Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rajesh Kumar Gandhirajan | -- | 5450 | 2023-05-05 04:41:33 | | | |
| 2 | Conner Chen | -1 word(s) | 5449 | 2023-05-08 03:26:58 | | | | |
| 3 | Conner Chen | Meta information modification | 5449 | 2023-05-08 03:27:26 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Murali, R.; Balasubramaniam, V.; Srinivas, S.; Sundaram, S.; Venkatraman, G.; Warrier, S.; Dharmarajan, A.; Gandhirajan, R.K. Deregulated Metabolic Pathways in Ovarian Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/43815 (accessed on 28 July 2026).
Murali R, Balasubramaniam V, Srinivas S, Sundaram S, Venkatraman G, Warrier S, et al. Deregulated Metabolic Pathways in Ovarian Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/43815. Accessed July 28, 2026.
Murali, Roopak, Vaishnavi Balasubramaniam, Satish Srinivas, Sandhya Sundaram, Ganesh Venkatraman, Sudha Warrier, Arun Dharmarajan, Rajesh Kumar Gandhirajan. "Deregulated Metabolic Pathways in Ovarian Cancer" Encyclopedia, https://encyclopedia.pub/entry/43815 (accessed July 28, 2026).
Murali, R., Balasubramaniam, V., Srinivas, S., Sundaram, S., Venkatraman, G., Warrier, S., Dharmarajan, A., & Gandhirajan, R.K. (2023, May 05). Deregulated Metabolic Pathways in Ovarian Cancer. In Encyclopedia. https://encyclopedia.pub/entry/43815
Murali, Roopak, et al. "Deregulated Metabolic Pathways in Ovarian Cancer." Encyclopedia. Web. 05 May, 2023.
Copy Citation
Cancer cells thrive on cellular metabolism to facilitate their growth, uncontrolled proliferation, invasiveness, and metastasis. In cancer cells, multiple metabolic pathways were altered compared to their normal counterparts so that these cancer cells survive and sustain themselves against the changing conditions in the tumor microenvironment.
ovarian cancer
tumor metabolism
oncogenes
1. Glycolysis
Glucose is an important molecule that plays a central role in the energy generation since the oxidation of glucose carbon dioxide and water results in a standard free-energy change of −2840 kJ/mol. Glycolysis is considered an important metabolic pathway that breaks down glucose into two three-carbon compounds. During this process, free energy is released in the form of high-energy deriving molecules such as adenosine triphosphate (ATP) and reduced form of Nicotinamide Adenine Dinucleotide (NADPH). The process of glycolysis is a series of enzyme-catalyzed reactions to yield two molecules of pyruvate. Glycolysis is initiated when glucose is phosphorylated at the hydroxyl group of C-6. It consists of two phases: (i) preparatory phase where two molecules of ATP are invested, and (ii) payoff phase where energy is gained. Glycolysis occurs in the cell cytoplasm under anaerobic conditions. Cancer cells undergo a modified form of glycolysis called aerobic glycolysis or the Warburg effect in which the cells rapidly proliferate and there is an increased glucose uptake and lactate production even in the presence of oxygen [1]. Figure 1 shows the differentially expressed proteins in the glycolysis pathway, represented as boxplots obtained from GEPIA web server uploaded by Zhang et al., 2017 [2]. Almost 60% of the ATP is generated by tumor cells in the presence of aerobic conditions through glycolysis [3]. The Warburg effect has been shown to play a role in progression of ovarian cancer. A study conducted by Teng et al. showed silencing of AKT2 (AKT serine/threonine kinase 2) and AKT3 (AKT serine/threonine kinase 3), isoforms of AKT which are a downstream mediator of PI3K signaling pathway, was determined to regulate the Warburg effect in EOC [4].
