 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ian Fraser Pryme | -- | 3787 | 2023-05-05 10:52:42 |
Video Upload Options
Insulin is the main metabolic regulator of fuel molecules in the diet, such as carbohydrates, lipids, and proteins. It does so by facilitating glucose influx from the circulation into the liver, adipose tissue, and skeletal myocytes. The outcome of which is subjected to glycogenesis in skeletal muscle and lipogenesis in adipose tissue, as well as in the liver. Metabolic syndrome is the congregation of abdominal obesity (visceral obesity), hypertension, hyperglycemia, hyperlipidemia (triglycerides), and low serum high-density lipoprotein (HDL). At least three of these criteria must exist for the diagnosis of this syndrome. Insulin resistance is indeed a component of this syndrome. When hyperglycemia falls below the threshold to diagnose diabetes mellitus, the condition is called prediabetes. Insulin resistance, prediabetes, and metabolic syndrome share a spectrum of an overlapping area, thus they are closely related conditions and are milestones of a spectrum of a huge metabolic disorder of energy utilization and storage. Metabolic syndrome is indeed a serious condition in that it is a potential risk factor for ischemic heart diseases and type 2 diabetes (T2D).
1. Type 2 Diabetes
|
Foodstuff and GI |
Examples of Food |
|---|---|
|
Food with a low GI range (55 or less) |
Monosaccharides: fructose; tagatose. Pulses (beans): black; kidney; lentil; chickpea; pinto. Seeds (small): sesame; flax; sunflower; poppy; pumpkin; hemp. Nuts: walnuts; cashew; peanuts. Grains: wheat (durum, spelt, kamut); millet; oat; rye; rice; barley. Sweet fruits: peaches; strawberries; mangos. Vegetables: most vegetables; unpeeled sweet potatoes and mushrooms. |
|
Food with medium GI ranges (56–69) |
Table sugar; regular ice cream; cranberry juice; grape juice. Enriched whole wheat; basmati rice; unpeeled potatoes; peeled sweet potatoes; pita bread. Raisins; prunes; pumpernickel bread. |
|
Food with high GI ranges (70 and above) |
Sugars: glucose: dextrose; grape sugar; high fructose corn syrup; maltose; maltodextrins. White bread (from endosperm). Most white rice (from endosperm). Peeled potatoes Extruded breakfast cereals; corn flakes. |
2. Obesity
3. Gestational Diabetes
4. Human Insulin in Brief
5. Glucose Transporters
5.1. GLUT1
5.2. GLUT2
5.3. GLUT3
5.4. GLUT4
5.5. GLUT14
6. Insulin as a First Messenger in Signal Transduction Cascade
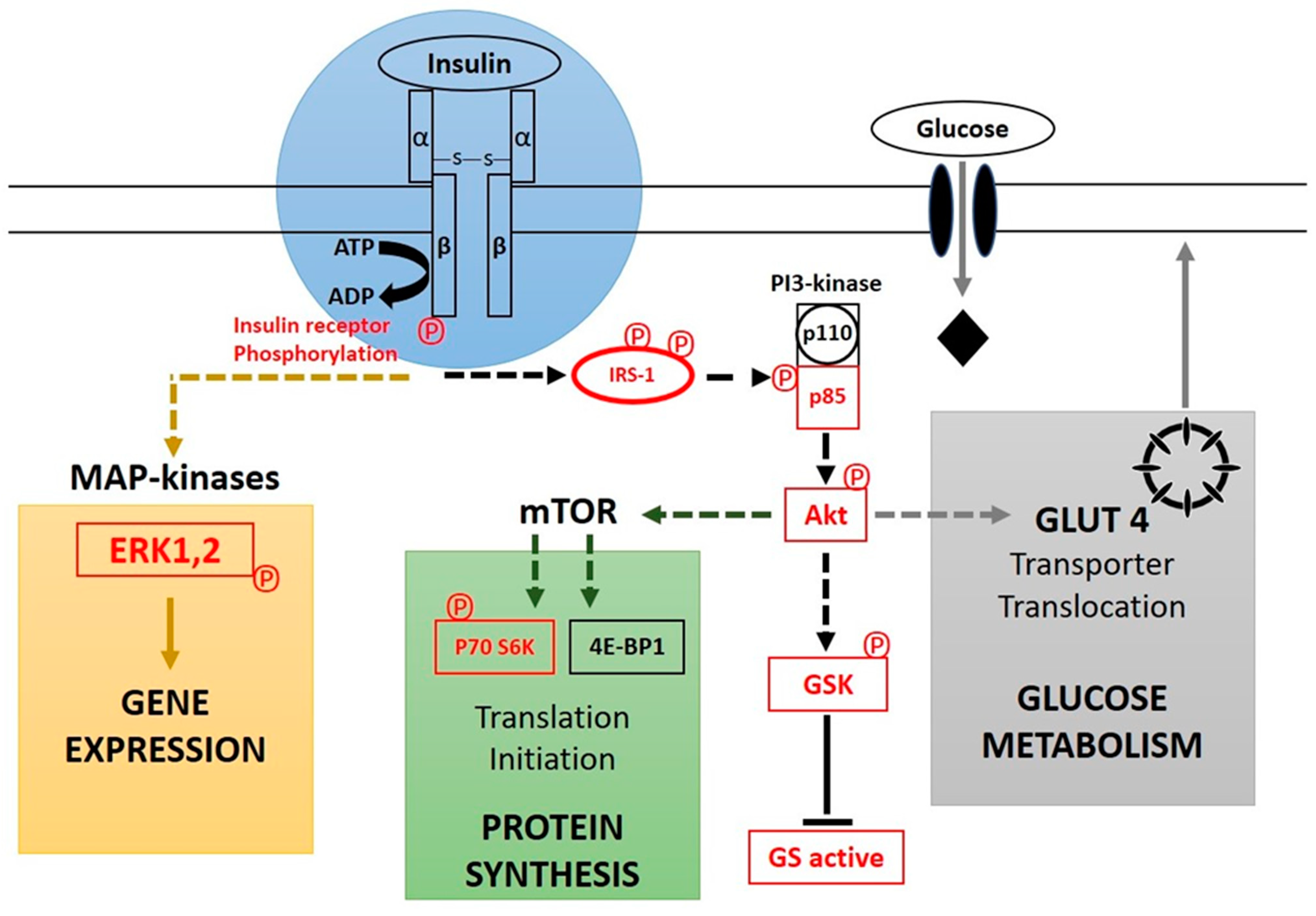
7. Physiological Effects of Insulin
|
