Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Martine Darwish | -- | 4648 | 2023-05-11 23:16:43 | | | |
| 2 | Rita Xu | -69 word(s) | 4579 | 2023-05-12 03:58:43 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Holz, E.; Darwish, M.; Tesar, D.B.; Shatz-Binder, W. Protein- and Peptide-Based Chemical Conjugates. Encyclopedia. Available online: https://encyclopedia.pub/entry/44175 (accessed on 27 July 2026).
Holz E, Darwish M, Tesar DB, Shatz-Binder W. Protein- and Peptide-Based Chemical Conjugates. Encyclopedia. Available at: https://encyclopedia.pub/entry/44175. Accessed July 27, 2026.
Holz, Emily, Martine Darwish, Devin B. Tesar, Whitney Shatz-Binder. "Protein- and Peptide-Based Chemical Conjugates" Encyclopedia, https://encyclopedia.pub/entry/44175 (accessed July 27, 2026).
Holz, E., Darwish, M., Tesar, D.B., & Shatz-Binder, W. (2023, May 11). Protein- and Peptide-Based Chemical Conjugates. In Encyclopedia. https://encyclopedia.pub/entry/44175
Holz, Emily, et al. "Protein- and Peptide-Based Chemical Conjugates." Encyclopedia. Web. 11 May, 2023.
Copy Citation
The complexity of molecular entities being advanced for therapeutic purposes has continued to evolve. A main propellent fueling innovation is the perpetual mandate within the pharmaceutical industry to meet the needs of novel disease areas and/or delivery challenges. As new mechanisms of action are uncovered, and as our understanding of existing mechanisms grows, the properties that are required and/or leveraged to enable therapeutic development continue to expand.
bioconjugation
protein therapeutics
protein–polymer conjugates
protein delivery
1. Introduction
Proteins and peptides have played a foundational role in the treatment of diseases for nearly a century, beginning with the first commercial use of insulin in 1923. Early protein-based therapeutics, developed prior to the introduction of recombinant DNA technology, were limited by their immunogenicity. The use of chemical conjugation to enhance the properties of proteins dates back to at least the 1970s, when Frank Davis hypothesized that conjugation of a hydrophilic polymer such as PEG could reduce the immunogenicity of non-native proteins and unexpectedly discovered that PEGylation improved the circulating half-life of proteins as well [1][2][3][4]. Since then, at least 30 polymer–protein and polymer–peptide conjugates have been approved by the FDA [5], each of which use polymer conjugation to improve their pharmacokinetics and shield them from antidrug antibody recognition. In addition, the 50-year span of research into the properties of polymer conjugates has unveiled many new properties in this class of therapeutics that have expanded both their prevalence and mechanisms of action in the clinical space.
While the protein represents the active pharmaceutical ingredient (API) for polymer conjugates, other chemically enhanced therapeutics take the opposite approach, using conjugation to proteins as the delivery strategy. Inspired by Paul Ehrlich’s “magic bullet” approach, conceptualized in 1907, antibody–drug conjugates (ADC) were developed to target malignant cells for the delivery of cytotoxic drugs. In this approach, the role of the antibody is to improve circulating half-life and cell-specific uptake of the drug in its target tissue, thereby improving its therapeutic index (TI). Since the first report in 1958 [6], 12 ADCs have been approved to date, while over 80 are currently undergoing clinical trials. Another example of using proteins for a purpose other than as API is in the case of protein-based vaccines, with the first approved hepatitis B vaccine some 30 years ago [7]. In this case, the protein is acting as an agonist to stimulate an immune response, thereby increasing the efficacy of the vaccine. Since then, various novel protein-based approaches have been developed to offer a wider variety of tools for effective vaccines against difficult pathogens. More recently, antibody conjugates have expanded beyond cytotoxic drugs to include novel payloads such as oligonucleotides, offering new and innovative ways to tackle what were previously thought to be undruggable targets. For example, the recent advancements of oligonucleotide-based therapies, designed to bind noncoding RNAs and toxic RNAs associated with disease pathogenesis, have greatly expanded the numbers and types of selectable targets.
The advancement of these novel modalities has nonetheless brought with it new challenges. For polymer–protein and polymer–peptide conjugates, recent progress has been directed towards the design of novel polymers and conjugation chemistries to retain or improve biological activity and evade immune recognition in vivo. Meanwhile, the refinement of both the payload and linker chemistries to improve safety and on-target delivery has been a major focus for ADCs. In the case of therapeutic oligonucleotides, the druggable space within the genome is quite expansive, and their efficacy is mostly limited by ineffective delivery to their intracellular targets. Thus, devising and evaluating an array of delivery strategies has become a critical component of the development of this emerging class of therapeutics. Likewise, the field of protein conjugate vaccines has also benefited from recent innovations leveraging modern protein engineering and chemical conjugation strategies to address the long-standing challenge of generating a robust immune response to difficult carbohydrate antigens.
2. Protein/Peptide Therapeutics That Are Enhanced through Chemical Modification
2.1. Introduction to Polymer–Protein Conjugates
Conventional polymer conjugates were developed to reduce immunogenicity and improve half-life of peptides and proteins with poor systemic PK. The increase in size afforded by polymer conjugation reduces renal filtration, while steric shielding by the polymer may allow proteins to resist opsonization, protease degradation, and antidrug antibody binding. The combined effect of these properties is a significant increase in the circulating half-life of polymer–protein and polymer–peptide conjugates, which sustains serum concentrations within the optimal therapeutic window for longer periods of time. The core principle in the design of polymer–protein conjugates is patient convenience; polymer conjugates are often designed as “biobetters”, leveraging established biology but decreasing the dosing frequency or improving the safety profile of existing therapeutics to enhance the overall patient experience during treatment.
An excellent case study exemplifying the benefits of polymer conjugation is pegfilgrastim, a PEGylated human granulocyte colony-stimulating factor (G-CSF) approved in 2002 for the prophylactic treatment of neutropenia during chemotherapy. Pegfilgrastim’s non-PEGylated predecessor, filgrastim, was limited by the short half-life of G-CSF, requiring a daily dosing regimen that placed a large burden on patients and healthcare systems. Covalent conjugation of a 20 kDa PEG to the N-terminus of filgrastim significantly extends its serum half-life (from a median half-life of 3.5–3.8 h with filgratism to 42 h with pegfilgratism) and enables administration of a single dose per chemotherapy cycle [8]. A retrospective comparison of pegfilgrastim and filgrastim use in breast cancer patients revealed that single-dose pegfilgrastim resulted not only in a lower patient burden, but improved therapeutic outcomes as well. A total of 72.4% of pegfilgrastim patients received their intended dose on time, compared to only 58% in the filgrastim group. Most notably, pegfilgrastim patients were nearly three times less likely to experience severe neutropenia [9]. The impact of pegfilgrastim on the healthcare system and patient burden is illustrated by its commercial success; pegfilgrastim recorded USD 69 billion in lifetime sales as of 2017, and six biosimilar products exist on the market today [10]. This narrative is mirrored in many other commercial polymer–protein conjugates; for example, weekly dosing of pegintron as a monotherapy in chronic hepatitis C patients led to a twofold higher incidence of a durable, complete response compared to the administration of the native IFN-α2b three times weekly [11]. Similarly, pegylation of asparaginase markedly reduced the frequency of hypersensitivity reactions in patients. As a consequence, although the cost per vial is higher for pegasparaginase, its superior safety profile leads to similar total payer costs relative to native asparaginase and a better treatment experience for patients [12].
Over the past few decades, monoclonal antibodies have comprised an increasingly large proportion of the biologics market. This has arguably reduced the need for systemic half-life extension through polymer conjugation, as FcRn recycling gives these molecules long circulation times with half-lives ranging from 6–32 days [13]. Genetic fusion of Fc, albumin, or albumin-binding domains have also become popular methods for extending the half-life of nonantibody proteins, such as cytokines and enzymes [14]. Nevertheless, the clinical landscape is shifting, with a greater emphasis on tissue-specific drug delivery, a need to access targets once considered “undruggable”, and a demand for new approaches to further reduce the treatment burden on patients and healthcare systems. Concurrently, advances in polymer–protein conjugates have revealed new functionalities beyond increased size that position them to have a broader impact on the design of next-generation, chemically enhanced peptides and biologics.
