Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francisco Jaime Bezerra Mendonça-Junior | -- | 3445 | 2023-05-16 14:30:31 | | | |
| 2 | Camila Xu | Meta information modification | 3445 | 2023-05-17 04:19:06 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Leite, F.F.; De Sousa, N.F.; De Oliveira, B.H.M.; Duarte, G.D.; Ferreira, M.D.L.; Scotti, M.T.; Filho, J.M.B.; Rodrigues, L.C.; De Moura, R.O.; Mendonça-Junior, F.J.B.; et al. Chalcones with Anticancer Activity. Encyclopedia. Available online: https://encyclopedia.pub/entry/44376 (accessed on 19 July 2026).
Leite FF, De Sousa NF, De Oliveira BHM, Duarte GD, Ferreira MDL, Scotti MT, et al. Chalcones with Anticancer Activity. Encyclopedia. Available at: https://encyclopedia.pub/entry/44376. Accessed July 19, 2026.
Leite, Fernando Ferreira, Natália Ferreira De Sousa, Bruno Hanrry Melo De Oliveira, Gabrielly Diniz Duarte, Maria Denise Leite Ferreira, Marcus Tullius Scotti, José Maria Barbosa Filho, Luís Cezar Rodrigues, Ricardo Olímpio De Moura, Francisco Jaime Bezerra Mendonça-Junior, et al. "Chalcones with Anticancer Activity" Encyclopedia, https://encyclopedia.pub/entry/44376 (accessed July 19, 2026).
Leite, F.F., De Sousa, N.F., De Oliveira, B.H.M., Duarte, G.D., Ferreira, M.D.L., Scotti, M.T., Filho, J.M.B., Rodrigues, L.C., De Moura, R.O., Mendonça-Junior, F.J.B., & Scotti, L. (2023, May 16). Chalcones with Anticancer Activity. In Encyclopedia. https://encyclopedia.pub/entry/44376
Leite, Fernando Ferreira, et al. "Chalcones with Anticancer Activity." Encyclopedia. Web. 16 May, 2023.
Copy Citation
Chalcones of natural origin present a pattern of phenolic hydroxyls that originate from the biosynthetic reactions of flavonoids.
chalcones
anticancer activity
in vitro
drug discovery
1. Introduction
Chalcones are direct precursors in the biosynthesis of flavonoids. They have an α,β-unsaturated carbonyl system (Figure 1), which gives them broad biological properties [1].
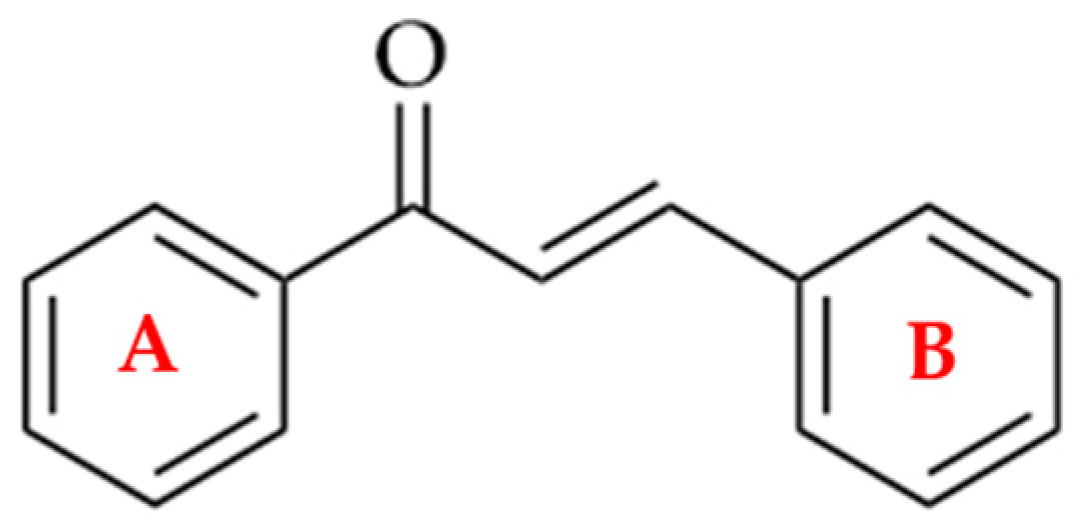
Figure 1. Chemical structure of a chalcone.
Chalcones of natural origin present a pattern of phenolic hydroxyls that originate from the biosynthetic reactions of flavonoids. As chalcones are metabolites derived from the mixed pathway, all natural chalcones have phenolic hydroxyls at positions 5 and 7 [2]. According to Leonte (2021) [3], this class of chalcones is very important, as they are crucial precursors in the biosynthesis of several metabolites with potential antitumor activity, such as flavones, flavanones, aurones, pyrazolines, pyrazoles, and epoxides.
Among the most used synthetic routes for the production of chalcones, the main route is through the Claisen–Schmidt condensation, which involves a condensation between a benzaldehyde and an acetophenone. They also have a conjugated ketone system, which can be functionalized through chemical reactions such as, for example, the addition of thiazole groups, producing a range of chalcone derivatives [3][4][5].
2. TNF-α
The TNF-α factor was discovered in the 1970s, and the function of a serum mediation of innate immunity that is responsible for the induction of hemorrhagic necrosis in tumors is attributed to this signaling pathway [6]. After a few years of studies, it was noticeable that this factor has a dual action in cancer, especially in breast cancer, in which TNF-α can be a target that causes the disease as well as a therapeutic agent [7].
Articles that address TNF-α and natural chalcones correspond to research carried out by Roh and collaborators (2020) [8], which addressed the structural optimization of natural chalcones: isoliquiritigenin, which corresponds to a trihydroxy chalcone (1), as well as butein (2), which represents a tetrahydroxychalcone. Compounds that have already been reported as good inhibitors of histone deacetylases (HDAC) [9] are also considered to be inhibitors of this inflammatory mediator, but their physical-chemical characteristic of solubility is inconvenient for their study and use, because despite having polar groups, these compounds have low solubility (79 mM, 21.0 mg/mL) and a partition coefficient (Log P) of only 0.42, in addition to demonstrating low potency (IC50, 43.3 mM) and insufficient efficacy, with only 21% inhibition at 20 mM in vitro. Due to these limitations, the authors carried out pro-drug optimization strategies by conducting structural modifications in the two mentioned chalcones, with the main objective of improving their pharmacokinetic properties. Based on this information, derivatives of butein were synthesized, corresponding to compounds 3, 4, and 5 (Figure 2), which showed the power to suppress up to 50% of TNF-α production in peritoneal macrophages of mice after stimulation with lipopolysaccharides. Compound 5 was shown to be the most potent inhibitor, with an in vitro IC50 of 14.6 mM and limb volume suppressed by 70% in a murine lymphedema model. Thus, the authors concluded that the pro-drug strategy allowed a six-fold increase in the kinetic solubility of compound 1 and five-fold higher levels of the active metabolite in the blood for compound 5 with oral administration in the pharmacokinetic study. When undergoing modifications in their structure in order to facilitate solubility and permeability, an increase in potency was equivalent to three times (17.3 mM) for anti-inflammatory effects in vitro than the unmodified compound (43.3 mM). Compound 4, despite its insufficient solubility (28 mg/mL), showed greater permeability and was able to suppress limb swelling by 53% orally (100 mg/kg/day). Compound 5 was the most potent (14.6 mM) and had five times greater solubility (136 mg/mL). As a pro-drug, compound 5 was rapidly converted to compound 2 by liver microsomes of three species, including mice, rats, and humans. Thus, the authors suggested that compound 5 could be developed as a potential therapeutic agent targeting anti-inflammatory activity to alleviate the progression of lymphedema.
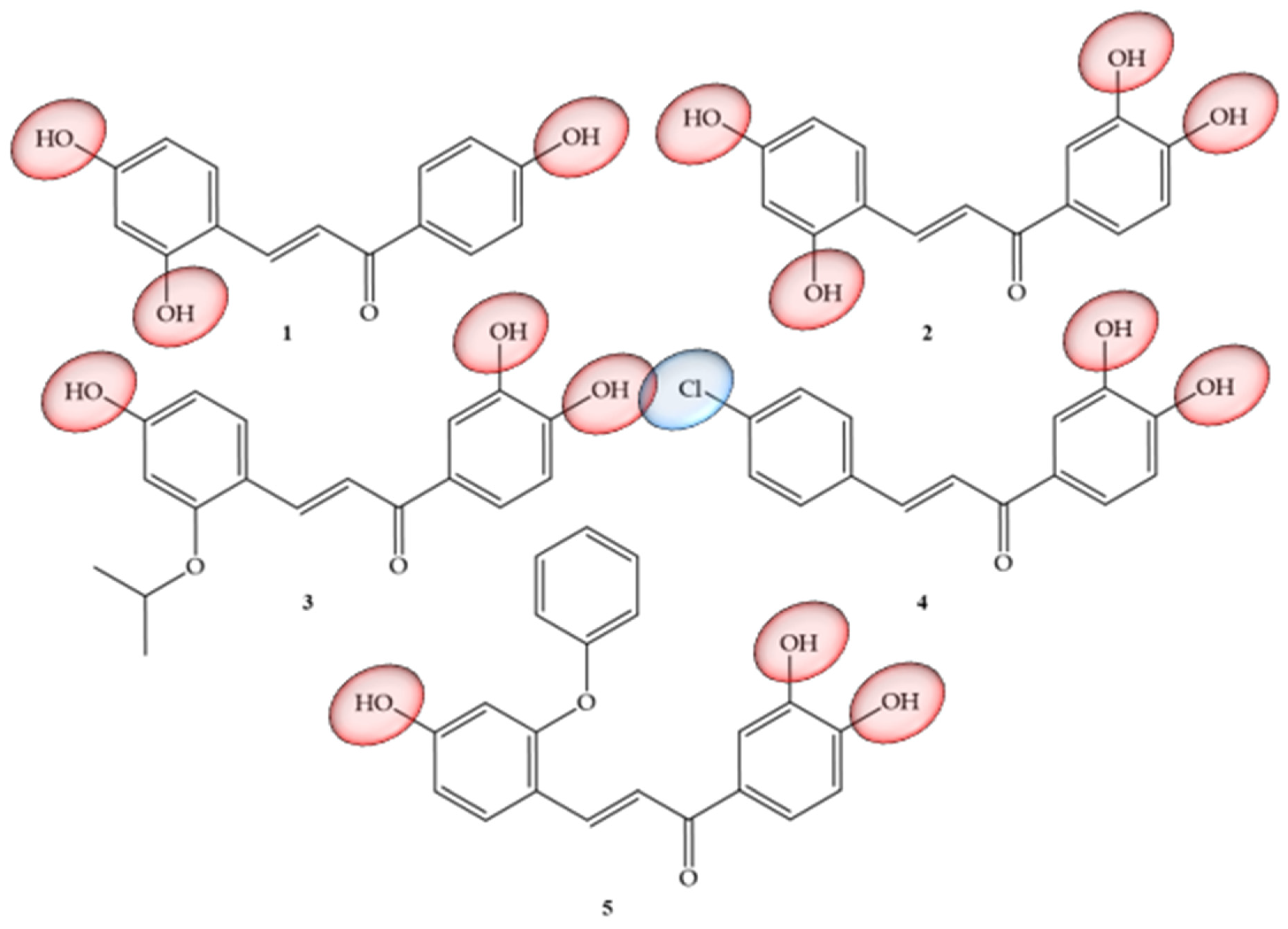
Figure 2. Chemical structure of TNF-α inhibitor compounds.