Parameter |
Physiological Function |
Improper Function/Decrease in Insulin |
|---|---|---|
|
Glucose |
Stimulates glucose uptake via insertion of GLUT4 in the membranes of myocytes and lipocytes. |
Increase in blood glucose concentration. |
|
Triglycerols (fat) |
Increases lipogenesis by forcing lipocytes to take in glucose. |
Decrease in lipogenesis and hyperglycemia. |
|
Fatty acids |
Increased esterification to triglycerides (neutral lipids). |
Lipolysis of triglycerides to fatty acids and glycerol. |
|
Lipolysis |
Decreases lipolysis and decreases free fatty acid and glycerol in the circulation. |
Hyperlipidemia |
|
Glycogen |
Induces glycogen synthesis, by activation of the hexokinase that activates glucose by adding a phosphate, a process that traps glucose inside the cell. |
Inhibits glycogen synthesis by reverse steps that induce glycogen synthesis. |
|
Inhibits glucose-6-phosphatase, which dephosphorylates glucose. |
||
|
Activates both phosphofructokinase and glycogen synthase, which are responsible for glycogen synthesis. |
||
|
Gluconeogenesis and glycogenolysis |
Decreases these two processes by decreasing glucose synthesis from noncarbohydrate biomolecules mainly in the liver. |
Gluconeogenesis in the liver from diverse substrate biomolecules. |
|
Protein |
Decreases protein breakdown |
Proteolysis is eased, as is the case in advanced cases of diabetes. |
|
Autophagy |
Deceleration of degradation of damaged organelles. |
Autophagy is accelerated. |
|
Arterial muscle tone |
Increases this, especially arterioles and micro-arteries, and thus increases blood flow. |
Reduces blood flow in these by allowing muscles to contract. |
|
Gastric chlorhydria |
Increases hydrochloric acid secretion by the gastric parietal cells. |
T he occurrence of the reverse process is expected. |
|
Potassium uptake |
Forces glycogen-synthesizing cells to absorb potassium with water from the extracellular fluids via translocation of the Na+/K+-ATPase to the membranes of skeletal myocytes. |
Inhibits potassium absorption. |
|
Renal sodium excretion |
Decreases excretion of renal sodium. |
The reverse process occurs. |
Based on [52][53][54][55][56][57][58][59], GLUT4: glucose transporter type 4, Na+/K+-ATPase: ATP dependent sodium/potassium pump.
8. Treatment
References
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262.
- Shoback, D.M.; Gardner, D.G. (Eds.) Chapter 17: Pancreatic hormones & diabetes mellitus. In Greenspan’s Basic & Clinical Endocrinology, 9th ed.; McGraw-Hill Medical: New York, NY, USA, 2011; ISBN 978-0-07-162243-1.