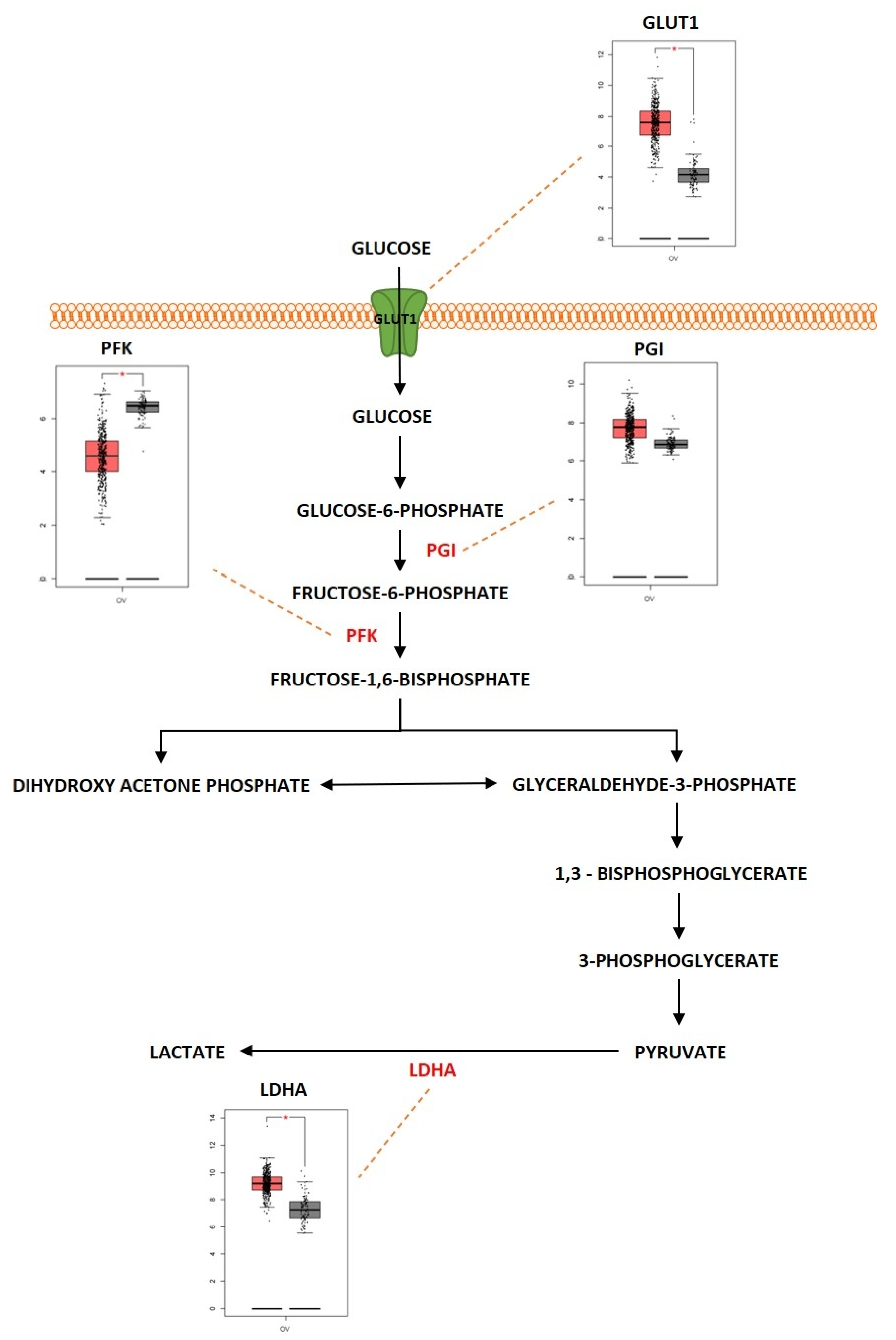
Figure 1. Differentially expressed proteins of the glycolysis pathway in ovarian cancer. Schematic presentation of glycolytic pathway in ovarian cancer and highlight of key differentially regulated genes (GLUT1; overexpressed, PFK; underexpressed, PGI; overexpressed, LDHA; overexpressed). Boxplot derived from TGCA expression datasets with tumor (red) and control (gray). * Represents statistical significance of p < 0.05.