2.2. Polymer Selection
2.2.1. PEG
PEG has an extensive history in protein formulations, with >1000 approved therapeutics using PEG as an excipient. The first PEGylated protein, Adagen, was approved in 1990, giving PEG conjugates more than 30 years of clinical experience [15]. Adagen consists of a PEGylated bovine enzyme, adenosine deaminase (AD). Non-site-specific attachment of a 5 kDa PEG onto the enzyme both increased the half-life of AD and decreased immunogenicity against the nonhuman protein [16]. Many product approvals have followed, each of which employ increased half-life and/or reduced immunogenicity as central design principles. In addition, PEGylation may improve the physical stability of proteins, including their solubility [17][18][19], colloidal stability [20], and melting temperature (Tm) [21][22][23][24][25]. Today, PEGylated proteins still comprise the majority of clinical polymer conjugate candidates. Despite this widespread use, PEG is known to have several drawbacks that have motivated a wealth of research into novel polymeric alternatives to PEG. Among these drawbacks include its polydispersity, lack of biodegradability, and potential immunogenicity.
Mounting evidence suggests the possibility of immunological responses to PEG such as allergic reactions and formation of anti-PEG antibodies (APAs), which may impact the toxicology profile or efficacy of a therapeutic. A subset of patients experience hypersensitivity reactions such as anaphylaxis and infusion reactions after administration with PEG-containing formulations. Furthermore, up to 72% of the human population may have pre-existing anti-PEG antibodies, although reported prevalences vary widely depending on the assay used [26][27]. In most cases, the relatively low concentrations of APAs prevent them from impacting efficacy, but low-dose therapeutics may suffer from accelerated blood clearance (ABC) initiated by anti-PEG antibodies. In a Phase 2 trial of pegylated uricase (pegloticase), 41% of patients developed high-titer antibodies against the PEG portion of pegloticase, resulting in lower serum concentrations of pegloticase and a poor response to treatment in these patients [28]. Similarly, serum anti-PEG antibody levels were associated with a loss of PEG-asparaginase activity in acute lymphoblastic leukemia patients [29]. The increasing exposure to PEG in therapeutics may also lead to increasing overall APA levels in the human population over time; for instance, a small study of patients receiving the COVID vaccines revealed that the PEG-containing mRNA-1273 vaccine increased plasma anti-PEG IgM and IgG antibody concentrations 68.5- and 13.1-fold, respectively [30]. More research is required to fully understand the impact of repeated exposure to PEG-containing products on the safety and efficacy of these therapeutics.
PEG is not biodegradable, resulting in an upper limit on the half-life extension achievable and potential concerns over its ability to accumulate in the body. PEGs up to 40 kDa have been used in the clinic, which is close to the renal filtration limit for PEG (roughly 50 kDa) [31]. PEGs in this size range also accumulate in the form of vacuoles in cells, but vacuolization has not been associated with any safety concerns in currently marketed therapeutics [32]. Finally, PEG is synthesized by ring-opening polymerization of ethylene oxide. The resulting product is polydisperse, which increases analytical complexity in both the PEG intermediate and the resulting conjugate. In addition, commercial PEGs are only functionalized at their termini, limiting their drug-loading capacity. These challenges have paved the way for the development of next-generation polymer chemistries (Figure 1A) and architectures (Figure 1B).
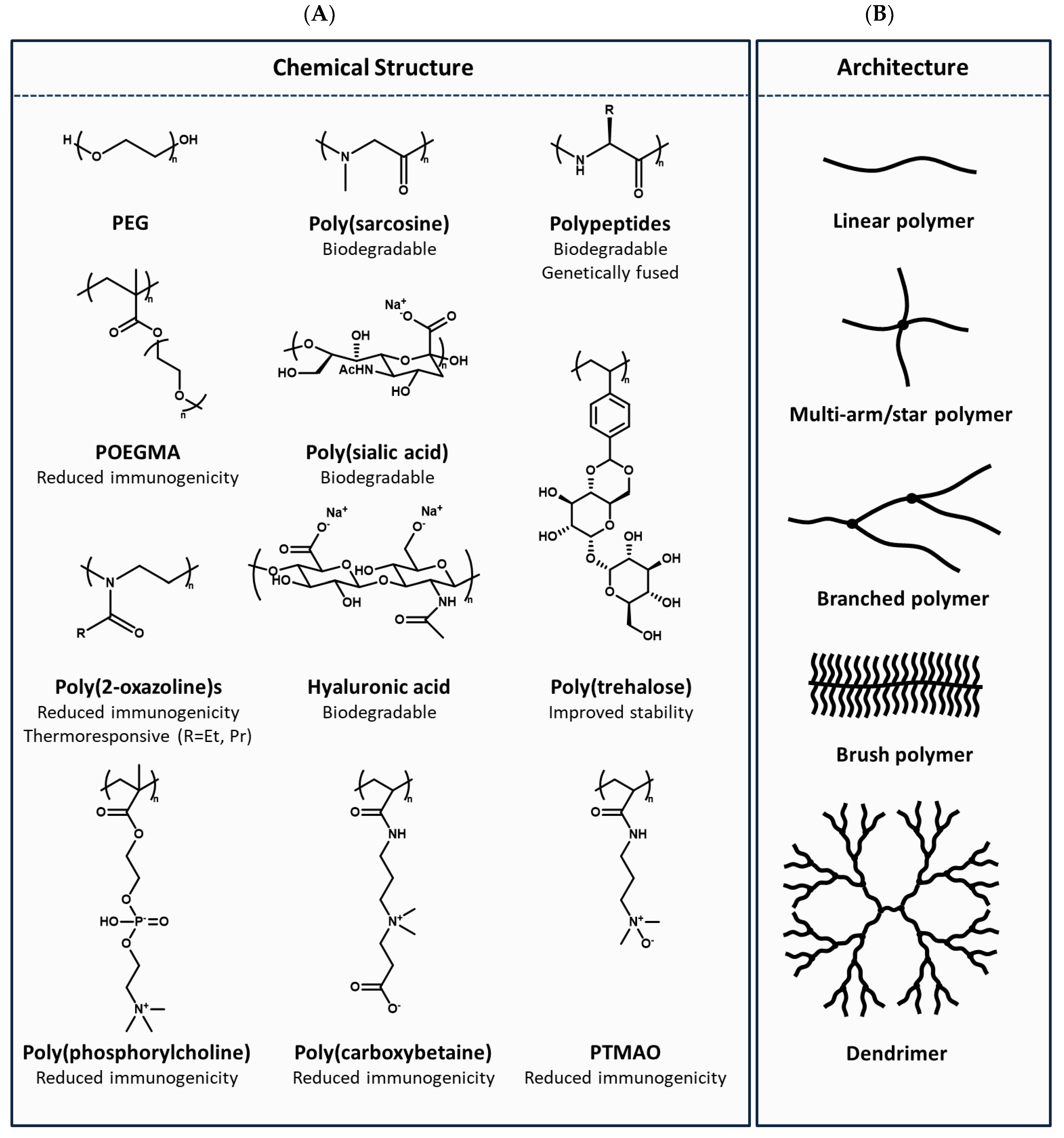
Figure 1. Overview of polymer (A) chemical structures and (B) architectures.
2.2.2. Next-Generation PEG Derivatives
PEG derivatives with modified architectures have been proposed as potential alternatives to linear PEGs. For instance, the use of multiarm PEGs allows multiple APIs to be loaded onto a single polymer. Branched architectures may have more favorable PK properties as well. In one example, comb-shaped PEG polymers (“PolyPEG”) were prepared by grafting pendant PEG chains onto a polymethacrylate backbone. Despite their smaller hydrodynamic size, PolyPEG-IFNa conjugates had longer serum half-lives in rats; this effect was attributed to the modified architecture, which may allow the conjugate to further resist renal filtration or proteolytic degradation. In addition, PolyPEG conjugates were less viscous than the corresponding linear PEG conjugates [33].