- American Diabetes Association. Classification and Diagnosis of Diabetes. Diabetes Care 2017, 40 (Suppl. 1), S11–S24.
- Carris, N.W.; Magness, R.R.; Labovitz, A.J. Prevention of Diabetes Mellitus in Patients With Prediabetes. Am. J. Cardiol. 2019, 123, 507–512.
- Ling, C.; Ronn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044.
- Laakso, M. Biomarkers for type 2 diabetes. Mol. Metab. 2019, 27S, S139–S146.
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Despres, J.P.; Hu, F.B. Sugar-sweetened beverages, obesity, type 2 diabetes mellitus, and cardiovascular disease risk. Circulation 2010, 121, 1356–1364.
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Despres, J.P.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: A meta-analysis. Diabetes Care 2010, 33, 2477–2483.
- Riserus, U.; Willett, W.C.; Hu, F.B. Dietary fats and prevention of type 2 diabetes. Prog. Lipid Res. 2009, 48, 44–51.
- Atkinson, F.S.; Foster-Powell, K.; Brand-Miller, J.C. International tables of glycemic index and glycemic load values: 2008. Diabetes Care 2008, 31, 2281–2283.
- Di Ciaula, A.; Garruti, G.; Fruhbeck, G.; De Angelis, M.; de Bari, O.; Wang, D.Q.; Lammert, F.; Portincasa, P. The Role of Diet in the Pathogenesis of Cholesterol Gallstones. Curr. Med. Chem. 2019, 26, 3620–3638.
- Hu, E.A.; Pan, A.; Malik, V.; Sun, Q. White rice consumption and risk of type 2 diabetes: Meta-analysis and systematic review. BMJ 2012, 344, e1454.
- Jenkins, D.J.; Wolever, T.M.; Taylor, R.H.; Barker, H.; Fielden, H.; Baldwin, J.M.; Bowling, A.C.; Newman, H.C.; Jenkins, A.L.; Goff, D.V. Glycemic index of foods: A physiological basis for carbohydrate exchange. Am. J. Clin. Nutr. 1981, 34, 362–366.
- Morales, P.E.; Bucarey, J.L.; Espinosa, A. Muscle Lipid Metabolism: Role of Lipid Droplets and Perilipins. J. Diabetes Res. 2017, 2017, 1789395.
- Meex, R.C.R.; Blaak, E.E.; van Loon, L.J.C. Lipotoxicity plays a key role in the development of both insulin resistance and muscle atrophy in patients with type 2 diabetes. Obes. Rev. 2019, 20, 1205–1217.
- Glycemic Research Institute. Glycemic Load Defined; Glycemic Research Institute: Washington, DC, USA, 2013.
- U.S. Department of Health and Human Services. National Diabetes Clearinghouse (NDIC): National Diabetes Statistics; U.S. Department of Health and Human Services: Washington, DC, USA, 2011.
- Soldavini, J. Krause’s Food & The Nutrition Care Process. J. Nutr. Educ. Behav. 2019, 51, 1225.
- Beilby, H.; Yang, F.; Gannon, B.; McIntyre, H.D. Cost-effectiveness of gestational diabetes screening including prevention of type 2 diabetes: Application of the GeDiForCE model in Australia. J. Matern. Fetal. Neonatal. Med. 2022, 35, 8286–8293.
- Li, Z.; Cheng, Y.; Wang, D.; Chen, H.; Chen, H.; Ming, W.K.; Wang, Z. Incidence Rate of Type 2 Diabetes Mellitus after Gestational Diabetes Mellitus: A Systematic Review and Meta-Analysis of 170,139 Women. J. Diabetes Res. 2020, 2020, 3076463.
- Amraei, M.; Mohamadpour, S.; Sayehmiri, K.; Mousavi, S.F.; Shirzadpour, E.; Moayeri, A. Effects of Vitamin D Deficiency on Incidence Risk of Gestational Diabetes Mellitus: A Systematic Review and Meta-analysis. Front. Endocrinol. 2018, 9, 7.
- Zhang, Y.; Gong, Y.; Xue, H.; Xiong, J.; Cheng, G. Vitamin D and gestational diabetes mellitus: A systematic review based on data free of Hawthorne effect. BJOG 2018, 125, 784–793.
- Managing & Treating Gestational Diabetes | NIDDK; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2019.
- Voet, D.; Voet, J.G. Biochemistry, 4th ed.; Wiley: New York, NY, USA, 2011.