In a study carried out in 2019, Hashid and collaborators [10] observed that chalcones that have hydroxy, methoxy, and chlorine groups in the ortho and para positions, have strong inhibitory actions against mechanisms of inflammatory action, such as inhibition of cyclooxygenase. It was also revealed in this study that treatment with these compounds was able to inhibit up to 90% of inflammatory edemas.
3. Colon Cancer
As potential therapeutic agents for colon cancer, articles that reported synthetic chalcones were observed with chalcones-ciprofloxacins linked to 1,2,3-triazole (6) (Figure 3), which showed an IC50 ranging from 2.53–8.67 mM, 8.67–62.47 mM, and 4.19–24.37 mM for HCT116, HT29, and Caco-2 cell lines, respectively; while doxorubicin showed IC50 values of 1.22, 0.88, and 4.15 mM. In addition, the compounds in studies still showed Topoisomerase I and II inhibitory activity [11]. Based on the properties of anthocyanidins and aglycones, Păușescu and collaborators (2022) [12] carried out a synthesis of derivatives of the flavilium cation (7) (Figure 3), these being evaluated for anticancer activities in HCT116 and HepG2 strains. The anticancer effect was influenced by the position (6-, 7-, or 8-) of the methoxy group on the β-ring for the methoxy-4′-hydroxy-3′ methoxyflavilyl cation. Thus, the authors concluded that the evaluation of the anticancer activities of derivatives containing methoxy groups in the flavilium cation in hepatocellular carcinoma cells (HepG2) and colon cells of the HCT116 lineage, showed greater efficiency even at low concentrations (26 µM). Formation of the inclusion complex of Compound 5 with the cyclodextrin derivative SBECD led to a 1.5-fold increase in water solubility, preserving 70% of the cytotoxic effect in HepG2 cells.
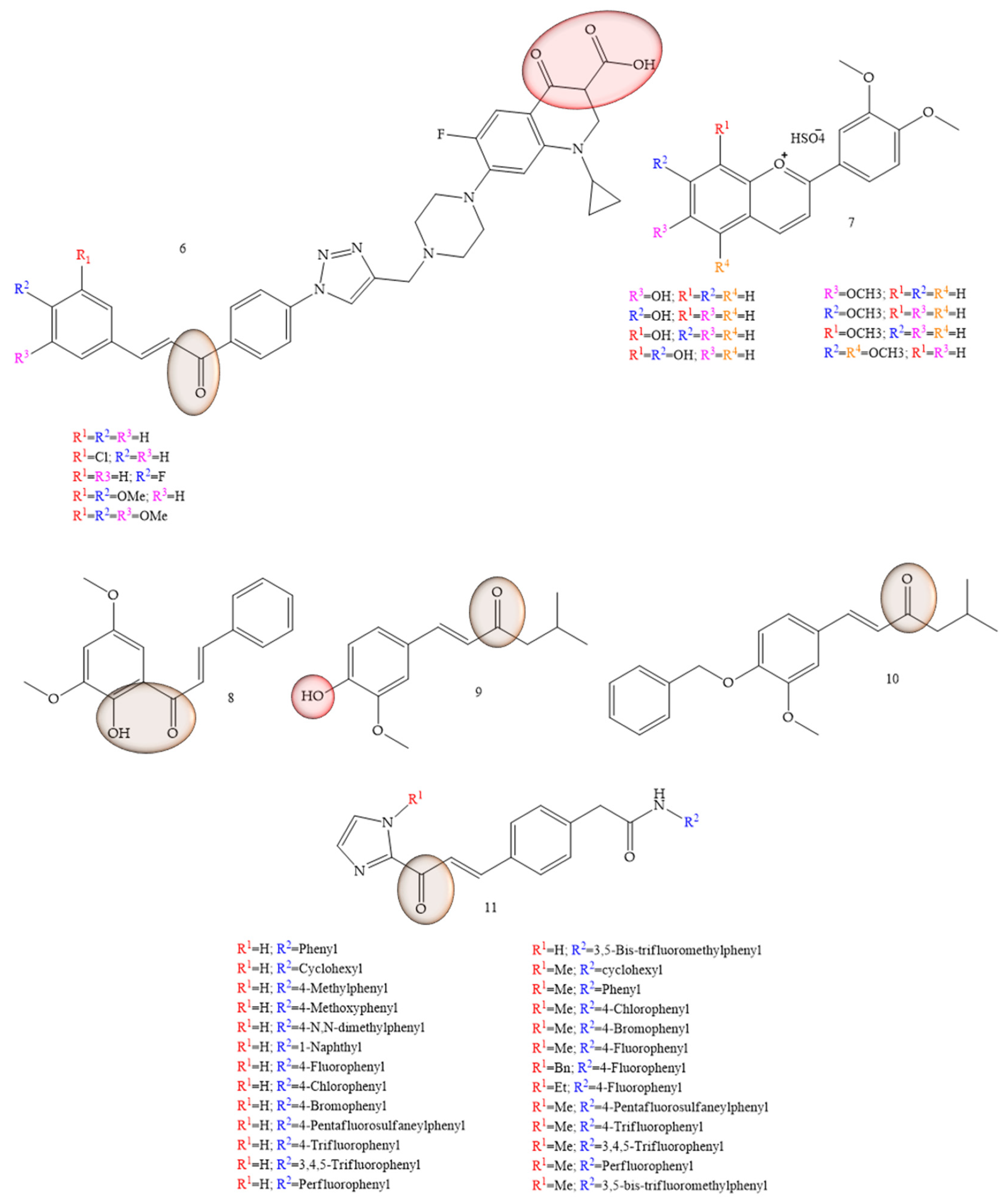
Figure 3. Chemical structure of colon cancer inhibitor compounds.
In a study conducted by Palko-Labuz and collaborators (2020) [13], the natural chalcone found in the Kawa plant, flavokawain B (3) was evaluated for its inhibitory capacity of LoVo/Dx cells. The results showed that the natural chalcone under study showed strong cytotoxic activity, as well as strongly inhibited cell proliferation of the strain under study. Furthermore, at low concentrations the chalcone flavokawain B (8) contributed to apoptosis, as it led to an increase in the expression of caspase-3 activity.
Vanillin-based chalcone analogues were discussed by Lukovic and collaborators (2020) [14]. The IC50 values observed in the HCT-116 cell models were equivalent to 6.85 ± 0.71 μg/mL for Compound 9 (Figure 3) and 7.9 ± 1.37 μg/mL for Compound 10 (Figure 3). Furthermore, vanillin-based chalcone analogues caused overexpression and activation of mitochondrial Bax protein and caspase-3 in HCT-116 cells, indicating that their antitumor mechanism of action was mediated by activation of the internal apoptotic pathway.
Inhibition of HCT-116 cells for evaluation of new tubulin inhibitors was performed by Hamashi and collaborators (2021) [15], through the aldol reaction of N-tosyl imidazolketone with the respective aldehyde group (Compound 11) (Figure 3), thus obtaining 26 compounds that obtained GI50 values corresponding to 5.14 ± 6.81 µM. Regarding the IC50 values of the enzyme histone deacetylase (HDAC), these were equivalent to 1.8 ± 9.0 µM.
The presence of groups that favor hydrogen bonds, as well as the presence of halogenated elements may be related to the activity, since among the compounds considered the most active, they have these characteristics in common. In contrast, these groups demonstrate large cytotoxic loads, as demonstrated by Palko-Lobuz and collaborators [13].
4. Lung Cancer
To obtain new therapeutic agents for lung cancer, Mphahlele and collaborators (2021) [16], carried out a sulfonylation reaction on compounds of the type 5-styryl-2-aminochalcones with p-toluenesulfonyl chloride in pyridine providing new hybrids of 5-styryl-2-sulfonamidochalcones. The in vitro results of the compounds (12) and (13) (Figure 4) against A549 and vero cell lines with LPS, showed a suppression capacity of up to 55% of ROS in vero cells and 35% in A549 cells. These compounds also reduced the cytotoxicity against the A549 cell line and did not affect the viability of vero cells. Another mechanism used for the discovery of drugs against lung cancer was addressed by Sherikar and collaborators (2021) [17]; in this work, the authors evaluated synthetic chalcone derivatives for blocking calcium channels, the study being carried out through pharmacophore modeling combined with experimental evaluation. Pharmacophore modeling revealed that hydrogen bonding receptors and hydrophobic groups are important features for calcium channel-blocking activity. The docking study showed the existence of hydrophobic interactions, hydrogen bonds, and Van der Waals interactions between the amino acid residues and the ligands. In vitro screening showed that compounds 14, 15, and 16 were potent, yielding an IC50 of 4756, 3608, and 5211 µM, respectively, while the standard drug, Nifedipine, showed an IC50 of 1.30 µM. Furthermore, it is important to mention that synthetic chalcone derivatives with NO (nitric oxide) donating capacity is promising for designing new calcium channel blockers.