Qi et al. extended this work to bottlebrush PEGs, a subclass of comb-shaped polymers characterized by very high graft densities [34] (Figure 1B). Dense bottlebrush architectures have been reported to be nonfouling, e.g., resistant to both cell and protein adsorption, which may enable them to evade APA recognition. Poly(oligo(ethylene glycol) methyl ether methacrylate) (POEGMA) bottlebrush polymer conjugates reduced APA-related immunogenicity and accelerated clearance; compared with two commercial PEG conjugates, Adagen and Krystexxa, POEGMA-exendin conjugates exhibited significantly lower binding to APAs in human plasma. Based on previous reports that the minimum antigenic PEG length is 6–7 ethylene glycol repeats, these authors further demonstrated that decreasing the average ethylene glycol side chain length to three eliminated APA reactivity entirely. These findings were then extended by Ozer et al. to include POEGMA conjugates with highly immunogenic uricase as a model protein. Administration of POEGMA–uricase conjugates eliminated both the accelerated blood clearance and the development of ADAs observed in groups treated with PEGylated uricase [35][36].
2.2.3. Poly(2-oxazoline)s
Poly(2-oxazoline) (POZ) polymers represent promising alternatives to PEG, with reported benefits including low immunogenicity, low viscosity, high drug-loading capability, and oxidative stability. POZ polymers were shown to be significantly less viscous than PEG solutions at the same molecular weight with only a slight reduction in hydrodynamic size [37]. Lower drug product viscosity results in lower pain during injection for patients and enables the use of smaller diameter needles for administration [38]. POZ conjugates were successfully prepared with a variety of proteins, including BSA, insulin, and uricase, and the loss in bioactivity upon polymer conjugation was similar to the corresponding PEG conjugates. Additionally, repeat administration of POZ–BSA conjugates generated lower anti-BSA antibody titers in rabbits when compared with the PEG–BSA group, suggesting that POZ conjugates were more effective than PEG at shielding BSA immunogenicity [37].
POZ polymers can be synthesized with diverse properties; for example, POZs exhibit thermoresponsive behavior that is tunable by altering the monomer hydrophobicity. Poly(2-ethyl-2-oxazoline) has a cloud point temperature (Tcp) of 61–69 °C, while the more hydrophobic poly(2-isopropyl-2-oxazoline) has a Tcp between 26–34 °C, and hydrophilic poly(2-methyl-2-oxazoline) is not thermoresponsive at any temperature [39]. In addition, conjugation handles can be incorporated directly into the polymer backbone during synthesis, allowing multiple drugs to be loaded onto a single polymer. Serina Therapeutics is in Phase 1 clinical trials with a POZ–rotigotine conjugate (SER-214), but to date, no large molecule POZ conjugates have entered the clinic.
2.2.4. Zwitterionic Polymers
Zwitterionic polymers are gaining attention as alternatives to PEG for their ability to stabilize proteins against denaturation and evade immune recognition while maintaining extended circulation in vivo. These polymers contain an equal number of positive and negative charges to form net-neutral polymers that interact strongly with water, and their high degree of solvation is reported to impart ultra-low fouling properties [40]. Examples of zwitterionic polymers used to prepare polymer–protein conjugates include poly(carboxybetaine) (pCB), poly(phosphorylcholine) (pPC) [41], poly(sulfobetaine) (pSB), and poly(trimethylamine N-oxide) (pTMAO). KSI-301, a conjugate between an 800 kDa branched phosphorylcholine polymer and an anti-VEGF IgG, represents the most advanced zwitterionic polymer–protein conjugate in the clinic.
In the preclinical space, pCB conjugates have been prepared for a variety of therapeutically relevant peptides and proteins, including uricase, insulin, glucagon-like peptide-1, and IFN-α2a [42][43][44][45]. The pCB-IFN-α2a conjugates mitigated the accelerated blood clearance and partially restored the activity loss observed in the PEGylated counterpart, and pCB conjugation was also shown to protect ⍺-chymotrypsin from thermal and chemical denaturation. More recently, pTMAO conjugates have been reported as potential next-generation zwitterionic polymers. Similar to pCB, pTMAO conjugates have been shown to generate significantly lower anticonjugate antibody titers in vivo and eliminate accelerated blood clearance observed in the PEGylated controls. For example, pTMAO–uricase conjugates maintained extended serum half-life and sustained in vivo efficacy after three consecutive injections in mice. In contrast, PEG–uricase-treated groups exhibited lower serum half-lives and reduced efficacy after repeat injections, consistent with the loss of efficacy observed in the clinic for pegloticase. pTMAO conjugation was also shown to be superior to PEG in stabilizing uricase against thermal treatment at 50–70 °C and after incubation with urea.
Molecular dynamics simulations revealed that, relative to PEG, pTMAO accepted more hydrogen bonds from water per monomer, each hydrogen bond had a longer lifetime on average, and water formed a contiguous hydration shell around the polymer. All of these attributes likely contribute to the ultra-low fouling characteristics in this new class of polymer conjugates [46]. Nevertheless, additional work is required to understand the specific design features that enable zwitterionic polymers to be nonfouling. For example, the Leckband group recently found evidence that linear pSB polymers interact with certain proteins, leading to decreases in Tm, while densely packed brush pSB polymers resist protein adsorption [47][48].
2.2.5. Amino-Acid-Based Polymers
One of the most elegant approaches to reduce the CMC complexity of polymer–protein conjugates is the genetic fusion of unstructured polypeptides to the N- or C-terminus of the protein. A major advantage of this approach is that it allows for direct expression of the conjugate, substantially reducing the manufacturing complexity by eliminating several conjugation process steps. Notably, this strategy also enables the production of monodisperse, chemically defined conjugates, which simplifies analytical characterization. Finally, polypeptide fusion proteins are fully biodegradable, mitigating concerns about accumulation in vivo.
Examples of these polymers include XTEN (unstructured, hydrophilic, biodegradable protein polymers), proline/alanine-rich sequences (PAS), and elastin-like polypeptides (ELP). XTEN represents the most advanced format, with multiple programs currently undergoing clinical trials. XTEN polypeptides are constructed from hydrophilic amino acid building blocks (A, E, G, P, S, and T), and they are designed to have high chemical and physical stability, a lack of secondary structure, and high solubility and expression yields [49]. XTEN polypeptides exhibit properties of random coil polymers, allowing them to adopt larger hydrodynamic radii for a given molecular weight relative to globular protein. As a result, XTENylation achieves robust half-life extension for a series of peptides and proteins, with improvements in half-life ranging from 13–125-fold over their unmodified counterparts [50]. Similarly, PAS polypeptides are composed of P, A, and S amino acids and form hydrophilic, disordered polymers with no secondary structure. A PASylated anti-TNF-α Fab’ achieved similar half-life extension to the marketed PEGylated version, certolizumab pegol, while mitigating the antigen-binding loss observed in the PEG-conjugated antibody [51]. These disordered polypeptides can also be used as multivalent polymer scaffolds via the incorporation of reactive amino acids such as cysteine into the sequence, enabling the development of multivalent arrays with precisely defined ligand spacing [52] and high-DAR antibody–drug conjugates [53].
Inspired by tropoelastin, ELPs are polypeptide chains composed of VPGXG repeats, where “X” can be any amino acid except proline. ELPs are unique in their ability to undergo a temperature-dependent phase transition from a random coil structure to globular aggregates; when designed appropriately, this thermoresponsive behavior can be programmed to promote self-assembly and depot formation upon injection [54]. ELP fusions have been used to attain zero-order release of GLP1 over 14 days in nonhuman primates, a sevenfold increase relative to Trulicity, a marketed long-acting GLP1 therapy [55]. PB1046, an ELP fusion to vasoactive intestinal peptide, reached Phase 2 clinical trials before the program was voluntarily terminated due to COVID-19-related enrollment and manufacturing challenges [56].