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; W. H. Freeman and Company: New York, NY, USA, 2001; pp. 858–859.
- Koeslag, J.H.; Saunders, P.T.; Terblanche, E. A reappraisal of the blood glucose homeostat which comprehensively explains the type 2 diabetes mellitus-syndrome X complex. J. Physiol. 2003, 549, 333–346.
- Rowlett, R. A Dictionary of Units of Measurement; The University of North Carolina at Chapel Hill: Chapel Hill, NC, USA, 2001.
- Iwase, H.; Kobayashi, M.; Nakajima, M.; Takatori, T. The ratio of insulin to C-peptide can be used to make a forensic diagnosis of exogenous insulin overdosage. Forensic Sci. Int. 2001, 115, 123–127.
- Hellman, B.; Gylfe, E.; Grapengiesser, E.; Dansk, H.; Salehi, A. Insulin oscillations--clinically important rhythm. Antidiabetics should increase the pulsative component of the insulin release. Lakartidningen 2007, 104, 2236–2239.
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145.
- Bell, G.I.; Kayano, T.; Buse, J.B.; Burant, C.F.; Takeda, J.; Lin, D.; Fukumoto, H.; Seino, S. Molecular biology of mammalian glucose transporters. Diabetes Care 1990, 13, 198–208.
- Berger, C.; Zdzieblo, D. Glucose transporters in pancreatic islets. Pflugers Arch. 2020, 472, 1249–1272.
- Mueckler, M.; Caruso, C.; Baldwin, S.A.; Panico, M.; Blench, I.; Morris, H.R.; Allard, W.J.; Lienhard, G.E.; Lodish, H.F. Sequence and structure of a human glucose transporter. Science 1985, 229, 941–945.
- Olson, A.L.; Pessin, J.E. Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu. Rev. Nutr. 1996, 16, 235–256.
- Gould, G.W.; Thomas, H.M.; Jess, T.J.; Bell, G.I. Expression of human glucose transporters in Xenopus oocytes: Kinetic characterization and substrate specificities of the erythrocyte, liver, and brain isoforms. Biochemistry 1991, 30, 5139–5145.
- Freitas, H.S.; Schaan, B.D.; Seraphim, P.M.; Nunes, M.T.; Machado, U.F. Acute and short-term insulin-induced molecular adaptations of GLUT2 gene expression in the renal cortex of diabetic rats. Mol. Cell Endocrinol. 2005, 237, 49–57.
- Kellett, G.L.; Brot-Laroche, E. Apical GLUT2: A major pathway of intestinal sugar absorption. Diabetes 2005, 54, 3056–3062.
- Vannucci, S.J.; Maher, F.; Simpson, I.A. Glucose transporter proteins in brain: Delivery of glucose to neurons and glia. Glia 1997, 21, 2–21.
- Brown, K.; Heller, D.S.; Zamudio, S.; Illsley, N.P. Glucose transporter 3 (GLUT3) protein expression in human placenta across gestation. Placenta 2011, 32, 1041–1049.
- Simpson, I.A.; Dwyer, D.; Malide, D.; Moley, K.H.; Travis, A.; Vannucci, S.J. The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E242–E253.
- James, D.E.; Brown, R.; Navarro, J.; Pilch, P.F. Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature 1988, 333, 183–185.
- Chadt, A.; Al-Hasani, H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflugers Arch. 2020, 472, 1273–1298.
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138.
- Buchberger, A.; Howard, M.J.; Proctor, M.; Bycroft, M. The UBX domain: A widespread ubiquitin-like module. J. Mol. Biol. 2001, 307, 17–24.
- Wu, X.; Freeze, H.H. GLUT14, a duplicon of GLUT3, is specifically expressed in testis as alternative splice forms. Genomics 2002, 80, 553–557.
- McManus, E.J.; Sakamoto, K.; Armit, L.J.; Ronaldson, L.; Shpiro, N.; Marquez, R.; Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005, 24, 1571–1583.
- Cross, D.A.; Watt, P.W.; Shaw, M.; van der Kaay, J.; Downes, C.P.; Holder, J.C.; Cohen, P. Insulin activates protein kinase B, inhibits glycogen synthase kinase-3 and activates glycogen synthase by rapamycin-insensitive pathways in skeletal muscle and adipose tissue. FEBS Lett. 1997, 406, 211–215.
- Stryer, L. Biochemistry, 4th ed.; W. H. Freeman and Company: New York, NY, USA, 1995; pp. 351–356, 494–495, 505, 605–606, 773–775. ISBN 0-7167-2009-4.