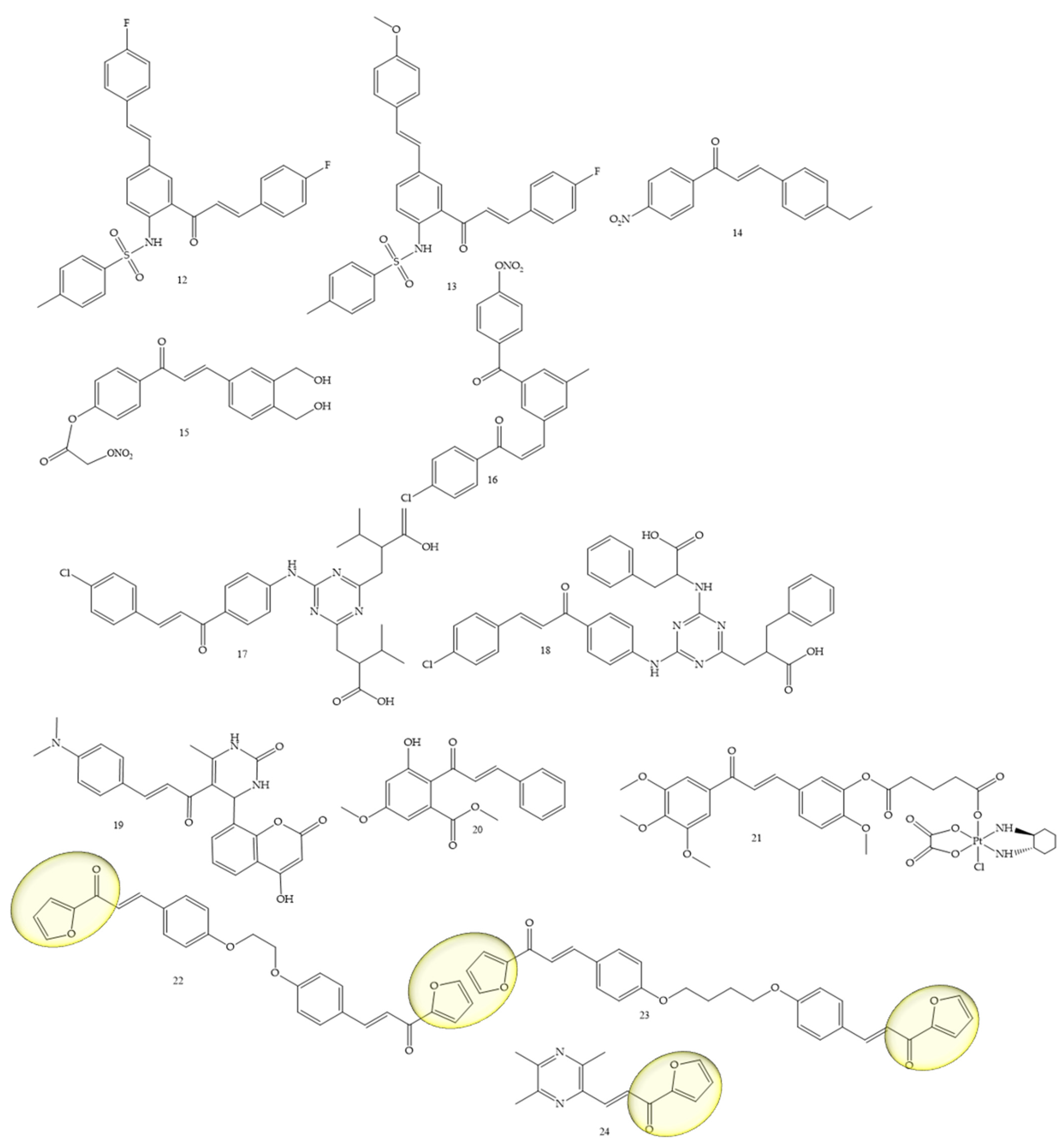
Figure 4. Chemical structure of lung cancer inhibitor compounds.
Another class under study was approached by El-wakil and collaborators (2020) [18], which refers to a series of 1,3,5-triazines linked to chalcones. The results showed that compounds (17) and (18) (Figure 4) significantly inhibited the viability of cancer cells of the A549 lineage, and their IC50 values were 24.5 and 17 µM, respectively, in reference to cisplatin (IC50 = 21.5 µM). Furthermore, mechanistic studies employing MALDI-TOF MS (matrix-assisted laser desorption ionization—time of flight mass spectrometry) and fluorescence spectroscopy using the EvaGreen probe inferred that (17) and (18) induced DNA double-stranded breaks, in contrast to cisplatin, which induces crosslinks between DNA strands’ DNA.
The functionalization of coumarins to chalcones as potent agents for lung cancer, among other activities, was mentioned by Kumar and collaborators (2021) [19]. In this article, the authors synthesized coumaryl–chalcone derivatives and it was observed that the compound lead (19) (Figure 4) is very potent against the A-549 (lung) strain, and also showed satisfactory results for the Jurkat (leukemia) and MCF-7 (breast). The IC50 values were, respectively, equivalent to 70.90, 79.34, and 79.13 μg/mL.
The natural chalcone flavokawain B (Compound 20) (Figure 4) was studied by Hseu and collaborators (2019) [20] as an inhibitor of A459 cells and NSCLC cells (H1299). IC50 values of 11 µg/mL were observed for A549 cells and 5.1 µg/mL for H1299 cells. Furthermore, at concentrations of 5–15 μg/mL, the chalcone under study induced apoptosis and autophagy in A-549 cells.
The platinum complex functionalization in the chalcone structure was a strategy adopted by Wang and collaborators (2021) [21]. Compound 21 (Figure 4) was the best performing complex, showing high apopytotic capacity, as well as IC50 values of 0.31 ± 0.09 and 0.71 ± 0.18 μM for the resistant strains A549/CDDP and SGC-7901/CDDP, while IC50 values for cisplatin corresponded to 35.05 ± 1.39 and 26.81 ± 1.73 μM, respectively.
Bischalcone derivatives linked to aliphatic ligands, with furan units in the A or B rings were synthesized by Fathi and collaborators (2021) [22]. Substances 22 and 23 (Figure 4) were considered the most promising, with IC50 (24.9 and 13.7 μg/mL, respectively) against A549, compared with the reference drug doxorubicin (IC50, 28.3 μg/mL), in addition to presenting IC50 (26.1 and 14.4 μg/mL, respectively) against A431 compared with the reference drug doxorubicin (IC50, 24.9 μg/mL).
A ligustrazine chalcone was synthesized by Bukhari (2022) [23]. Compound 24 (Figure 4) showed an IC50 of 5.11 µM, as well as an inhibitory potential of other strains such as MCF-7.
A study led by Gaur and collaborators [24] pointed out that compounds of the chalcone class which have indole and furan groups in their structure, present high cytotoxicity against cell lines of this specific type of cancer, capable of combating these cells with an IC50 of 1 µg/mL, as well as cyclized chalcones with groups of diazoles, as reported by Bracke et al., 2008 [25].
5. Breast Cancer
Breast cancer was one of the most mentioned pathologies, Wang and collaborators (2020) [26] reported the synthesis of new chalcones containing a diaryl ether moiety (25) (Figure 5). The results showed that among the synthesized compounds, the compound (25) with the 4-methoxy substitution on the right aromatic ring was considered the most active in cancer for MCF-7, HepG2, and HCT116 cell lines, showing IC50 values of 3.44 ± 0.19, 4.64 ± 0.23, and 6.31 ± 0.27 μM, respectively. In vitro tests showed that the compound (25) can effectively inhibit tubulin polymerization. Further studies of the mechanism of action revealed that the compound (25) was able to induce G2/M phase arrest and cell apoptosis. Furthermore, molecular docking studies revealed that compound (25) interacts and binds at the colchicine binding site of tubulin.