Hepatocyte nuclear factor 1β (HNF1β) is a transcription factor involved in the development of kidney and pancreatic beta cells. Overexpression of HNF1β was associated with altered glucose metabolism in OCCC by promoting increased glucose uptake and increased aerobic glycolysis [5]. The tumor growth in cancer is associated with metabolic reprogramming of nitric oxide (NO), especially in cancers such as ovarian cancer. Caneba et al. showed that in ovarian cancer, NO is involved in regulating tumor growth and inhibits mitochondrial respiration, shifting these cells towards glycolysis so that production of ATP is maintained. NO was also discovered to decrease reactive oxygen species (ROS) levels by increasing the levels of NADPH and glutathione [6]. Glucose transporter 1 (GLUT1), which is the first component of glycolysis, is an isoform of glucose transporters that plays an important role in transporting glucose into cells. GLUT1 was overexpressed in different tumors and especially in ovarian cancer. In addition, the increased expression of GLUT1 was associated with poor survival rate in ovarian cancer patients [7]. Ovarian cancer was reported to rely upon the GLUT1 transporter to regulate glycolysis and tumor growth. A study showed that silencing GLUT1 expression blocks stress-regulated glycolysis and anchorage-dependent and independent growth of ovarian cancer cells [8]. Forkhead box protein M1 (FOXM1), a transcription factor, was associated with glycolysis. FOXM1, GLUT1 and hexokinase 2 (HK2) was upregulated in EOC. Molecular investigations show that FOXM1 binds directly to the GLUT1 and HK2 promoter regions and regulates the expression of the genes at the transcriptional level. Knockdown of FOXM1 significantly reduced the expression of GLUT1 and HK2 genes and also downregulated aerobic glycolysis as well as cell proliferation [9]. Targeting GLUT1 by microRNA (miR)-144 and silencing it exhibited a metabolic shift in glucose uptake, proving that miR-144 can regulate GLUT1 expression and aerobic glycolysis [10]. Hexokinase 2 (HK2) is an important enzyme that catalyzes the conversion of glucose to glucose-6-phosphate, which is one of the initial steps in glycolysis. HK2 was also shown to have a critical role in ovarian carcinogenesis. HK2 was overexpressed in ovarian cancer. The expression of HK2 is associated with advanced stage and high-grade cancers. HK2 was also discovered to regulate lactate production and was also linked with cancer metastasis [11]. Upregulation of miR-603 was reported to diminish the malignant behavior of ovarian cancer cells by targeting aerobic glycolysis. The miR-603 directly targets HK2 to target cellular metabolism and inhibit malignancy by acting as a tumor suppressor [12]. DNA methyltransferase 3A (DNMT3A) is a de novo methyltransferase that functions by methylating the unmethylated CpG sites. DNMT3A was overexpressed in ovarian cancer tissues when compared with normal ovary tissues. The overexpression of DNMT3A was associated with miR-603 as DNMT3A inhibited the expression of the microRNA and promoted aerobic glycolysis, cell proliferation, migration, and invasion of ovarian cancer [13]. Lactate dehydrogenase A (LDHA) is another important enzyme that is involved in glycolysis as it catalyzes the reduction in pyruvate. LDHA was upregulated in ovarian cancer tissues when compared to normal ovarian tissues [14]. Since LDHA is a key player in the conversion of pyruvate to lactate and in the maintenance of glycolysis, targeting LDHA was shown to reduce tumor growth by suppressing glycolysis. Qiu et al. showed that suppressing LDHA could inhibit lactate production, and as a result there is decreased energy supply to ovarian tumors [15]. miR-383 is an miRNA that functions as a tumor suppressor in different types of cancers. Since it functions as a tumor suppressor, this miRNA was downregulated in most tumors, and especially in ovarian cancer [16]. Ectopic expression of miR-383 induced apoptosis and inhibited cell proliferation and migration in ovarian cancer. The miR-383 was negatively correlated with LDHA in ovarian cancer tissues and it was determined to suppress LDHA by directly targeting the 3′-UTR of LDHA gene. Overexpression of LDHA was determined to reverse the inhibitory effect of miR-383 in ovarian cancer [17]. A study conducted by Xintaropoulou et al. evaluated the expression of different glycolytic enzymes (GLUT1, HK2 and LDHA) and their role in promoting ovarian cancer. There was significantly higher expression of GLUT1 and HK2 in high-grade serous ovarian carcinoma (HGSOC) when compared to non-HGSOC and it was also associated with advanced stages of ovarian cancer. This study also showed that when the glycolytic pathway was inhibited by using different glycolytic inhibitors, it suppressed cell growth and proliferation in ovarian cancer [18]. Therefore, these studies prove that the glycolytic pathway is an important metabolic determinant for the survival and progression of ovarian cancer and that targeting the glycolytic pathway is a potential therapeutic strategy for the treatment of ovarian cancer.