2.2.6. Dendrimers
Dendrimers represent a unique class of nearly monodisperse polymers, with compact structures, a high density of functional groups, and a large range of accessible surface chemistries. Dendrimers are primarily synthesized using the divergent approach, in which branched monomers are iteratively installed from a central core to form successive generations with exponentially increased branching (Figure 1B). The result is a dense and highly branched polymer with a high drug-loading capacity and physical properties that are largely governed by the identity of the terminal branches [57][58]. The use of a hydrophobic core enables noncovalent encapsulation of hydrophobic drugs, while surface functionalization enables targeting or multivalent display. Commonly used chemistries include poly(amidoamine) (PAMAM) [59], poly-2,2-bis(hydroxymethyl)propionic acid (bis-MPA) [60], and poly(L-lysine) dendrimers [61], but monodisperse, degradable PEG dendrimers have been recently reported as well [62][63]. Starpharma has several small molecule candidates in the clinic using their poly(L-lysine) dendrimer platform, but there are no protein or peptide dendrimer conjugates in the clinic to date.
2.2.7. Biodegradable Polymers
A range of biodegradable polymers are being explored for the production of polymer–protein conjugates, including polysialic acid (PSA) [64][65], hyaluronic acid (HA) [66][67], and polysarcosine (pSar) [68]. Biodegradable polymers may address concerns over the potential accumulation of nondegradable polymers such as PEG in vivo, and they allow access to much larger size ranges while ensuring that these compounds can still be metabolized. HA and PSA are anionic biopolymers, while pSAR is a nonionic, hydrophilic polymer.
Polysialic acid is a biodegradable polymer of sialic acid, a component of cell membranes and glycoproteins. A PSA-rhFVIII recently completed Phase 1 clinical trials but was discontinued for lack of efficacy [69]. HA is an endogeneous polysaccharide that is degraded via hyaluronidases present in many tissues, including in the liver and kidneys. This semirigid polymer has a long estimated persistence length of 4 nm [70], giving it a significantly larger hydrodynamic size for a given molecular weight when compared with more flexible polymers such as PEG. HA is also internalized into cells through binding to CD44; this property has been exploited for delivery to CD44-overexpressing tumor cells [71][72].
Polysarcosine is a polypeptoid derived from sarcosine, an endogenous, noncoded amino acid. The polymer’s solution properties, including its solubility, hydrodynamic size, and interactions with proteins, are similar to those of PEG [73][74], but pSar-IFN-α conjugates exhibited lower antidrug antibody (ADA) levels and better antitumor efficacy than the PEGylated comparators after multiple administrations in mice [75]. Monodisperse, short pSAR chains have also been used to improve the physicochemical properties of ADCs [76].
2.2.8. Trehalose-Based Glycopolymers
Proteins are susceptible to aggregation in aqueous formulations, and this physical instability often limits their shelf lives and storage temperatures. As many protein aggregates have been reported to be immunogenic [77], the levels of aggregates must be tightly controlled during the manufacturing and long-term storage of the drug product. The majority of therapeutic proteins must be stored frozen or refrigerated to maintain their physicochemical stability; these cold-chain requirements increase the cost and complexity of the supply chain and preclude global access to these therapeutics. Thus, strategies that permit the long-term storage of therapeutic proteins at room temperature remain highly desirable.
Inspired by commonly-used excipients such as trehalose and sucrose, the Maynard group demonstrated the ability of trehalose-based glycopolymers to protect proteins against environmental stresses such as thermal and agitation stress. Conjugation of the trehalose polymer stabilized lysozyme against successive lyophilization cycles, while conjugation to insulin stabilized the protein against agitation stress [78][79]. These properties may permit the removal of agitation-stabilizing surfactants such as polysorbates in poly(trehalose)-conjugated protein and peptide formulations, which are prone to instability and particle formation in aqueous formulations [80]. Similarly, lyophilized formulations may have greater flexibility to remove osmolality-increasing excipients such as monomeric trehalose or sucrose, which are included to protect against freezing and desiccation stresses during lyophilization. Multiple poly(trehalose) conjugates also showed improved stability during thermal stress at high temperatures, including 90 °C stress for lysozyme and insulin as well as 75 °C stress for Herceptin and its Fab fragment [81]. While these results are promising, no studies have evaluated the impact of polymer conjugation on protein stability at relevant storage temperatures, such as room temperature or 2–8 °C, where thermal unfolding and Tm are often poor predictors of stability [82]. This dataset will be needed to understand the broader potential of polymers such as poly(trehalose) to improve drug product shelf life.
2.3. Advances in Conjugation Chemistries
Advances in conjugation chemistries for the synthesis of polymer–protein conjugates are extensively covered in several excellent reviews [83][84][85][86] and will only be reviewed briefly here. Conjugation to peptides is relatively straightforward, as solid-phase peptide synthesis allows for the facile incorporation of functional handles into the peptide sequence.
2.3.1. Grafting To
All commercial polymer–protein conjugates follow the “grafting to” approach to produce the final drug product (DP), in which the polymer is synthesized and functionalized prior to conjugation to the protein. This approach enables the use of mild, protein-compatible reaction conditions, but it often requires large molar excesses of polymer to drive conversion and the development of subsequent purification steps to remove the residual unreacted polymer. First-generation protein–polymer conjugates were prepared using nonspecific conjugation to lysine amino groups within the protein via PEGs terminated with activated esters such as NHS esters. This strategy typically generated heterogeneous conjugates with diminished activity due to the lack of control over the conjugation site [87].
Second-generation conjugates used more targeted, site-specific conjugation strategies to reduce heterogeneity and minimize activity loss. Rebinyn utilizes enzymatic conjugation with sialidase to conjugate a 40 kDa PEG to one of the two N-linked glycans in recombinant human factor IX [88]. Other conjugates were prepared through selective conjugation to the α-amino group on the N-terminus, which often has higher reactivity relative to lysine side chains due to its lower pKa. For example, pegfilgrastim is selectively PEGylated at the α-amino group on the N-terminus via reductive alkylation with PEG-aldehyde [89]. However, in many instances, including in mAbs, the N-terminus of the protein is in close proximity to the binding site. An alternative strategy to minimize the loss of binding in these molecules involves the introduction of engineered cysteines into the antibody sequence for conjugation to maleimides, pyridyl disulfides, or vinyl sulfones. Cimzia, a PEGylated Fab, incorporates an engineered cysteine at the C-terminus of the protein for site-specific conjugation to a maleimide-functionalized PEG via Michael addition [90].
Despite its prevalence in clinical and commercial conjugates, thiol–maleimide chemistry suffers from several CMC challenges, including gradual deconjugation and the potential for disulfide scrambling during the conjugation process. These shortcomings have motivated the development of next-generation, site-specific conjugation chemistries, which range from the use of noncanonical amino acids to enzymatic ligation onto specific recognition sequences engineered into the protein. Popular enzymatic conjugation methods include transglutaminase and sortase A. Transglutaminase catalyzes the formation of a stable isopeptide bond between a primary amine and a glutamine-containing sequence in the protein, while sortase A catalyzes the formation of an amide bond between a LPXTG sequence in the protein and an N-terminal oligoglycine [91][92][93][94]. While enzymatic approaches have shown promise for site-specific modification of proteins, the need to source an additional protein as an intermediate and subsequently purify it from the reaction mixture adds CMC complexity to the bioconjugation process.
The incorporation of noncanonical amino acids into the protein sequence significantly expands the repertoire of accessible conjugation chemistries. Since the pioneering work in this space by the Schultz lab, hundreds of non-natural amino acids have been incorporated into proteins during expression, enabling site-specific conjugation with a variety of biorthogonal chemistries [95][96]. In particular, noncanonical amino acids facilitate the use of click chemistries such as strain-promoted click chemistry; these bioconjugation reactions are advantageous as they typically proceed with rapid kinetics, high yield, and under mild conditions. SAR444245 is currently in Phase 2 clinical trials and uses a non-natural N6-(2-azidoethoxy)-carbonyl-L-lysine amino acid for site-selective conjugation to dibenzocyclooctyne-functionalized PEG. While the methods for incorporating non-natural amino acids into protein sequences, readers are referred to several recent reviews on this topic [97][98].