- Hou, J.C.; Pessin, J.E. Ins (endocytosis) and outs (exocytosis) of GLUT4 trafficking. Curr. Opin Cell Biol. 2007, 19, 466–473.
- Najjar, S. Insulin Action: Molecular Basis of Diabetes. In Encyclopedia of Life Sciences; John Wiley & Sons: Hoboken, NJ, USA, 2001; ISBN 978-0470016176.
- Duckworth, W.C.; Bennett, R.G.; Hamel, F.G. Insulin degradation: Progress and potential. Endocr. Rev. 1998, 19, 608–624.
- Physiologic Effects of Insulin. Available online: www.vivo.colostate.edu (accessed on 1 June 2017).
- Benziane, B.; Chibalin, A.V. Frontiers: Skeletal muscle sodium pump regulation: A translocation paradigm. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E553–E558.
- Bergamini, E.; Cavallini, G.; Donati, A.; Gori, Z. The role of autophagy in aging: Its essential part in the anti-aging mechanism of caloric restriction. Ann. N. Y. Acad. Sci. 2007, 1114, 69–78.
- Clausen, T. Regulatory role of translocation of Na+-K+ pumps in skeletal muscle: Hypothesis or reality? Am. J. Physiol. Endocrinol. Metab. 2008, 295, E727–E728.
- Dimitriadis, G.; Mitrou, P.; Lambadiari, V.; Maratou, E.; Raptis, S.A. Insulin effects in muscle and adipose tissue. Diabetes Res. Clin. Pract 2011, 93 (Suppl. 1), S52–S59.
- Gupta, A.K.; Clark, R.V.; Kirchner, K.A. Effects of insulin on renal sodium excretion. Hypertension 1992, 19, I78–I82.
- Kreitzman, S.N.; Coxon, A.Y.; Szaz, K.F. Glycogen storage: Illusions of easy weight loss, excessive weight regain, and distortions in estimates of body composition. Am. J. Clin. Nutr. 1992, 56, 292S–293S.
- Zheng, C.; Liu, Z. Vascular function, insulin action, and exercise: An intricate interplay. Trends Endocrinol. Metab. 2015, 26, 297–304.
- Rhea, E.M.; Nirkhe, S.; Nguyen, S.; Pemberton, S.; Bammler, T.K.; Beyer, R.; Niehoff, M.L.; Morley, J.E.; Farr, S.A.; Banks, W.A. Molecular Mechanisms of Intranasal Insulin in SAMP8 Mice. J. Alzheimers Dis. 2019, 71, 1361–1373.
- Benedict, C.; Brede, S.; Schioth, H.B.; Lehnert, H.; Schultes, B.; Born, J.; Hallschmid, M. Intranasal insulin enhances postprandial thermogenesis and lowers postprandial serum insulin levels in healthy men. Diabetes 2011, 60, 114–118.
- Sliwowska, J.H.; Fergani, C.; Gawalek, M.; Skowronska, B.; Fichna, P.; Lehman, M.N. Insulin: Its role in the central control of reproduction. Physiol. Behav. 2014, 133, 197–206.
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Glycolysis Chapter 16. In Biochemistry, 5th ed.; W. H. Freeman and Company: New York, NY, USA, 2002; pp. 688–690. Available online: www.whfreeman.com/biochem5 (accessed on 1 June 2017).
- Eurich, D.T.; McAlister, F.A.; Blackburn, D.F.; Majumdar, S.R.; Tsuyuki, R.T.; Varney, J.; Johnson, J.A. Benefits and harms of antidiabetic agents in patients with diabetes and heart failure: Systematic review. BMJ. 2007, 335, 497.
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585.
- Bloomgarden, Z.T. Developments in diabetes and insulin resistance. Diabetes Care 2006, 29, 161–167.
- Landau, Z.; Raz, I.; Wainstein, J.; Bar-Dayan, Y.; Cahn, A. The role of insulin pump therapy for type 2 diabetes mellitus. Diabetes Metab. Res. Rev. 2017, 33, e2822.
- Costanzo, P.; Cleland, J.G.; Pellicori, P.; Clark, A.L.; Hepburn, D.; Kilpatrick, E.S.; Perrone-Filardi, P.; Zhang, J.; Atkin, S.L. The obesity paradox in type 2 diabetes mellitus: Relationship of body mass index to prognosis: A cohort study. Ann. Intern. Med. 2015, 162, 610–618.
- Mikstas, C. U.S. Department of Agriculture FoodData Central. 9 November 2020. Available online: https://fdc.nal.usda.gov/docs/Foundation_Foods_Documentation_Apr2021.pdf (accessed on 1 June 2017).