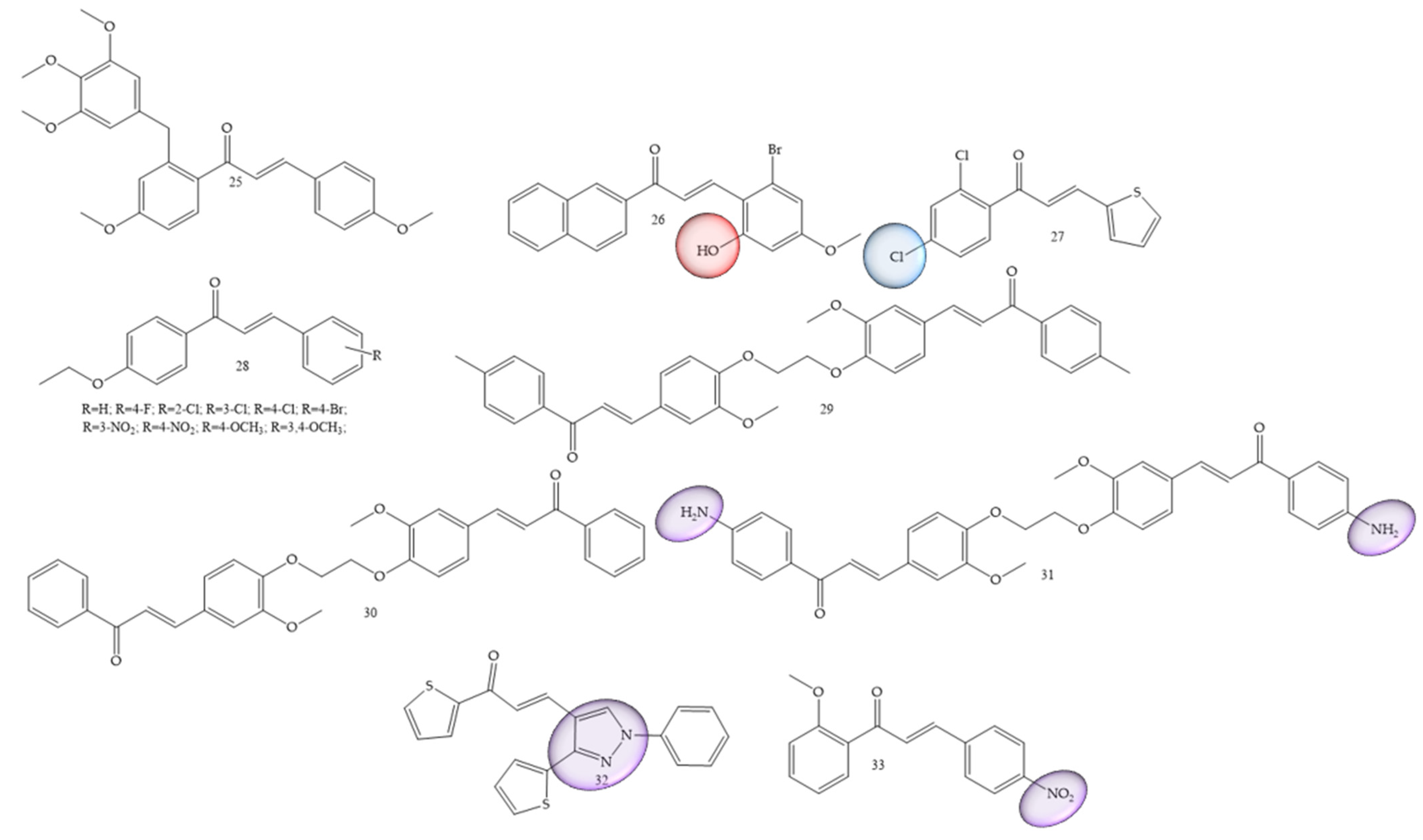
Figure 5. Chemical structure of breast cancer inhibitor compounds.
In another work Guruswamy and Jayarama (2020) [27] carried out synthesis of chalcone derivatives by the Claisen–Schmidt method. The resulting compound (26) (Figure 5) demonstrated incredible anticancer potential in MCF7 cells, with IC50 estimates of 6.55–10.14 µM. In addition, this induced apoptosis, since the increase in the expression level of Caspase 9 and Caspase 3 was noticeable. These results demonstrated expressive apoptotic activity.
The authors Homerin and collaborators (2020) [28], carried out synthesis of chalcones with thienyl groups as potential inhibitors of the farnesyltransferase enzyme as well as of the MCF-7 cell line. The bis(thienyl) chalcone (compound 27) (Figure 5) was the most active compound in the series (IC50 = 7.4 µM), thus showing antiproliferative potential against the MCF7 cancer cell lines.
Another reported class refers to ethoxychalcones; these were studied by Harshitha and collaborators (2020) [29] and were obtained by classic aldol condensation. The best compound was derivative (28) (Figure 5), which showed IC50 value = 53.47 μM for breast cancer cell line MDA-MB-231 and metastatic melanoma cells (A-375).
Al-kaabi and collaborators (2021) [30] synthesized chalcones from 1,2-bis(2-methoxy-4-vinylphenoxy)ethane. Compounds (29), (30) and (31) (Figure 5) showed the highest inhibition rate against the human breast cancer cell line Cal51, 64.1% 60.2%, and 50.4%, respectively.
Chalcones with thiophene groups were synthesized by Mangoud and collaborators (2020) [31]. The results showed that chalcone 32 (Figure 5) had an inhibition percentage of 56.90% against T-47 D.
Polymethoxylated chalcones substituted with nitro groups (NO2) were synthesized by Ahn and collaborators (2022) [32]. IC50 values for the MCF-7 cell line were around 1.33 and 172.20 μM, with compound 33 (Figure 5) being considered the best performer.
6. Oral Cancer
In cases where the search for new therapeutic agents for oral cancer was mentioned, this was related to the suppression of inflammatory mediators such as interleukins. Rajeswari and collaborators (2022) [33], performed the functionalization of chalcones with other natural products belonging to the coumarin class, which are considered pharmacophoric groups and which have already reported activity against oral cancer. In addition, pyrazolone aldehydes were produced using the Vilsmeier–Haack reaction, via the reaction between aldehydes and ketones in an alcoholic medium with sodium hydroxide. The in vitro assays were combined with in silico simulations that demonstrated high affinity of the compounds under study with mediators such as interleukins, as it was also possible to observe that the in vitro cell viability studies of the series show that chalcones (34), (35), and (36) (Figure 6) presented IC50 values of 2.96, 2.97, and 2.82 µM against CAl27 oral cancer cell lines.
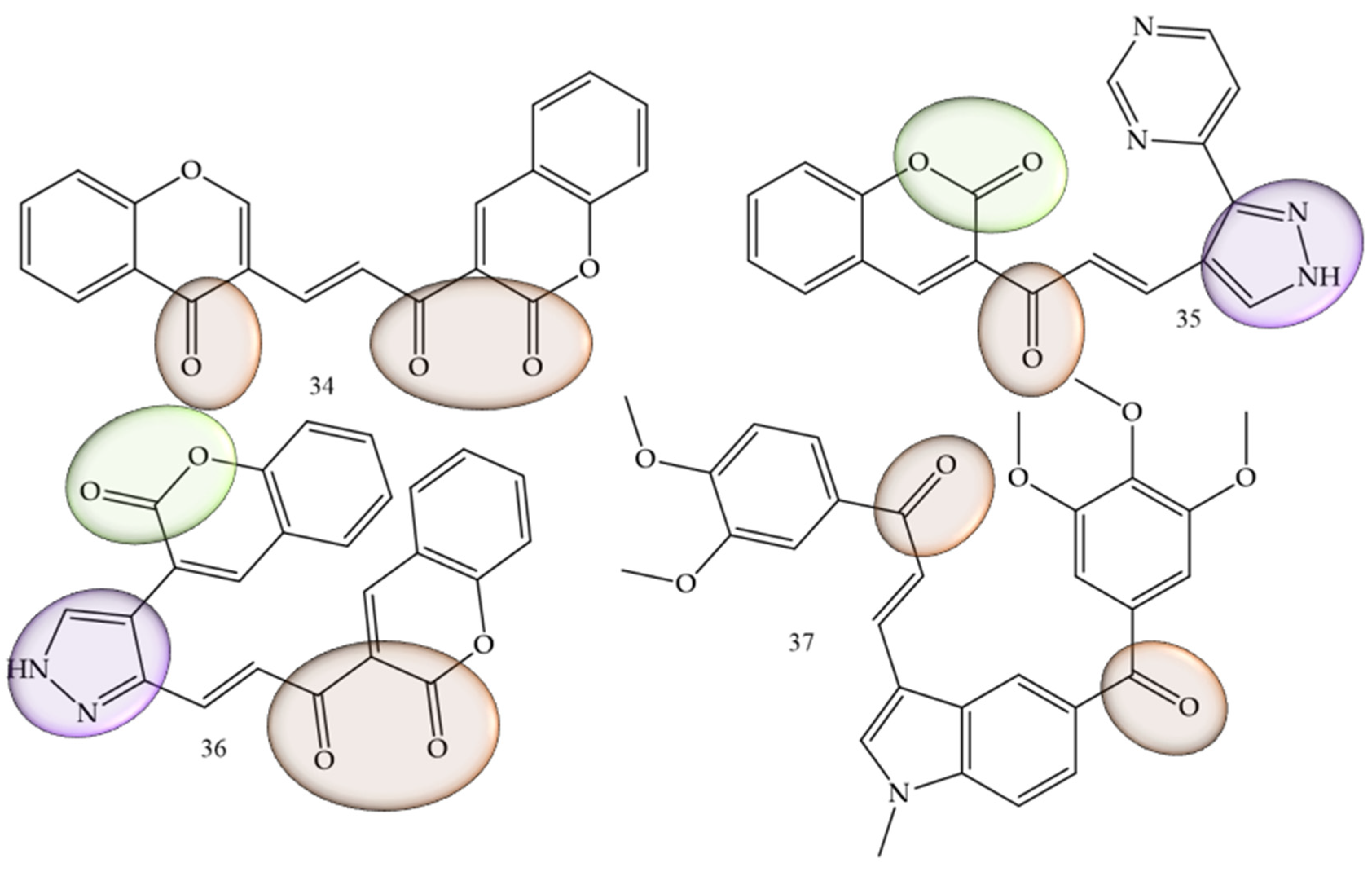
Figure 6. Chemical structure of oral cancer inhibitor compounds.
The inhibitory activity of the AW13516 cell line was studied by Kode and collaborators (2020) [34]. In this work, the authors synthesized the compound 37 (E)-1-(3,4-dimethoxyphenyl)-3-(1-methyl-5-(3,4,5-trimethoxybenzoyl)-1H-indol-3-yl)prop -2-in-1-one (37) (Figure 6). This derivative has 1-methyl, 2 and 3-methoxy substituents on the aromatic ring and was effective in inhibiting the AW13516 strain, showing GI50 values of 0.96 µM.
A study guided by Gul and collaborators [35] demonstrated that chalcone-like compounds have a greater selectivity for cells of the oral cancer lineage when they are trimethoxylated. In addition to the selectivity, based on in vitro experiments the authors concluded that these groups increase the potency of the compounds, showing promise in the fight against this type of cancer.
7. Leukemia
Leukemia was also one of the most prevalent pathologies; synthetic derivatives of chalcones were reported in research by Mphahlele and collaborators (2022) [36], a series of 2-hydroxy-3-nitrochalcones substituted by 5-methyl, 5-bromo, and 5-chloro were synthesized. The results showed that chalcones 38 to 45 (Figure 7) exhibited inhibitory effect against α-glucosidase and α-amylase enzymes, in addition, it exhibited minimal cytotoxicity against Raw-264.7 macrophage cells (murine) in comparison with the anticancer drug, curcumin. For acute lymphoblastic leukemia, Kudličková and collaborators (2020) [37] performed the synthesis of 20 chalcone derivatives substituted with nitro groups (NO2) and (CF3). Of the synthesized compounds, four derivatives (compounds 46, 47, 48, and 49), presented IC50 values between 6.1 and 8.9 μM for the inhibition of T lymphocytes, thus demonstrating a high antiproliferative role.