2.3.2. Grafting From
While all commercial polymer–protein conjugates utilize the “grafting to” approach for conjugation, “grafting from” has recently emerged as an alternative to reduce purification process complexity and improve overall yields. This method uses protein-compatible controlled radical polymerization techniques, most commonly atom-transfer radical polymerization (ATRP) [99][100] or reversible addition-fragmentation chain transfer polymerization (RAFT) [101], to polymerize directly from the protein after the introduction of an initiator or chain transfer agent onto the protein. Because it uses small molecule initiators and monomers, the “grafting to” method can overcome steric barriers that may otherwise prevent the conjugation of a polymer to a protein, allowing for conjugation to sites with lower solvent exposure or denser packing of polymer chains near the protein surface. These monomers can also be readily separated from the protein via high-throughput purification techniques such as tangential flow filtration (TFF) [102].
Importantly, the “grafting from” strategy still requires modification of the protein with a reactive handle for polymerization; thus, the identification of site-selective conjugation chemistries remains vital to the successful development of polymer–protein conjugates. In addition, polymerization conditions must be carefully optimized to be compatible with proteins, which require the use of aqueous solvents and low temperatures; as a result, achieving a balance between mild polymerization conditions and low polymer dispersity is often challenging. Finally, ATRP uses transition metal catalysts such as copper, which is a potential concern as metal ions can bind to proteins and trigger chemical degradation, including oxidation and fragmentation [103][104].
References
- Abuchowski, A.; McCoy, J.R.; Palczuk, N.C.; van Es, T.; Davis, F.F. Effect of Covalent Attachment of Polyethylene Glycol on Immunogenicity and Circulating Life of Bovine Liver Catalase. J. Biol. Chem. 1977, 252, 3582–3586.
- Abuchowski, A.; van Es, T.; Palczuk, N.C.; Davis, F.F. Alteration of Immunological Properties of Bovine Serum Albumin by Covalent Attachment of Polyethylene Glycol. J. Biol. Chem. 1977, 252, 3578–3581.
- Savoca, M.P.; Tonoli, E.; Atobatele, A.G.; Verderio, E.A.M. Biocatalysis by Transglutaminases: A Review of Biotechnological Applications. Micromachines 2018, 9, 562.
- Davis, F.F. The Origin of Pegnology. Adv. Drug Deliv. Rev. 2002, 54, 457–458.
- FDA Approved PEGylated Drugs Up To 2022. Available online: https://www.biochempeg.com/article/58.html (accessed on 12 December 2022).
- Mathe, G.; Ba, L.O.C.T.; Bernard, J. Effect on Mouse Leukemia 1210 of a Combination by Diazo-Reaction of Amethopterin and Gamma-Globulins from Hamsters Inoculated with Such Leukemia by Heterografts. Comptes Rendus Hebd. Des Seances De L’academie Des. Sci. 1958, 246, 1626–1628.
- Berinstein, N.L.; Spaner, D. Therapeutic Cancer Vaccines. In Vaccines (Fifth Edition); Plotkin, S.A., Orenstein, W.A., Offit, P.A., Eds.; Saunders Elsevier: Philadelphia, Pennsylvania, 2008; pp. 1135–1145. ISBN 9781416036111.
- Molineux, G. The Design and Development of Pegfilgrastim (PEG-RmetHuG-CSF, Neulasta). Curr. Pharm. Des. 2004, 10, 1235–1244.
- Kourlaba, G.; Dimopoulos, M.A.; Pectasides, D.; Skarlos, D.V.; Gogas, H.; Pentheroudakis, G.; Koutras, A.; Fountzilas, G.; Maniadakis, N. Comparison of Filgrastim and Pegfilgrastim to Prevent Neutropenia and Maintain Dose Intensity of Adjuvant Chemotherapy in Patients with Breast Cancer. Support Care Cancer 2015, 23, 2045–2051.
- Staton, T. The Top 10 Patent Losses of 2015. Available online: https://www.fiercepharma.com/special-report/top-10-patent-losses-of-2015 (accessed on 15 December 2022).
- Wang, Y.-S.; Youngster, S.; Grace, M.; Bausch, J.; Bordens, R.; Wyss, D.F. Structural and Biological Characterization of Pegylated Recombinant Interferon Alpha-2b and Its Therapeutic Implications. Adv. Drug Deliver. Rev. 2002, 54, 547–570.
- Kurre, H.A.; Ettinger, A.G.; Veenstra, D.L.; Gaynon, P.S.; Franklin, J.; Sencer, S.F.; Reaman, G.H.; Lange, B.J.; Holcenberg, J.S. A Pharmacoeconomic Analysis of Pegaspargase Versus Native Escherichia Coli L-Asparaginase for the Treatment of Children with Standard-Risk, Acute Lymphoblastic Leukemia: The Children’s Cancer Group Study (CCG-1962). J. Pediatr. Hematol. Oncol. 2002, 24, 175–181.
- Grevys, A.; Frick, R.; Mester, S.; Flem-Karlsen, K.; Nilsen, J.; Foss, S.; Sand, K.M.K.; Emrich, T.; Fischer, J.A.A.; Greiff, V.; et al. Antibody Variable Sequences Have a Pronounced Effect on Cellular Transport and Plasma Half-Life. Iscience 2022, 25, 103746.
- Strohl, W.R. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. Biodrugs 2015, 29, 215–239.
- Claassen, J.L.; Kobayashi, R.H.; Kobayashi, A.L.; Hershfield, M.S.; Schiff, R.I.; Buckley, R.H. 276 Antigen-Specific Humoral and Cellular Immune Responses during Treatment of Adenosine Deaminase Deficient Severe Combined Immunodeficiency (ADA-SCID) with Polyethylene Glycol-Modified Bovine Adenosine Deaminase (PEG-ADA). J. Allergy Clin. Immun. 1988, 81, 237.
- Davis, S.; Abuchowski, A.; Park, Y.K.; Davis, F.F. Alteration of the Circulating Life and Antigenic Properties of Bovine Adenosine Deaminase in Mice by Attachment of Polyethylene Glycol. Clin. Exp. Immunol. 1981, 46, 649–652.
- Basu, A.; Yang, K.; Wang, M.; Liu, S.; Chintala, R.; Palm, T.; Zhao, H.; Peng, P.; Wu, D.; Zhang, Z.; et al. Structure−Function Engineering of Interferon-β-1b for Improving Stability, Solubility, Potency, Immunogenicity, and Pharmacokinetic Properties by Site-Selective Mono-PEGylation. Bioconjugate Chem. 2006, 17, 618–630.
- Katre, N.V.; Knauf, M.J.; Laird, W.J. Chemical Modification of Recombinant Interleukin 2 by Polyethylene Glycol Increases Its Potency in the Murine Meth A Sarcoma Model. Proc. Natl. Acad. Sci. USA 1987, 84, 1487–1491.
- Rajan, R.S.; Li, T.; Aras, M.; Sloey, C.; Sutherland, W.; Arai, H.; Briddell, R.; Kinstler, O.; Lueras, A.M.K.; Zhang, Y.; et al. Modulation of Protein Aggregation by Polyethylene Glycol Conjugation: GCSF as a Case Study. Protein Sci. 2006, 15, 1063–1075.
- Roque, C.; Sheung, A.; Rahman, N.; Ausar, S.F. Effect of Polyethylene Glycol Conjugation on Conformational and Colloidal Stability of a Monoclonal Antibody Antigen-Binding Fragment (Fab′). Mol. Pharmaceut. 2015, 12, 562–575.
- Rodríguez-Martínez, J.A.; Solá, R.J.; Castillo, B.; Cintrón-Colón, H.R.; Rivera-Rivera, I.; Barletta, G.; Griebenow, K. Stabilization of A-chymotrypsin upon PEGylation Correlates with Reduced Structural Dynamics. Biotechnol. Bioeng. 2008, 101, 1142–1149.
- Nie, Y.; Zhang, X.; Wang, X.; Chen, J. Preparation and Stability of N-Terminal Mono-PEGylated Recombinant Human Endostatin. Bioconjugate Chem. 2006, 17, 995–999.
- Narasimhan, D.; Collins, G.T.; Nance, M.R.; Nichols, J.; Edwald, E.; Chan, J.; Ko, M.-C.; Woods, J.H.; Tesmer, J.J.G.; Sunahara, R.K. Subunit Stabilization and Polyethylene Glycolation of Cocaine Esterase Improves In Vivo Residence Time. Mol. Pharm. 2011, 80, 1056–1065.