Outcome of Somatic Driver Mutations in Glycolysis
As previously described, Her-2/neu overexpression has been observed in different types of ovarian cancer. The overexpression of HER2 was associated with regulating tumor growth by upregulating the mTOR pathway activity and by activating the metabolic shift towards glycolysis [19]. GLUT-1 expression was positively associated with the expression of HER2 [20]. The increased expression of HER2 was also reported to elevate the expression G6PD (Glucose 6 Phosphate Dehydrogenase) [21]. HER2 overexpression also upregulates the expression of LDH-A, and targeting LDH-A using inhibitors was reported to reduce cell proliferation and survival in HER2 overexpressing tumors [22]. c-MYC is another oncogene that is significantly amplified and has high copy number variations in ovarian cancer and plays an important role in deregulating glycolysis. c-MYC was reported to upregulate the expression of GLUT1, leading to increased uptake of glucose. c-MYC also upregulated other important genes that are involved in glycolysis such as LDHA, phosphoglucose isomerase (PGI), phosphofructokinase (PFK), glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate kinase, and enolase as well [23][24]. KRAS was identified as a commonly mutated oncogene in different subtypes of ovarian cancer. KRAS was determined to deregulate the glycolytic pathway and its components in KRAS-driven cancers [25]. KRAS mutation was reported to trigger the overexpression of GLUT1. The presence of a KRAS mutation and overexpression of GLUT1 worsened the survival rate of cancer patients [26]. In order to survive glucose deprivation, the KRAS mutant colorectal cancer cells, were found to upregulate the expression of GLUT1 and during this glucose deprivation, the wild type cells were found to acquire new mutations in the KRAS gene [27]. KRAS mutant pancreatic cancer cells were also identified to upregulate HK2 expression as well as LDHA expression [28][29][30]. BTAK also played an important role in deregulating glycolytic metabolism. Aurora-A kinase induced a metabolic shift towards glycolysis and altered the expression of glucose metabolic genes such as LDHA and HK2 by participating and influencing the SOX8/FOXK1 signaling axis in ovarian cancer [31]. Aurora-A kinase also regulates glycolysis by stabilizing the Myc protein [32]. Aurora-A kinase is also involved in regulating the expression of metabolic genes such as GLUT1, LDHA and HK2 expressions. Inhibiting Aurora-A kinase downregulates the expressions of these genes [33]. Aurora-A kinase was also shown to interact directly by phosphorylating LDHB, a subunit of LDH. Phosphorylation of LDHB serine 162 significantly increased its activity in reducing pyruvate to lactate, which then efficiently promoted NAD+ regeneration, glycolytic flux, lactate production and biosynthesis with glycolytic intermediates [34]. Similarly, tumor suppressor genes also play an important role in regulating the glycolytic metabolic pathway in ovarian cancer. TP53 mutations have been observed with a high frequency rate in ovarian cancer. TP53 was shown to mediate metabolic activities such as glycolysis in cells under both physiological and pathological conditions. TP53 was determined to regulate glycolysis via regulating the expression of TP53-induced glycolysis regulator (TIGAR). TIGAR downregulated glycolysis by degrading fructose-2,6-bisphosphate, an allosteric effector of the glycolytic enzyme 6-phosphofructo-kinase-1 (PFK-1). TIGAR was also involved in switching the glycolytic pathway into the pentose phosphate pathway, thereby decreasing ROS generation, and promoting glutathione production [35]. Suppressing TP53 was shown to increase the expression of GLUT1 to promote glycolysis [36]. Mutation of TP53 was also reported to activate the expression of HK2 and phosphoglycerate mutase (PGM). Mutant TP53 was discovered to upregulate the expression of HK2 gene and thus increase the glycolytic state in cancer cells [37]. Normal p53 expression was shown to have an inhibitory effect on expression of PGM by mediating and inhibiting the expressions of transporters GLUT1 and GLUT4 [38][39]. BRCA1 is another major tumor suppressor gene that is reported to mutate in ovarian cancer and was determined to regulate glycolysis. A study conducted by Chiyoda et al. showed that silencing BRCA1 increased the rate of glycolysis in ovarian surface epithelial and fallopian tube cells. The deleterious mutations in BRCA1 caused the increased expression of HK2 and thereby promoted glycolysis [40]. BRCA1 was also determined to regulate GLUT1. A higher expression of GLUT1 was observed in cancer cells that were carrying germline mutations for BRCA1 [41]. Cancer cells containing mutant BRCA1 were also determined to suppress glycolysis by repressing the genes GLUT1, HK1, HK2, and LDHA. This study also showed that BRCA1 in turn increased the tricarboxylic acid (TCA) cycle and oxidative phosphorylation activity [42]. The tumor suppressor PTEN was also involved in regulation of glycolysis. Studies have identified PI3k/AKT pathway as a key regulator of GLUT1 expression [43][44]. PTEN was discovered to physically interact with AKT and cause its dephosphorylation; as a result, there is a limited expression of GLUT1 at the plasma membrane in ovarian cancer cells [45].