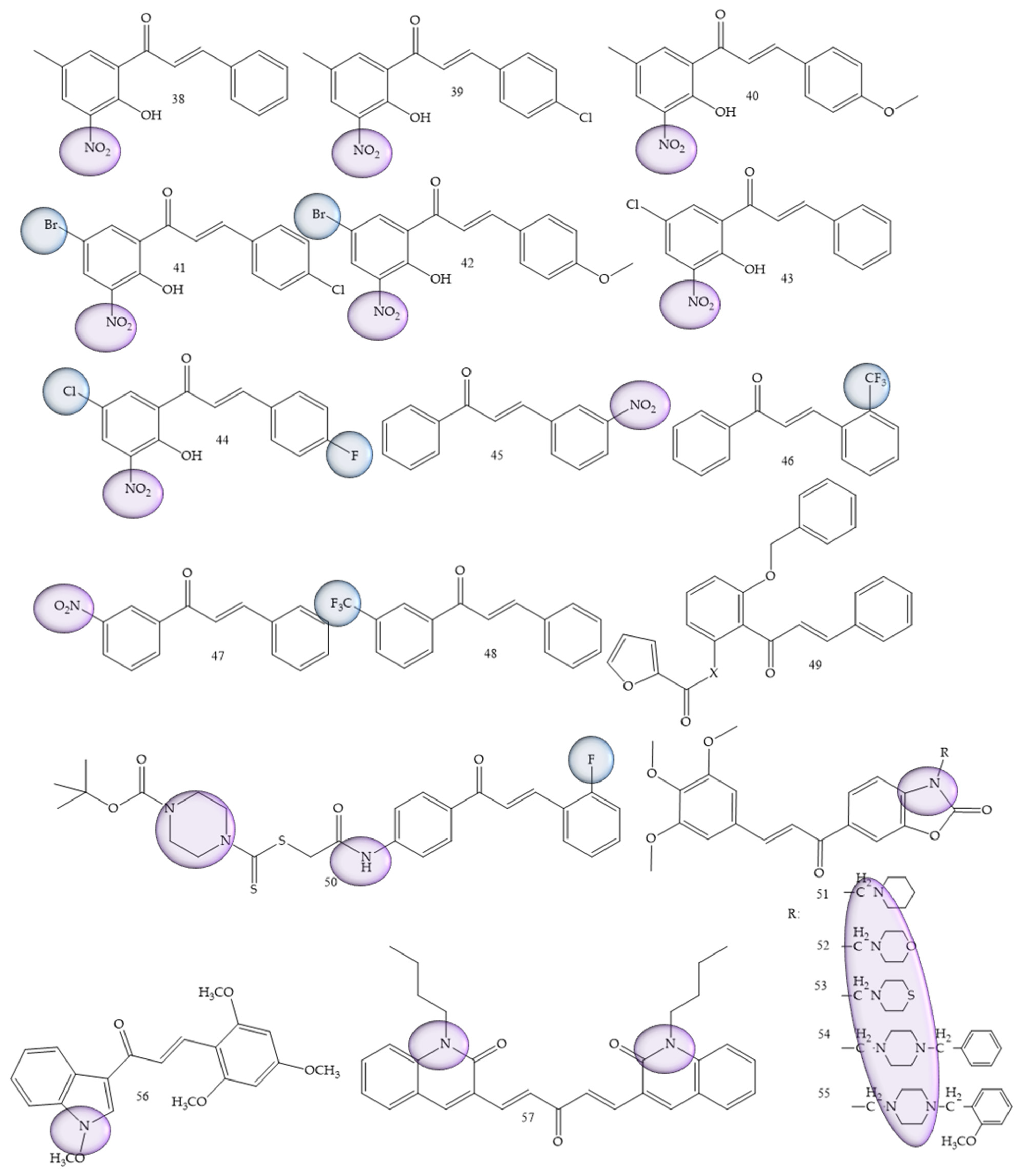
Figure 7. Chemical structure of leukemia inhibitor compounds.
Del Rosário and collaborators (2022) [38], carried out the synthesis of 50 chalcones through a standard aldol condensation reaction of three acetophenones with three benzaldehydes. These compounds were evaluated by means of the inhibition capacity of the strains HL-60 and U-937. Chalcone 2,2′-furoyloxy-4-methoxychalcone (compound 50) (Figure 7) was the most active compound, with IC50 values corresponding to 4.9 ± 1.3 μM.
In a differentiated methodology, Li and collaborators (2021) [39] aimed to search for new inhibitors of the specific histone lysine demethylase 1 (LSD1) enzyme, for which the synthesis was carried out through the aldol condensation of acetophenone and benzaldehyde. Compound (51) (Figure 7), characterized as a piperidine oxazole chalcone, presented IC50 values corresponding to 0.14 μM, thus being about 100 times more potent than its precursor, in addition to representing a highly promising compound for the treatment of leukemia.
Petrov and collaborators (2020) [40], carried out the synthesis of benzoxazolone derivatives to evaluate the inhibition of human leukemia strains. Compound (52) (Figure 7) demonstrated dose-dependent effect of cytotoxicity, being more sensitive for BV-173, SKW-3 and HL-60 strains (IC50 = 3.6–10.7 μM). The introduction of the aminomethyl group at position 3 of benzoxazolone was considered a structural prerequisite for the cytotoxic activity of the synthesized molecules.
For acute lymphoblastic leukemia, Kudličková and collaborators (2021) [37] performed the synthesis of 20 chalcone derivatives substituted with nitro groups (NO2) and (CF3). Of the synthesized compounds, four derivatives (compounds 46, 47, 48, and 49), presented IC50 values between 6.1 and 8.9 μM for the inhibition of T lymphocytes, thus demonstrating a high antiproliferative role. The greatest efficacy was demonstrated by chalcones against Jurkat leukemic cells, which are rapidly proliferative and more sensitive cells. IC50 values (excluding three compounds) ranged from 3.9 to 15 µM. The best results were obtained by compound 53 (Figure 7).
Bis-quinolinyl-chalcone compounds were synthesized by Insuasty and collaborators (2020) [41]. Compound 54 (Figure 7) showed significant activity against leukemia cells K-562 (GI50 = 0.88 µM), RPMI-8226 (GI50 = 0.32 µM) and SR (GI50 = 0.32 µM).
Studies led by Vrontaki (2017) [42] and Mercader (2012) [43] showed that nitrogenated and halogenated chalcones have high cytotoxicity against leukemic cells, being able to limit their growth, in addition to inducing the process of cell apoptosis.
8. Hepatocarcinoma
A study conducted by Wang and collaborators (2022) [44] showed two series of chalcone derivatives containing aminoguanidine or bis-chalcone that were designed, synthesized, and screened for their cytotoxicity, proliferation inhibition, and apoptosis-promoting activity in vitro. The results showed that 2-((E)-4-((E)-3-oxo-3-(p-tolyl)prop-1-en-1-yl)benzylidene)hydrazine-1-carboximidamide (58) (Figure 8) was the most potent compound, with IC50 values of 7.17 μM and 3.05 μM of in vitro anti-proliferative activity against HepG2 human hepatocarcinoma cells and SMMC-7721 cells, respectively. This result showed that the compound had a certain degree of selectivity for human hepatocellular carcinoma cells, especially for SMMC-7721, affirming this compound as a potential drug candidate.
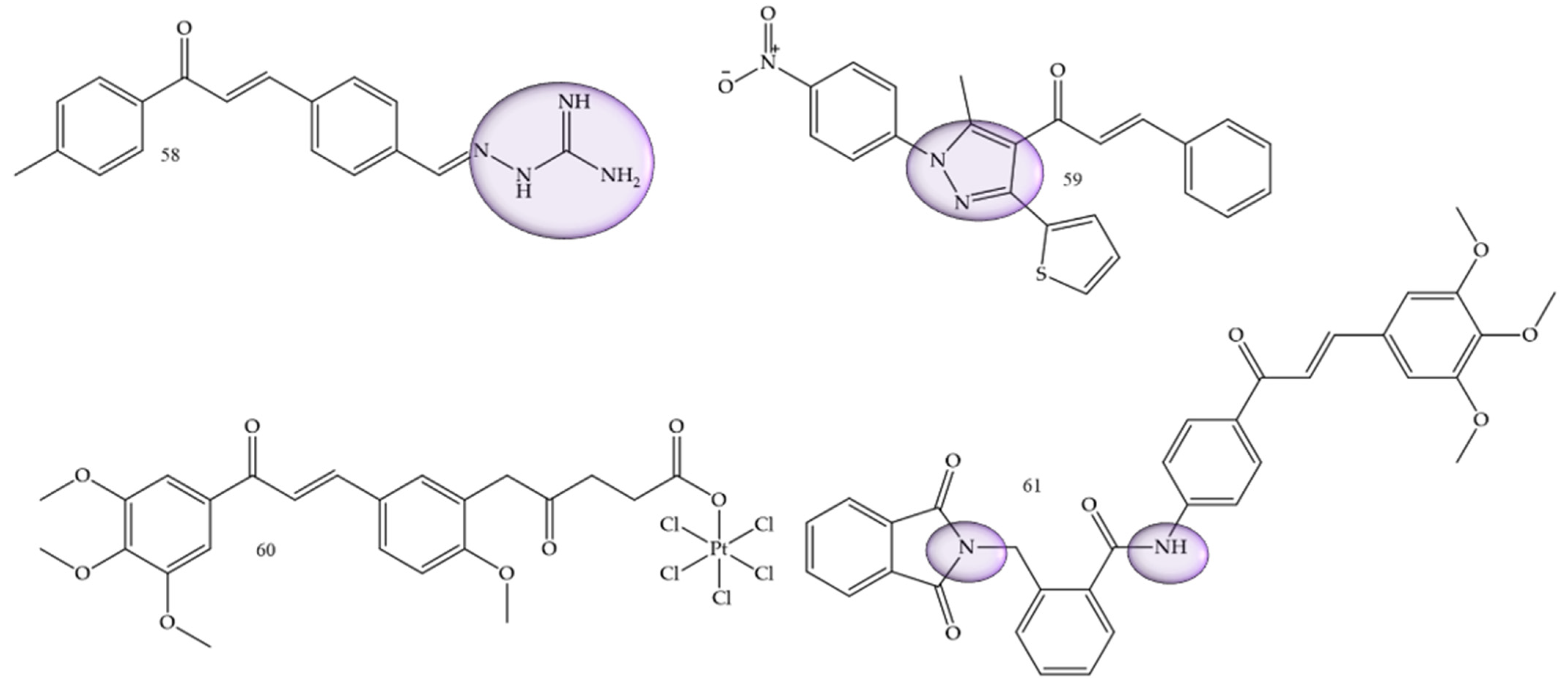
Figure 8. Chemical structure of hepatocarcinoma inhibitor compounds.