- Grigoletto, A.; Mero, A.; Zanusso, I.; Schiavon, O.; Pasut, G. Chemical and Enzymatic Site Specific PEGylation of HGH: The Stability and in Vivo Activity of PEG-N-Terminal-hGH and PEG-Gln141-hGH Conjugates. Macromol. Biosci. 2016, 16, 50–56.
- Natalello, A.; Ami, D.; Collini, M.; D’Alfonso, L.; Chirico, G.; Tonon, G.; Scaramuzza, S.; Schrepfer, R.; Doglia, S.M. Biophysical Characterization of Met-G-CSF: Effects of Different Site-Specific Mono-Pegylations on Protein Stability and Aggregation. PLoS ONE 2012, 7, e42511.
- Chen, B.-M.; Cheng, T.-L.; Roffler, S.R. Polyethylene Glycol Immunogenicity: Theoretical, Clinical, and Practical Aspects of Anti-Polyethylene Glycol Antibodies. ACS Nano 2021, 15, 14022–14048.
- Yang, Q.; Jacobs, T.M.; McCallen, J.D.; Moore, D.T.; Huckaby, J.T.; Edelstein, J.N.; Lai, S.K. Analysis of Pre-Existing IgG and IgM Antibodies against Polyethylene Glycol (PEG) in the General Population. Anal. Chem. 2016, 88, 11804–11812.
- Lipsky, P.E.; Calabrese, L.H.; Kavanaugh, A.; Sundy, J.S.; Wright, D.; Wolfson, M.; Becker, M.A. Pegloticase Immunogenicity: The Relationship between Efficacy and Antibody Development in Patients Treated for Refractory Chronic Gout. Arthritis Res. 2014, 16, R60.
- Armstrong, J.K.; Hempel, G.; Koling, S.; Chan, L.S.; Fisher, T.; Meiselman, H.J.; Garratty, G. Antibody against Poly(Ethylene Glycol) Adversely Affects PEG-asparaginase Therapy in Acute Lymphoblastic Leukemia Patients. Cancer 2007, 110, 103–111.
- Ju, Y.; Lee, W.S.; Pilkington, E.H.; Kelly, H.G.; Li, S.; Selva, K.J.; Wragg, K.M.; Subbarao, K.; Nguyen, T.H.O.; Rowntree, L.C.; et al. Anti-PEG Antibodies Boosted in Humans by SARS-CoV-2 Lipid Nanoparticle MRNA Vaccine. ACS Nano 2022, 16, 11769–11780.
- Yamaoka, T.; Tabata, Y.; Ikada, Y. Distribution and Tissue Uptake of Poly(Ethylene Glycol) with Different Molecular Weights after Intravenous Administration to Mice. J. Pharm. Sci. 1994, 83, 601–606.
- Baumann, A.; Tuerck, D.; Prabhu, S.; Dickmann, L.; Sims, J. Pharmacokinetics, Metabolism and Distribution of PEGs and PEGylated Proteins: Quo Vadis? Drug Discov. Today 2014, 19, 1623–1631.
- Podobnik, B.; Helk, B.; Smilović, V.; Škrajnar, S.; Fidler, K.; Jevševar, S.; Godwin, A.; Williams, P. Conjugation of PolyPEG to Interferon Alpha Extends Serum Half-Life While Maintaining Low Viscosity of the Conjugate. Bioconjugate Chem. 2015, 26, 452–459.
- Qi, Y.; Simakova, A.; Ganson, N.J.; Li, X.; Luginbuhl, K.M.; Ozer, I.; Liu, W.; Hershfield, M.S.; Matyjaszewski, K.; Chilkoti, A. A Brush-Polymer/Exendin-4 Conjugate Reduces Blood Glucose Levels for up to Five Days and Eliminates Poly(Ethylene Glycol) Antigenicity. Nat. Biomed. Eng. 2016, 1, 2.
- Ozer, I.; Kelly, G.; Gu, R.; Li, X.; Zakharov, N.; Sirohi, P.; Nair, S.K.; Collier, J.H.; Hershfield, M.S.; Hucknall, A.M.; et al. Polyethylene Glycol-Like Brush Polymer Conjugate of a Protein Drug Does Not Induce an Antipolymer Immune Response and Has Enhanced Pharmacokinetics than Its Polyethylene Glycol Counterpart. Adv. Sci. 2022, 9, 2103672.
- Yoshikawa, C.; Qiu, J.; Huang, C.-F.; Shimizu, Y.; Suzuki, J.; Bosch, E. van den Non-Biofouling Property of Well-Defined Concentrated Polymer Brushes. Colloids Surf. B Biointerfaces 2015, 127, 213–220.
- Viegas, T.X.; Bentley, M.D.; Harris, J.M.; Fang, Z.; Yoon, K.; Dizman, B.; Weimer, R.; Mero, A.; Pasut, G.; Veronese, F.M. Polyoxazoline: Chemistry, Properties, and Applications in Drug Delivery. Bioconjugate Chem. 2011, 22, 976–986.
- Berteau, C.; Filipe-Santos, O.; Wang, T.; Rojas, H.E.; Granger, C.; Schwarzenbach, F. Evaluation of the Impact of Viscosity, Injection Volume, and Injection Flow Rate on Subcutaneous Injection Tolerance. Med. Devices Auckl. N. Z. 2015, 8, 473–484.
- Hoogenboom, R.; Schlaad, H. Thermoresponsive Poly(2-Oxazoline)s, Polypeptoids, and Polypeptides. Polym. Chem. 2016, 8, 24–40.
- Ladd, J.; Zhang, Z.; Chen, S.; Hower, J.C.; Jiang, S. Zwitterionic Polymers Exhibiting High Resistance to Nonspecific Protein Adsorption from Human Serum and Plasma. Biomacromolecules 2008, 9, 1357–1361.
- Lewis, A.; Tang, Y.; Brocchini, S.; Choi, J.; Godwin, A. Poly(2-Methacryloyloxyethyl Phosphorylcholine) for Protein Conjugation. Bioconjugate Chem. 2008, 19, 2144–2155.
- Han, Y.; Yuan, Z.; Zhang, P.; Jiang, S. Zwitterlation Mitigates Protein Bioactivity Loss in Vitro over PEGylation. Chem. Sci. 2018, 9, 8561–8566.
- Liu, S.; Jiang, S. Zwitterionic Polymer-Protein Conjugates Reduce Polymer-Specific Antibody Response. Nano Today 2016, 11, 285–291.
- Tsao, C.; Zhang, P.; Yuan, Z.; Dong, D.; Wu, K.; Niu, L.; McMullen, P.; Luozhong, S.; Hung, H.-C.; Cheng, Y.-H.; et al. Zwitterionic Polymer Conjugated Glucagon-like Peptide-1 for Prolonged Glycemic Control. Bioconjugate Chem. 2020, 31, 1812–1819.
- Xie, J.; Lu, Y.; Wang, W.; Zhu, H.; Wang, Z.; Cao, Z. Simple Protein Modification Using Zwitterionic Polymer to Mitigate the Bioactivity Loss of Conjugated Insulin. Adv. Health Mater. 2017, 6, 1601428.
- Li, B.; Jain, P.; Ma, J.; Smith, J.K.; Yuan, Z.; Hung, H.-C.; He, Y.; Lin, X.; Wu, K.; Pfaendtner, J.; et al. Trimethylamine N-Oxide–Derived Zwitterionic Polymers: A New Class of Ultralow Fouling Bioinspired Materials. Sci. Adv. 2019, 5, eaaw9562.
- Kisley, L.; Miller, K.A.; Davis, C.M.; Guin, D.; Murphy, E.A.; Gruebele, M.; Leckband, D.E. Soluble Zwitterionic Poly(Sulfobetaine) Destabilizes Proteins. Biomacromolecules 2018, 19, 3894–3901.
- Ahmed, S.T.; Leckband, D.E. Protein Adsorption on Grafted Zwitterionic Polymers Depends on Chain Density and Molecular Weight. Adv. Funct. Mater. 2020, 30, 2000757.
