimulating factor in colon cancer. Sci. Rep. 2017, 7, 8265.
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.J.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132.
- Gao, X.; Mi, Y.; Guo, N.; Xu, H.; Xu, L.; Gou, X.; Jin, W. Cytokine-induced killer cells as pharmacological tools for cancer immunotherapy. Front. Immunol. 2017, 8, 774.
- Galdiero, M.R.; Marone, G.; Mantovani, A. Cancer inflammation and cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028662.
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363.
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the treatment of cancer. J. Interferon Cytokine Res. 2019, 39, 6–21.
- Silk, A.W.; Margolin, K. Cytokine therapy. Hematol. Oncol. Clin. 2019, 33, 261–274.
- Xue, D.; Hsu, E.; Fu, Y.-X.; Peng, H. Next-generation cytokines for cancer immunotherapy. Antibody Ther. 2021, 4, 123–133.
- Qiu, Y.; Su, M.; Liu, L.; Tang, Y.; Pan, Y.; Sun, J. Clinical application of cytokines in cancer immunotherapy. Drug Des. Dev. Ther. 2021, 2269–2287.
- Donnelly, R.P.; Young, H.A.; Rosenberg, A.S. An overview of cytokines and cytokine antagonists as therapeutic agents. Ann. N. Y. Acad. Sci. 2009, 1182, 1–13.
- Tagawa, M. Cytokine therapy for cancer. Curr. Pharm. Des. 2000, 6, 681–699.
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68.
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921.
- Zhang, W.-y.; Wang, Y.; Guo, Y.-l.; Dai, H.-r.; Yang, Q.-m.; Zhang, Y.-j.; Zhang, Y.; Chen, M.-x.; Wang, C.-m.; Feng, K.-c. Treatment of CD20-directed chimeric antigen receptor-modified T cells in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: An early phase IIa trial report. Signal Transduct. Target. Ther. 2016, 1, 16002.
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554.
- Wang, Y.; Zhang, W.-y.; Han, Q.-w.; Liu, Y.; Dai, H.-r.; Guo, Y.-l.; Bo, J.; Fan, H.; Zhang, Y.; Zhang, Y.-j. Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. Clin. Immunol. 2014, 155, 160–175.
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol. Ther. 2017, 25, 249–258.
- Siegler, E.L.; Kenderian, S.S. Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: Insights into mechanisms and novel therapies. Front. Immunol. 2020, 11, 1973.
- Murthy, H.; Iqbal, M.; Chavez, J.C.; Kharfan-Dabaja, M.A. Cytokine release syndrome: Current perspectives. ImmunoTargets Ther. 2019, 8, 43–52.
- Lythgoe, M.P.; Liu, D.S.K.; Annels, N.E.; Krell, J.; Frampton, A.E. Gene of the month: Lymphocyte-activation gene 3 (LAG-3). J. Clin. Pathol. 2021, 74, 543–547.
- Yu, X.; Huang, X.; Chen, X.; Liu, J.; Wu, C.; Pu, Q.; Wang, Y.; Kang, X.; Zhou, L. Characterization of a novel anti-human lymphocyte activation gene 3 (LAG-3) antibody for cancer immunotherapy. mAbs 2019, 11, 1139–1148.
- Atkinson, V.; Khattak, A.; Haydon, A.; Eastgate, M.; Roy, A.; Prithviraj, P.; Mueller, C.; Brignone, C.; Triebel, F. Eftilagimod alpha, a soluble lymphocyte activation gene-3 (LAG-3) protein plus pembrolizumab in patients with metastatic melanoma. J. Immunother. Cancer 2020, 8, e001681.
- Zhao, L.; Wang, H.; Xu, K.; Liu, X.; He, Y. Update on lymphocyte-activation gene 3 (LAG-3) in cancers: From biological properties to clinical applications. Chin. Med. J. 2022, 135, 1203–1212.
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602.
- Ji, G.; Zhang, Y.; Si, X.; Yao, H.; Ma, S.; Xu, Y.; Zhao, J.; Ma, C.; He, C.; Tang, Z. Biopolymer Immune Implants’ Sequential Activation of Innate and Adaptive Immunity for Colorectal Cancer Postoperative Immunotherapy. Adv. Mater. 2021, 33, 2004559.
- Deng, J.; Zhao, S.; Zhang, X.; Jia, K.; Wang, H.; Zhou, C.; He, Y. OX40 (CD134) and OX40 ligand, important immune checkpoints in cancer. OncoTargets Ther. 2019, 12, 7347.
- Sun, H.; Dong, Y.; Feijen, J.; Zhong, Z. Peptide-decorated polymeric nanomedicines for precision cancer therapy. J. Control. Release 2018, 290, 11–27.
- Song, H.; Yang, P.; Huang, P.; Zhang, C.; Kong, D.; Wang, W. Injectable polypeptide hydrogel-based co-delivery of vaccine and immune checkpoint inhibitors improves tumor immunotherapy. Theranostics 2019, 9, 2299.
- Shen, K.-Y.; Song, Y.-C.; Chen, I.-H.; Chong, P.; Liu, S.-J. Depletion of tumor-associated macrophages enhances the anti-tumor immunity induced by a Toll-like receptor agonist-conjugated peptide. Hum. Vaccines Immunother. 2014, 10, 3241–3250.
- Choi, Y.J.; Park, S.-J.; Park, Y.-S.; Park, H.S.; Yang, K.M.; Heo, K. EpCAM peptide-primed dendritic cell vaccination confers significant anti-tumor immunity in hepatocellular carcinoma cells. PLoS ONE 2018, 13, e0190638.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
4.6K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
30 May 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No