Helmy and collaborators (2022) [45], synthesized 4-acetyl-5-furan/thiophene-pyrazole derivatives. Compound (59) (Figure 8) showed to be the most promising compound, with IC50 = 26.6 µg/mL against HepG2 cells compared with the reference drug doxorubicin (IC50 = 21.6 µg/mL), and with IC50 = 27.7 µg/mL against A549 cells compared with the reference drug doxorubicin (IC50 = 28.3 µg/mL).
Huang and collaborators (2020) [46], reported functionalized platinum complexes to chalcones. Chalcone 60 (Figure 8) showed the best IC50 value for the HepG-2 strain IC50 of 0.33 μM, as well as other good values 0.41 μM, 0.30 μM, 0.45 μM, and 11.85 µM for HeLa, MGC-803, NCI-H460, and HL-7702, respectively.
α-phthalimido-chalcones were synthesized by Mourad and collaborators (2020) [47]. Trimethoxy derivative 61 (Figure 8) demonstrated the most potent anticancer activity, with an IC50 of 1.62 µM for Hep G2 and 1.88 µM for MCF-7.
Thiophenic, nitrogenous, and diazolic chalcones have a strong antioxidant power, in addition to acting significantly against breast cancer and hepatocellular carcinoma cells, as reported in studies led by Zahrani in 2020 [48].
9. Cervical Cancer
Methoxylation and hydroxylation reactions in chalcones were the strategies used by Sangpheak and collaborators (2019) [49] for the elaboration of new topoisomerase enzyme inhibitors. The synthesized compound corresponded to a derivative that had 2,4-dimethoxy and 6-hydroxy groups in ring A and 3′,4′,5′-trimethoxy in ring B (Compound 62) (Figure 9), and showed the highest cytotoxicity in in vitro against HeLa, HT-1376, and MCF-7 strains, whose IC50 values corresponded to 3.2, 10.8, and 21.1 µM. In addition, it was demonstrated that the test compound had a high affinity for the topoisomerase enzyme, which was confirmed by molecular docking and molecular dynamics simulations.
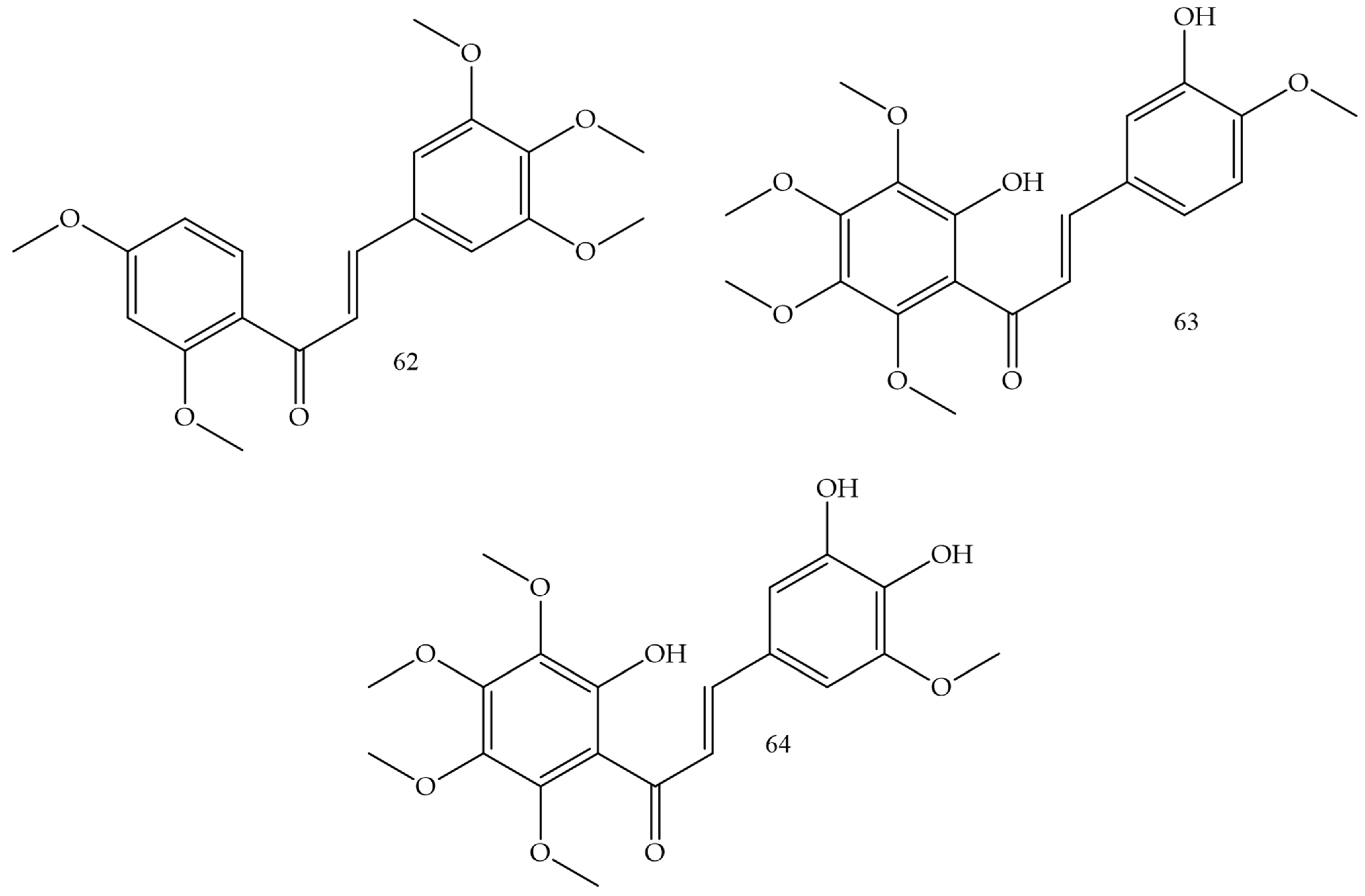
Figure 9. Chemical structure of cervical cancer inhibitor compounds.
HeLa cell inhibition was evaluated by Vongdeth and collaborators (2019) [50]. In this work, polyhydroxychalcones were synthesized by the classic Claisen–Schmidt condensation pathway of 2-hydroxy-4,6-dimethoxyacetophenone with several aldehydes. Compound (63) (Figure 9) was the most potent, with greater selectivity for HeLa cells (IC50 1.44 µM) and SK-OV-3 cells (IC50 1.60 µM).
10. Glioblastoma
Medanha and collaborators (2021) [51] reported the synthesis of new chalcones as promising inhibitors of human glioblastoma lines U98 and GL261. Chalcone 65 (Figure 10) was the compound with the best performance and reduced cell proliferation by 40% for U87 cells and 25% for GL261, not dependent on exposure time. The number of viable cells was significantly lower in treated cells and the invasive capacity of U87 cells was reduced by 50% after treatment with chalcone 65. These results demonstrate chalcone 65 as a promising agent.
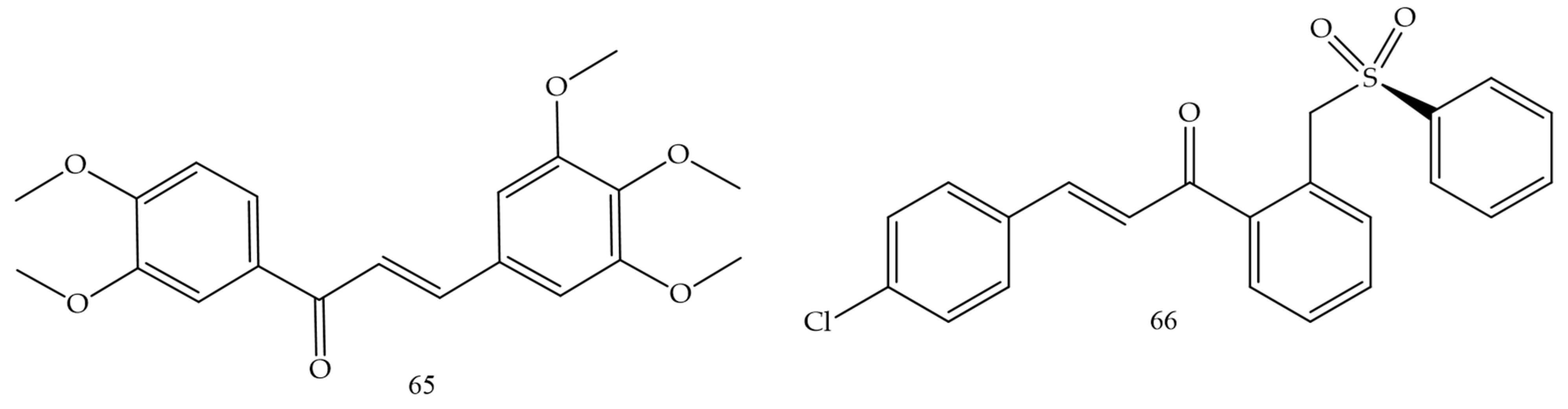
Figure 10. Chemical structure of glioblastoma inhibitor compounds.