- Schellenberger, V.; Wang, C.; Geething, N.C.; Spink, B.J.; Campbell, A.; To, W.; Scholle, M.D.; Yin, Y.; Yao, Y.; Bogin, O.; et al. A Recombinant Polypeptide Extends the in Vivo Half-Life of Peptides and Proteins in a Tunable Manner. Nat. Biotechnol. 2009, 27, 1186–1190.
- Podust, V.N.; Balan, S.; Sim, B.-C.; Coyle, M.P.; Ernst, U.; Peters, R.T.; Schellenberger, V. Extension of in Vivo Half-Life of Biologically Active Molecules by XTEN Protein Polymers. J. Control. Release 2016, 240, 52–66.
- Mazaheri, S.; Talebkhan, Y.; Mahboudi, F.; Nematollahi, L.; Cohan, R.A.; Ardakani, E.M.; Bayat, E.; Sabzalinejad, M.; Sardari, S.; Torkashvand, F. Improvement of Certolizumab Fab′ Properties by PASylation Technology. Sci. Rep. 2020, 10, 18464.
- Ding, S.; Song, M.; Sim, B.-C.; Gu, C.; Podust, V.N.; Wang, C.-W.; McLaughlin, B.; Shah, T.P.; Lax, R.; Gast, R.; et al. Multivalent Antiviral XTEN–Peptide Conjugates with Long in Vivo Half-Life and Enhanced Solubility. Bioconjugate Chem. 2014, 25, 1351–1359.
- Zacharias, N.; Podust, V.N.; Kajihara, K.K.; Leipold, D.; Rosario, G.D.; Thayer, D.; Dong, E.; Paluch, M.; Fischer, D.; Zheng, K.; et al. A Homogeneous High-DAR Antibody–Drug Conjugate Platform Combining THIOMAB Antibodies and XTEN Polypeptides. Chem. Sci. 2022, 13, 3147–3160.
- Gilroy, C.A.; Roberts, S.; Chilkoti, A. Fusion of Fibroblast Growth Factor 21 to a Thermally Responsive Biopolymer Forms an Injectable Depot with Sustained Anti-Diabetic Action. J. Control. Release 2018, 277, 154–164.
- Luginbuhl, K.M.; Schaal, J.L.; Umstead, B.; Mastria, E.M.; Li, X.; Banskota, S.; Arnold, S.; Feinglos, M.; D’Alessio, D.; Chilkoti, A. One-Week Glucose Control via Zero-Order Release Kinetics from an Injectable Depot of Glucagon-like Peptide-1 Fused to a Thermosensitive Biopolymer. Nat. Biomed. Eng. 2017, 1, 78.
- Long-Term, Open Label Extension Study of Pemziviptadil (PB1046) in PAH Subjects Following Completion of Study PB1046-PT-CL-0004 (VIP Extend). Available online: https://www.clinicaltrials.gov/ct2/show/NCT03795428 (accessed on 12 December 2022).
- Tomalia, D.A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. A New Class of Polymers: Starburst-Dendritic Macromolecules. Polym. J. 1985, 17, 117–132.
- Abbasi, E.; Aval, S.F.; Akbarzadeh, A.; Milani, M.; Nasrabadi, H.T.; Joo, S.W.; Hanifehpour, Y.; Nejati-Koshki, K.; Pashaei-Asl, R. Dendrimers: Synthesis, Applications, and Properties. Nanoscale Res. Lett. 2014, 9, 247.
- Esfand, R.; Tomalia, D.A. Poly(Amidoamine) (PAMAM) Dendrimers: From Biomimicry to Drug Delivery and Biomedical Applications. Drug Discov. Today 2001, 6, 427–436.
- Feliu, N.; Walter, M.V.; Montañez, M.I.; Kunzmann, A.; Hult, A.; Nyström, A.; Malkoch, M.; Fadeel, B. Stability and Biocompatibility of a Library of Polyester Dendrimers in Comparison to Polyamidoamine Dendrimers. Biomaterials 2012, 33, 1970–1981.
- Boyd, B.J.; Kaminskas, L.M.; Karellas, P.; Krippner, G.; Lessene, R.; Porter, C.J.H. Cationic Poly-l-Lysine Dendrimers: Pharmacokinetics, Biodistribution, and Evidence for Metabolism and Bioresorption after Intravenous Administration to Rats. Mol. Pharmaceut 2006, 3, 614–627.
- Zhang, Y.; Mesa-Antunez, P.; Fortuin, L.; Andrén, O.C.J.; Malkoch, M. Degradable High Molecular Weight Monodisperse Dendritic Poly(Ethylene Glycols). Biomacromolecules 2020, 21, 4294–4301.
- Wang, J.; Li, B.; Qiu, L.; Qiao, X.; Yang, H. Dendrimer-Based Drug Delivery Systems: History, Challenges, and Latest Developments. J. Biol. Eng. 2022, 16, 18.
- Gregoriadis, G.; Fernandes, A.; Mital, M.; McCormack, B. Polysialic Acids: Potential in Improving the Stability and Pharmacokinetics of Proteins and Other Therapeutics. Cell Mol. Life Sci. Cmls 2000, 57, 1964–1969.
- Jain, S.; Hreczuk-Hirst, D.H.; McCormack, B.; Mital, M.; Epenetos, A.; Laing, P.; Gregoriadis, G. Polysialylated Insulin: Synthesis, Characterization and Biological Activity in Vivo. Biochimica Et Biophysica Acta Bba Gen. Subj. 2003, 1622, 42–49.
- Kong, J.-H.; Oh, E.J.; Chae, S.Y.; Lee, K.C.; Hahn, S.K. Long Acting Hyaluronate–Exendin 4 Conjugate for the Treatment of Type 2 Diabetes. Biomaterials 2010, 31, 4121–4128.
- Mero, A.; Pasqualin, M.; Campisi, M.; Renier, D.; Pasut, G. Conjugation of Hyaluronan to Proteins. Carbohyd. Polym. 2013, 92, 2163–2170.
- Birke, A.; Ling, J.; Barz, M. Polysarcosine-Containing Copolymers: Synthesis, Characterization, Self-Assembly, and Applications. Prog. Polym. Sci. 2018, 81, 163–208.
- Tiede, A.; Allen, G.; Bauer, A.; Chowdary, P.; Collins, P.; Goldstein, B.; Jiang, H.J.; Köck, K.; Takács, I.; Timofeeva, M.; et al. SHP656, a Polysialylated Recombinant Factor VIII (PSA-rFVIII): First-in-human Study Evaluating Safety, Tolerability and Pharmacokinetics in Patients with Severe Haemophilia A. Haemophilia 2020, 26, 47–55.
- Cleland, R.L. The Persistence Length of Hyaluronic Acid: An Estimate from Small-Angle X-Ray Scattering and Intrinsic Viscosity. Arch. Biochem. Biophys. 1977, 180, 57–68.
- Luo, Z.; Dai, Y.; Gao, H. Development and Application of Hyaluronic Acid in Tumor Targeting Drug Delivery. Acta Pharm. Sinica B 2019, 9, 1099–1112.
- Montagner, I.M.; Merlo, A.; Carpanese, D.; Pietà, A.D.; Mero, A.; Grigoletto, A.; Loregian, A.; Renier, D.; Campisi, M.; Zanovello, P.; et al. A Site-Selective Hyaluronan-Interferonα2a Conjugate for the Treatment of Ovarian Cancer. J. Control. Release 2016, 236, 79–89.
- Settanni, G.; Schäfer, T.; Muhl, C.; Barz, M.; Schmid, F. Poly-Sarcosine and Poly(Ethylene-Glycol) Interactions with Proteins Investigated Using Molecular Dynamics Simulations. Comput. Struct. Biotechnol. J. 2018, 16, 543–550.
- Weber, B.; Birke, A.; Fischer, K.; Schmidt, M.; Barz, M. Solution Properties of Polysarcosine: From Absolute and Relative Molar Mass Determinations to Complement Activation. Macromolecules 2018, 51, 2653–2661.
- Hu, Y.; Hou, Y.; Wang, H.; Lu, H. Polysarcosine as an Alternative to PEG for Therapeutic Protein Conjugation. Bioconjugate Chem. 2018, 29, 2232–2238.
- Conilh, L.; Fournet, G.; Fourmaux, E.; Murcia, A.; Matera, E.-L.; Joseph, B.; Dumontet, C.; Viricel, W. Exatecan Antibody Drug Conjugates Based on a Hydrophilic Polysarcosine Drug-Linker Platform. Pharmaceuticals 2021, 14, 247.
- Moussa, E.M.; Panchal, J.P.; Moorthy, B.S.; Blum, J.S.; Joubert, M.K.; Narhi, L.O.; Topp, E.M. Immunogenicity of Therapeutic Protein Aggregates. J. Pharm. Sci. 2016, 105, 417–430.
- Liu, Y.; Lee, J.; Mansfield, K.M.; Ko, J.H.; Sallam, S.; Wesdemiotis, C.; Maynard, H.D. Trehalose Glycopolymer Enhances Both Solution Stability and Pharmacokinetics of a Therapeutic Protein. Bioconjugate Chem. 2017, 28, 836–845.
- Mancini, R.J.; Lee, J.; Maynard, H.D. Trehalose Glycopolymers for Stabilization of Protein Conjugates to Environmental Stressors. J. Am. Chem. Soc. 2012, 134, 8474–8479.
- Tomlinson, A.; Demeule, B.; Lin, B.; Yadav, S. Polysorbate 20 Degradation in Biopharmaceutical Formulations: Quantification of Free Fatty Acids, Characterization of Particulates, and Insights into the Degradation Mechanism. Mol. Pharmaceut. 2015, 12, 3805–3815.
- Forsythe, N.L.; Maynard, H.D. Synthesis of Disulfide-Bridging Trehalose Polymers for Antibody and Fab Conjugation Using a Bis-Sulfone ATRP Initiator. Polym. Chem. 2021, 12, 1217–1223.
- Thiagarajan, G.; Semple, A.; James, J.K.; Cheung, J.K.; Shameem, M. A Comparison of Biophysical Characterization Techniques in Predicting Monoclonal Antibody Stability. Mabs 2016, 8, 1088–1097.
- Boutureira, O.; Bernardes, G.J.L. Advances in Chemical Protein Modification. Chem. Rev. 2015, 115, 2174–2195.
- Dozier, J.K.; Distefano, M.D. Site-Specific PEGylation of Therapeutic Proteins. Int. J. Mol. Sci. 2015, 16, 25831–25864.
- Messina, M.S.; Messina, K.M.M.; Bhattacharya, A.; Montgomery, H.R.; Maynard, H.D. Preparation of Biomolecule-Polymer Conjugates by Grafting-from Using ATRP, RAFT, or ROMP. Prog. Polym. Sci. 2020, 100, 101186.
- Sadiki, A.; Vaidya, S.R.; Abdollahi, M.; Bhardwaj, G.; Dolan, M.E.; Turna, H.; Arora, V.; Sanjeev, A.; Robinson, T.D.; Koid, A.; et al. Site-Specific Conjugation of Native Antibody. Antib 2020, 3, 271–284.
- Zalipsky, S. Chemistry of Polyethylene Glycol Conjugates with Biologically Active Molecules. Adv. Drug Deliver. Rev. 1995, 16, 157–182.
- Østergaard, H.; Bjelke, J.R.; Hansen, L.; Petersen, L.C.; Pedersen, A.A.; Elm, T.; Møller, F.; Hermit, M.B.; Holm, P.K.; Krogh, T.N.; et al. Prolonged Half-Life and Preserved Enzymatic Properties of Factor IX Selectively PEGylated on Native N-Glycans in the Activation Peptide. Blood 2011, 118, 2333–2341.
- Arvedson, T.; O’Kelly, J.; Yang, B.-B. Design Rationale and Development Approach for Pegfilgrastim as a Long-Acting Granulocyte Colony-Stimulating Factor. Biodrugs 2015, 29, 185–198.
- Pasut, G. Pegylation of Biological Molecules and Potential Benefits: Pharmacological Properties of Certolizumab Pegol. Biodrugs 2014, 28, 15–23.
- Mao, H.; Hart, S.A.; Schink, A.; Pollok, B.A. Sortase-Mediated Protein Ligation: A New Method for Protein Engineering. J. Am. Chem. Soc. 2004, 126, 2670–2671.
- Chen, L.; Cohen, J.; Song, X.; Zhao, A.; Ye, Z.; Feulner, C.J.; Doonan, P.; Somers, W.; Lin, L.; Chen, P.R. Improved Variants of SrtA for Site-Specific Conjugation on Antibodies and Proteins with High Efficiency. Sci. Rep. 2016, 6, 31899.
- Rachel, N.M.; Toulouse, J.L.; Pelletier, J.N. Transglutaminase-Catalyzed Bioconjugation Using One-Pot Metal-Free Bioorthogonal Chemistry. Bioconjugate Chem. 2017, 28, 2518–2523.
- Lin, C.-W.; Ting, A.Y. Transglutaminase-Catalyzed Site-Specific Conjugation of Small-Molecule Probes to Proteins in Vitro and on the Surface of Living Cells. J. Am. Chem. Soc. 2006, 128, 4542–4543.
- Park, H.-S.; Hohn, M.J.; Umehara, T.; Guo, L.-T.; Osborne, E.M.; Benner, J.; Noren, C.J.; Rinehart, J.; Söll, D. Expanding the Genetic Code of Escherichia Coli with Phosphoserine. Science 2011, 333, 1151–1154.
- de la Torre, D.; Chin, J.W. Reprogramming the Genetic Code. Nat. Rev. Genet. 2021, 22, 169–184.
- Adhikari, A.; Bhattarai, B.R.; Aryal, A.; Thapa, N.; KC, P.; Adhikari, A.; Maharjan, S.; Chanda, P.B.; Regmi, B.P.; Parajuli, N. Reprogramming Natural Proteins Using Unnatural Amino Acids. RSC Adv. 2021, 11, 38126–38145.
- Lang, K.; Chin, J.W. Cellular Incorporation of Unnatural Amino Acids and Bioorthogonal Labeling of Proteins. Chem. Rev. 2014, 114, 4764–4806.
- Bontempo, D.; Heredia, K.L.; Fish, B.A.; Maynard, H.D. Cysteine-Reactive Polymers Synthesized by Atom Transfer Radical Polymerization for Conjugation to Proteins. J. Am. Chem. Soc. 2004, 126, 15372–15373.
- Lele, B.S.; Murata, H.; Matyjaszewski, K.; Russell, A.J. Synthesis of Uniform Protein−Polymer Conjugates. Biomacromolecules 2005, 6, 3380–3387.
- Liu, J.; Bulmus, V.; Herlambang, D.L.; Barner-Kowollik, C.; Stenzel, M.H.; Davis, T.P. In Situ Formation of Protein–Polymer Conjugates through Reversible Addition Fragmentation Chain Transfer Polymerization. Angew. Chem. Int. Ed. 2007, 46, 3099–3103.
- Wang, C.; Zhang, C.; Li, Z.; Yin, S.; Wang, Q.; Guo, F.; Zhang, Y.; Yu, R.; Liu, Y.; Su, Z. Extending Half Life of H-Ferritin Nanoparticle by Fusing Albumin Binding Domain for Doxorubicin Encapsulation. Biomacromolecules 2018, 19, 773–781.
- Glover, Z.K.; Basa, L.; Moore, B.; Laurence, J.S.; Sreedhara, A. Metal Ion Interactions with MAbs: Part 1. Mabs 2015, 7, 901–911.
- Glover, Z.K.; Wecksler, A.; Aryal, B.; Mehta, S.; Pegues, M.; Chan, W.; Lehtimaki, M.; Luo, A.; Sreedhara, A.; Rao, V.A. Physicochemical and Biological Impact of Metal-Catalyzed Oxidation of IgG1 Monoclonal Antibodies and Antibody-Drug Conjugates via Reactive Oxygen Species. Mabs 2022, 14, 2122957.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
827
Revisions:
2 times
(View History)
Update Date:
12 May 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No