Sulfonamide groups in chalcones were reported by Custodio and collaborators (2020) [52]. Compound (66) (Figure 10) had the lowest values of IC50 (2.1 e 2.4 µg.mL−1 against SF-295 e PC-3, respectively).
11. Melanoma
Castano and collaborators (2022) [53], synthesized chalcone-sulfonamide hybrids. Since the compound (67) (Figure 11) presented the best inhibition profile for LOX IMVI (melanoma) with IC50 = 0.34 µM, it also showed good results for MCF7 and MDA-MB-468 (breast cancer) with IC50 values of 0.97 and 1.20 µM, respectively; K-562 (leukemia) with IC50 = 1.50 µM, and HCT-116 (colon cancer) with IC50 = 1.49 µM.
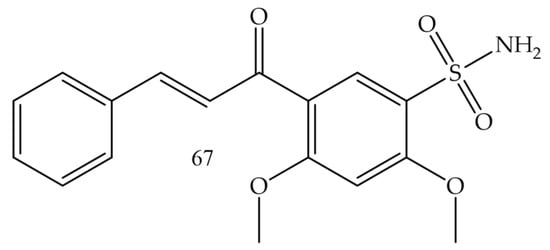
Figure 11. Chemical structure of melanoma inhibitor compounds.
References
- Neto, R.A.d.M.; Santos, C.B.R.; Henriques, S.V.C.; Machado, L.d.O.; Cruz, J.N.; da Silva, C.H.T.d.P.; Federico, L.B.; de Oliveira, E.H.C.; de Souza, M.P.C.; da Silva, P.N.B. Novel Chalcones Derivatives with Potential Antineoplastic Activity Investigated by Docking and Molecular Dynamics Simulations. J. Biomol. Struct. Dyn. 2022, 40, 2204–2216.
- Elkanzi, N.A.A.; Hrichi, H.; Alolayan, R.A.; Derafa, W.; Zahou, F.M.; Bakr, R.B. Synthesis of Chalcones Derivatives and Their Biological Activities: A Review. ACS Omega 2022, 7, 27769–27786.
- Leonte, D.; Coman, F.-M.; Fotso, G.W.; Ngadjui, B.T.; Zaharia, V.; Ngameni, B. Heterocycles 49. Synthesis, Chemical Behaviour and Biological Properties of Heterocyclic Chalcones. Review from Our Research. Farmacia 2021, 69, 821–836.
- Yadav, G.D.; Wagh, D.P. Claisen-Schmidt Condensation Using Green Catalytic Processes: A Critical Review. ChemistrySelect 2020, 5, 9059–9085.
- Liu, B.; Zhao, D.; Xu, D.; Xu, Z. Facile Aldol Reaction between Unmodified Aldehydes and Ketones in Bronsted Acid Ionic Liquids. Chem. Res. Chinese Univ. 2007, 23, 549–553.
- Josephs, S.F.; Ichim, T.E.; Prince, S.M.; Kesari, S.; Marincola, F.M.; Escobedo, A.R.; Jafri, A. Unleashing Endogenous TNF-Alpha as a Cancer Immunotherapeutic. J. Transl. Med. 2018, 16, 242.
- Cruceriu, D.; Baldasici, O.; Balacescu, O.; Berindan-Neagoe, I. The Dual Role of Tumor Necrosis Factor-Alpha (TNF-α) in Breast Cancer: Molecular Insights and Therapeutic Approaches. Cell. Oncol. 2020, 43, 1–18.
- Koh, J.-J.; Lin, S.; Aung, T.T.; Lim, F.; Zou, H.; Bai, Y.; Li, J.; Lin, H.; Pang, L.M.; Koh, W.L.; et al. Amino Acid Modified Xanthone Derivatives: Novel, Highly Promising Membrane-Active Antimicrobials for Multidrug-Resistant Gram-Positive Bacterial Infections. J. Med. Chem. 2015, 58, 739–752.
- Verza, F.A.; Das, U.; Fachin, A.L.; Dimmock, J.R.; Marins, M. Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy. Cancers 2020, 12, 1664.
- Rashid, H.U.; Xu, Y.; Ahmad, N.; Muhammad, Y.; Wang, L. Promising Anti-Inflammatory Effects of Chalcones via Inhibition of Cyclooxygenase, Prostaglandin E2, Inducible NO Synthase and Nuclear Factor Κb Activities. Bioorganic Chem. 2019, 87, 335–365.
- Mohammed, H.H.H.; Abd El-Hafeez, A.A.; Ebeid, K.; Mekkawy, A.I.; Abourehab, M.A.S.; Wafa, E.I.; Alhaj-Suliman, S.O.; Salem, A.K.; Ghosh, P.; Abuo-Rahma, G.E.-D.A. New 1, 2, 3-Triazole Linked Ciprofloxacin-Chalcones Induce DNA Damage by Inhibiting Human Topoisomerase I& II and Tubulin Polymerization. J. Enzym. Inhib. Med. Chem. 2022, 37, 1346–1363.
- Păușescu, I.; Kántor, I.; Babos, G.; May, Z.; Fodor-Kardos, A.; Miskolczy, Z.; Biczók, L.; Péter, F.; Medeleanu, M.; Feczkó, T. Halochromic Behavior and Anticancer Effect of New Synthetic Anthocyanidins Complexed with β-Cyclodextrin Derivatives. Int. J. Mol. Sci. 2022, 23, 8103.
- Palko-Łabuz, A.; Kostrzewa-Susłow, E.; Janeczko, T.; Środa-Pomianek, K.; Poła, A.; Uryga, A.; Michalak, K. Cyclization of Flavokawain B Reduces Its Activity against Human Colon Cancer Cells. Hum. Exp. Toxicol. 2020, 39, 262–275.
- Luković, J.; Mitrović, M.; Popović, S.; Milosavljević, Z.; Stanojević-Pirković, M.; Anđelković, M.; Zelen, I.; Šorak, M.; Muškinja, J.; Ratković, Z. Antitumor Effects of Vanillin Based Chalcone Analogues In Vitro. Acta Pol. Pharm. Res. 2020, 77, 57–67.
- Al-Hamashi, A.A.; Koranne, R.; Dlamini, S.; Alqahtani, A.; Karaj, E.; Rashid, M.S.; Knoff, J.R.; Dunworth, M.; Pflum, M.K.H.; Casero Jr, R.A. A New Class of Cytotoxic Agents Targets Tubulin and Disrupts Microtubule Dynamics. Bioorganic Chem. 2021, 116, 105297.
- Mphahlele, M.J.; Gildenhuys, S.; Agbo, E.N. In Vitro Evaluation and Docking Studies of 5-Oxo-5H-Furo Chromene-6-Carbaldehyde Derivatives as Potential Anti-Alzheimer’s Agents. Int. J. Mol. Sci. 2019, 20, 5451.
- Sherikar, A.S.; Bhatia, M.S.; Dhavale, R.P. Identification and Investigation of Chalcone Derivatives as Calcium Channel Blockers: Pharmacophore Modeling, Docking Studies, In Vitro Screening, and 3D-QSAR Analysis. Curr. Comput. Aided. Drug Des. 2021, 17, 676–686.
- El-Wakil, M.H.; Khattab, S.N.; El-Yazbi, A.F.; El-Nikhely, N.; Soffar, A.; Khalil, H.H. New Chalcone-Tethered 1, 3, 5-Triazines Potentiate the Anticancer Effect of Cisplatin against Human Lung Adenocarcinoma A549 Cells by Enhancing DNA Damage and Cell Apoptosis. Bioorganic Chem. 2020, 105, 104393.
- Kumar, K.S.; Kotra, V.; Kola, P.K.; Devi, C.H.B.P.; Anusha, N.; Babu, B.H.; Adil, S.F.; Shaik, M.R.; Khan, M.; Al-Warthan, A. ZnCl2 Catalyzed New Coumarinyl-Chalcones as Cytotoxic Agents. Saudi J. Biol. Sci. 2021, 28, 386–394.
- Hseu, Y.; Huang, Y.; Thiyagarajan, V.; Mathew, D.C.; Lin, K.; Chen, S.; Liu, J.; Hsu, L.; Li, M.; Yang, H. Anticancer Activities of Chalcone Flavokawain B from Alpinia Pricei Hayata in Human Lung Adenocarcinoma (A549) Cells via Induction of Reactive Oxygen Species-mediated Apoptotic and Autophagic Cell Death. J. Cell. Physiol. 2019, 234, 17514–17526.
- Wang, M.; Liu, Z.; Huang, X.; Chen, Y.; Wang, Y.; Kong, J.; Yang, Y.; Yu, C.; Li, J.; Wang, X. Dual-Target Platinum (IV) Complexes Exhibit Antiproliferative Activity through DNA Damage and Induce ER-Stress-Mediated Apoptosis in A549 Cells. Bioorganic Chem. 2021, 110, 104741.
- Fathi, E.M.; Sroor, F.M.; Mahrous, K.F.; Mohamed, M.F.; Mahmoud, K.; Emara, M.; Elwahy, A.H.M.; Abdelhamid, I.A. Design, Synthesis, In Silico and In Vitro Anticancer Activity of Novel Bis-Furanyl-Chalcone Derivatives Linked through Alkyl Spacers. ChemistrySelect 2021, 6, 6202–6211.
- Bukhari, S.N.A. Synthesis and Evaluation of New Chalcones and Oximes as Anticancer Agents. RSC Adv. 2022, 12, 10307–10320.
- Gaur, R.; Yadav, D.K.; Kumar, S.; Darokar, M.P.; Khan, F.; Singh Bhakuni, R. Molecular Modeling Based Synthesis and Evaluation of in Vitro Anticancer Activity of Indolyl Chalcones. Curr. Top. Med. Chem. 2015, 15, 1003–1012.
- Parmar, V.S.; Bracke, M.E.; Vanhoecke, B.W.A.; Derycke, L.; Bolca, S.; Possemiers, S.; Heyerick, A.; Stevens, C.V.; Keukeleire, D.D.; Depypere, H.T. Plant Polyphenolics as Anti-Invasive Cancer Agents. Anti-Cancer Agents Med. Chem. 2008, 8, 171–185.
- Wang, G.; Liu, W.; Gong, Z.; Huang, Y.; Li, Y.; Peng, Z. Design, Synthesis, Biological Evaluation and Molecular Docking Studies of New Chalcone Derivatives Containing Diaryl Ether Moiety as Potential Anticancer Agents and Tubulin Polymerization Inhibitors. Bioorganic Chem. 2020, 95, 103565.
- Guruswamy, D.K.M.; Jayarama, S. Proapoptotic and Anti-Angiogenic Activity of (2E)-3-(2-Bromo-6-Hydroxy-4-Methoxyphenyl)-1-(Naphthalene-2-Yl) Prop-2-En-1-One in MCF7 Cell Line. Chem. Pap. 2020, 74, 2229–2237.
- Homerin, G.; Nica, A.S.; Farce, A.; Dubois, J.; Ghinet, A. Ultrasounds-Mediated 10-Seconds Synthesis of Chalcones as Potential Farnesyltransferase Inhibitors. Bioorganic Med. Chem. Lett. 2020, 30, 127149.
- Harshitha, K.R.; Sarojini, B.K.; Narayana, B.; Lobo, A.G.; Kalal, B.S. Molecular Docking of 4-Ethoxychalcones on Oxidoreductase/Pirin Inhibitors and Cytotoxic Evaluation on Breast/Skin Cancer Cell Lines. Lett. Drug Des. Discov. 2020, 17, 1245–1260.
- Al-Kaabi, M.M.; Rady Al-Hazam, H.A.; Arwa, H.M. Microwave Assisted Synthesis, Characterization and Biochemical Study of New Chalcones. Egypt. J. Chem. 2021, 64, 4027–4035.
- Mangoud, M.M.; Hussein, M.Z.; El-Bordany, E.A. Design and Synthesis of Novel Pyrazoles, Pyrazolines, and Pyridines from Chalcone Derivatives with Evaluation of Their In Vitro Anticancer Activity Against T-47D and UACC-257 Cell Lines. Egypt. J. Chem. 2020, 63, 5203–5218.
- Ahn, S.; Truong, V.N.-P.; Kim, B.; Yoo, M.; Lim, Y.; Cho, S.K.; Koh, D. Design, Synthesis, and Biological Evaluation of Chalcones for Anticancer Properties Targeting Glycogen Synthase Kinase 3 Beta. Appl. Biol. Chem. 2022, 65, 17.
- Rajeswari, K.; Manturthi, S.; Sirisha, K.; Velidandi, A.N. Anchoring and Hydrophobic Nature of Coumarin in Newer Coumarin Based Chalcones: Synthesis, In Silico, and In Vitro Cell Viability Studies. Russ. J. Bioorganic Chem. 2022, 48, 636–642.
- Kode, J.; Kovvuri, J.; Nagaraju, B.; Jadhav, S.; Barkume, M.; Sen, S.; Kasinathan, N.K.; Chaudhari, P.; Mohanty, B.S.; Gour, J. Synthesis, Biological Evaluation, and Molecular Docking Analysis of Phenstatin Based Indole Linked Chalcones as Anticancer Agents and Tubulin Polymerization Inhibitors. Bioorganic Chem. 2020, 105, 104447.
- Gul, H.I.; Yamali, C.; Gunesacar, G.; Sakagami, H.; Okudaira, N.; Uesawa, Y.; Kagaya, H. Cytotoxicity, Apoptosis, and QSAR Studies of Phenothiazine Derived Methoxylated Chalcones as Anticancer Drug Candidates. Med. Chem. Res. 2018, 27, 2366–2378.
- Mphahlele, M.J.; Maluleka, M.M.; Choong, Y.S.; Monchusi, B.A.; Mbazima, V.G. An In Vitro Study of the 5-Methyl-and 5-Bromo/Chloro Substituted 2-Hydroxy-3-Nitrochalcones as α-Glucosidase and/or α-Amylase Inhibitors with Potential Anti-Inflammatory Activity. Med. Chem. Res. 2022, 31, 2243–2259.
- Kudličková, Z.; Takáč, P.; Sabolová, D.; Vilková, M.; Baláž, M.; Béres, T.; Mojžiš, J. Novel 1-Methoxyindole-and 2-Alkoxyindole-Based Chalcones: Design, Synthesis, Characterization, Antiproliferative Activity and DNA, BSA Binding Interactions. Med. Chem. Res. 2021, 30, 897–912.
- Del Rosario, H.; Saavedra, E.; Brouard, I.; González-Santana, D.; García, C.; Spínola-Lasso, E.; Tabraue, C.; Quintana, J.; Estévez, F. Structure-Activity Relationships Reveal a 2-Furoyloxychalcone as a Potent Cytotoxic and Apoptosis Inducer for Human U-937 and HL-60 Leukaemia Cells. Bioorganic Chem. 2022, 127, 105926.
- Li, Y.; Sun, Y.; Zhou, Y.; Li, X.; Zhang, H.; Zhang, G. Discovery of Orally Active Chalcones as Histone Lysine Specific Demethylase 1 Inhibitors for the Treatment of Leukaemia. J. Enzym. Inhib. Med. Chem. 2021, 36, 207–217.
- Petrov, O.I.; Ivanova, Y.B.; Gerova, M.S.; Momekov, G.T. Synthesis and Cytotoxicity of New Mannich Bases of 6--2 (3H)-Benzoxazolone. Lett. Drug Des. Discov. 2020, 17, 512–517.
- Insuasty, D.; García, S.; Abonia, R.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Cobo, J.; Borosky, G.L.; Laali, K.K. Design, Synthesis, and Molecular Docking Study of Novel Quinoline-based Bis-chalcones as Potential Antitumor Agents. Arch. Pharm. 2021, 354, 2100094.
- Vrontaki, E.; Melagraki, G.; Voskou, S.; Phylactides, M.S.; Mavromoustakos, T.; Kleanthous, M.; Afantitis, A. Development of a Predictive Pharmacophore Model and a 3D-QSAR Study for an In Silico Screening of New Potent Bcr-Abl Kinase Inhibitors. Mini Rev. Med. Chem. 2017, 17, 188–204.
- Mercader, A.G.; Pomilio, A.B. (Iso)Flav(an)Ones, Chalcones, Catechins, and Theaflavins as Anticarcinogens: Mechanisms, Anti-Multidrug Resistance and QSAR Studies. Curr. Med. Chem. 2012, 19, 4324–4347.
- Wang, Y.; Li, L.; Ma, T.; Cheng, X.; Liu, D. Design, Synthesis, and Apoptosis-Promoting Effect Evaluation of Chalcone Derivatives Containing Aminoguanidine Units. Anti-Cancer Agents Med. Chem. 2022, 22, 2116–2124.
- Helmy, M.T.; Sroor, F.M.; Mahrous, K.F.; Mahmoud, K.; Hassaneen, H.M.; Saleh, F.M.; Abdelhamid, I.A.; Mohamed Teleb, M.A. Anticancer Activity of Novel 3-(Furan-2-yl) Pyrazolyl and 3-(Thiophen-2-yl) Pyrazolyl Hybrid Chalcones: Synthesis and In Vitro Studies. Arch. Pharm. 2022, 355, 2100381.
- Huang, X.; Liu, Z.; Wang, M.; Yin, X.; Wang, Y.; Dai, L.; Wang, H. Platinum (IV) Complexes Conjugated with Chalcone Analogs as Dual Targeting Anticancer Agents: In Vitro and In Vivo Studies. Bioorganic Chem. 2020, 105, 104430.
- Mourad, A.A.E.; Mourad, M.A.E.; Jones, P.G. Novel HDAC/Tubulin Dual Inhibitor: Design, Synthesis and Docking Studies of α-Phthalimido-Chalcone Hybrids as Potential Anticancer Agents with Apoptosis-Inducing Activity. Drug Des. Devel. Ther. 2020, 14, 3111–3130.
- Al Zahrani, N.A.; El-Shishtawy, R.M.; Elaasser, M.M.; Asiri, A.M. Synthesis of Novel Chalcone-Based Phenothiazine Derivatives as Antioxidant and Anticancer Agents. Molecules 2020, 25, 4566.
- Sangpheak, K.; Mueller, M.; Darai, N.; Wolschann, P.; Suwattanasophon, C.; Ruga, R.; Chavasiri, W.; Seetaha, S.; Choowongkomon, K.; Kungwan, N. Computational Screening of Chalcones Acting against Topoisomerase IIα and Their Cytotoxicity towards Cancer Cell Lines. J. Enzym. Inhib. Med. Chem. 2019, 34, 134–143.
- Vongdeth, K.; Han, P.; Li, W.; Wang, Q.-A. Synthesis and Antiproliferative Activity of Natural and Non-Natural Polymethoxychalcones and Polymethoxyflavones. Chem. Nat. Compd. 2019, 55, 11–17.
- Mendanha, D.; Vieira de Castro, J.; Moreira, J.; Costa, B.M.; Cidade, H.; Pinto, M.; Ferreira, H.; Neves, N.M. A New Chalcone Derivative with Promising Antiproliferative and Anti-Invasion Activities in Glioblastoma Cells. Molecules 2021, 26, 3383.
- Custodio, J.M.F.; Moura, A.F.; de Moraes, M.O.; Perez, C.N.; Napolitano, H.B. On the In Silico and In Vitro Anticancer Activity of Sulfonamide Chalcones: Potential JNKK3 Inhibitors. New J. Chem. 2020, 44, 3294–3309.
- Castaño, L.F.; Quiroga, J.; Abonia, R.; Insuasty, D.; Vidal, O.M.; Seña, R.; Rubio, V.; Puerto, G.; Nogueras, M.; Cobo, J. Synthesis, Anticancer and Antitubercular Properties of New Chalcones and Their Nitrogen-Containing Five-Membered Heterocyclic Hybrids Bearing Sulfonamide Moiety. Int. J. Mol. Sci. 2022, 23, 12589.
More
Information
Subjects:
Others
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
17 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No