2. Tricarboxylic Acid Cycle
Tricarboxylic acid (TCA) cycle, also called the citric acid cycle or Kreb’s cycle, is a series of metabolic reactions occurring in mitochondria and acting as a critical source of energy for cells in the presence of aerobic conditions. The TCA cycle is present in the core of energy metabolism and is involved in macromolecule synthesis and maintaining redox balance. The TCA cycle is responsible for the production of NADH and FADH2 that fuels the electron transport chain in mitochondria for the generation of ATP. The TCA cycle is initiated when pyruvate generated from glycolysis is oxidized into acetyl-CoA by pyruvate dehydrogenase complex. It consists of eight successive reaction steps in a cyclical manner, and at the end of the cycle, one molecule of oxaloacetate becomes regenerated. The energy produced during these reactions is conserved when three NAD+ and one FAD are reduced and by the production of one ATP or GTP. The TCA cycle was deregulated in different diseases ranging from metabolic disease such as obesity to neurodegenerative disease such as Alzheimer’s disease. The TCA cycle was deregulated in ovarian cancer as well (Figure 2). Studies showed that cancer cells rely on glutamine as a fuel instead of using the pyruvate that is generated during glycolysis. In addition, when there is impaired mitochondrial pyruvate transport, glutamine is used to regulate the TCA cycle and to meet the cells’ increased metabolic needs [46][47]. In ovarian cancer, invasiveness is correlated with glutamine dependence. Low-invasive ovarian cancer was glutamine-independent, whereas invasive ovarian cancer was dependent on glycine [48]. Sometimes, the TCA cycle is also dependent on β-oxidation of fatty acids since acetyl-CoA acts as a converging point for both TCA and fatty acid metabolism [49]. Fatty Acid Synthase (FASN) is an important enzyme that converts acetyl-CoA into saturated fatty acid. FASN was highly expressed in ovarian cancer and was associated with poor survival rate [50]. In different types of cancer, and especially in ovarian cancer, the genes encoding for the enzymes aconitase, isocitrate dehydrogenase (IDH), succinate dehydrogenase (SDH) and citrate synthase (CS) were deregulated. Alterations in the genes of TCA enzymes cause the ectopic expression of different oncometabolites. Isocitrate dehydrogenase is an enzyme coded by the IDH1 gene. This enzyme helps in conversion of isocitrate to α—ketoglutarate by oxidative decarboxylation. Isoforms of IDH were identified to undergo missense mutation in different types of tumors which include grade II/III gliomas and secondary glioblastomas (GBM), chondrosarcomas, and acute myeloid leukemia [51]. In ovarian cancer, wild-type IDH1 was upregulated TCA cycle metabolism. The upregulation caused increase in the amount of α-ketoglutarate and NADPH production and also provided increased levels of reducing equivalents to sustain lipid biosynthesis and redox homeostasis [52]. Dahl et al. identified that HGSOC utilized glucose from TCA preferentially rather than from aerobic glycolysis. They also reported that IDH1 was upregulated in ovarian cancer and was associated with reduced progression free survival. Targeting IDH1 modifies the histone epigenetic landscape and this was discovered to induce senescence [53]. Bcl2-like-10 (Bcl2l10) is a member of the Bcl-2 family of genes that plays a key role in mediating apoptosis. Bcl2l10 was identified as a tumor suppressor gene as knocking down of the gene improved cell viability, motility, and proliferation [54]. Knocking down of Bcl2l10 was reported to deregulate the TCA cycle as some of the components of the TCA cycle acted as a downstream target of Bcl2l10. Succinate dehydrogenase complex subunit D (SDHD) and IDH1 were regulated by Bcl2l10. Knocking down Bcl2l10 downregulated IDH1 and SDHD and led to the accumulation of oncometabolites such as succinate and isocitrate, and therefore lead to the promotion and progression of ovarian cancer [55]. Succinate dehydrogenase is another enzyme that is a part of the TCA cycle and is primarily involved in the catalytic conversion of succinate to fumarate by oxidation [56]. SDH acts as a tumor suppressor gene and consists of six subunits that encode SDHA, SDHB, SDHC, SDHD, SDHAF1, and SDHAF2 [57]. Mutations were identified in SDH in different types of cancer. Amplification of SDH was identified with a high probability rate of occurrence in HGSOC. SDHB showed a high rate of amplification compared to the other subunits [58]. Chen et al. showed that silencing SDHB promoted cell proliferation, migration, and invasion, whereas SDHB overexpression suppressed cell proliferation and promoted apoptosis. Silencing of SDHB was shown to promote overexpression of HIF-1α, a tumor-promoting factor [59]. Another study showed that knocking down SDHB promoted epithelial mesenchymal transition (EMT) by increased H3K27 methylation. SDHB knockdown also led to altered glucose and glutamine utilization and caused mitochondrial dysfunction [60]. Citrate synthase (CS) is an enzyme involved in the TCA cycle that catalyzes the reaction between oxaloacetic acid and acetyl coenzyme A to produce citrate. CS was overexpressed in malignant ovarian tumors compared to benign tumors. Knocking down CS using RNAi mechanism resulted in reduced proliferation, migration, and invasion in in vitro studies using ovarian cancer cell lines [61].
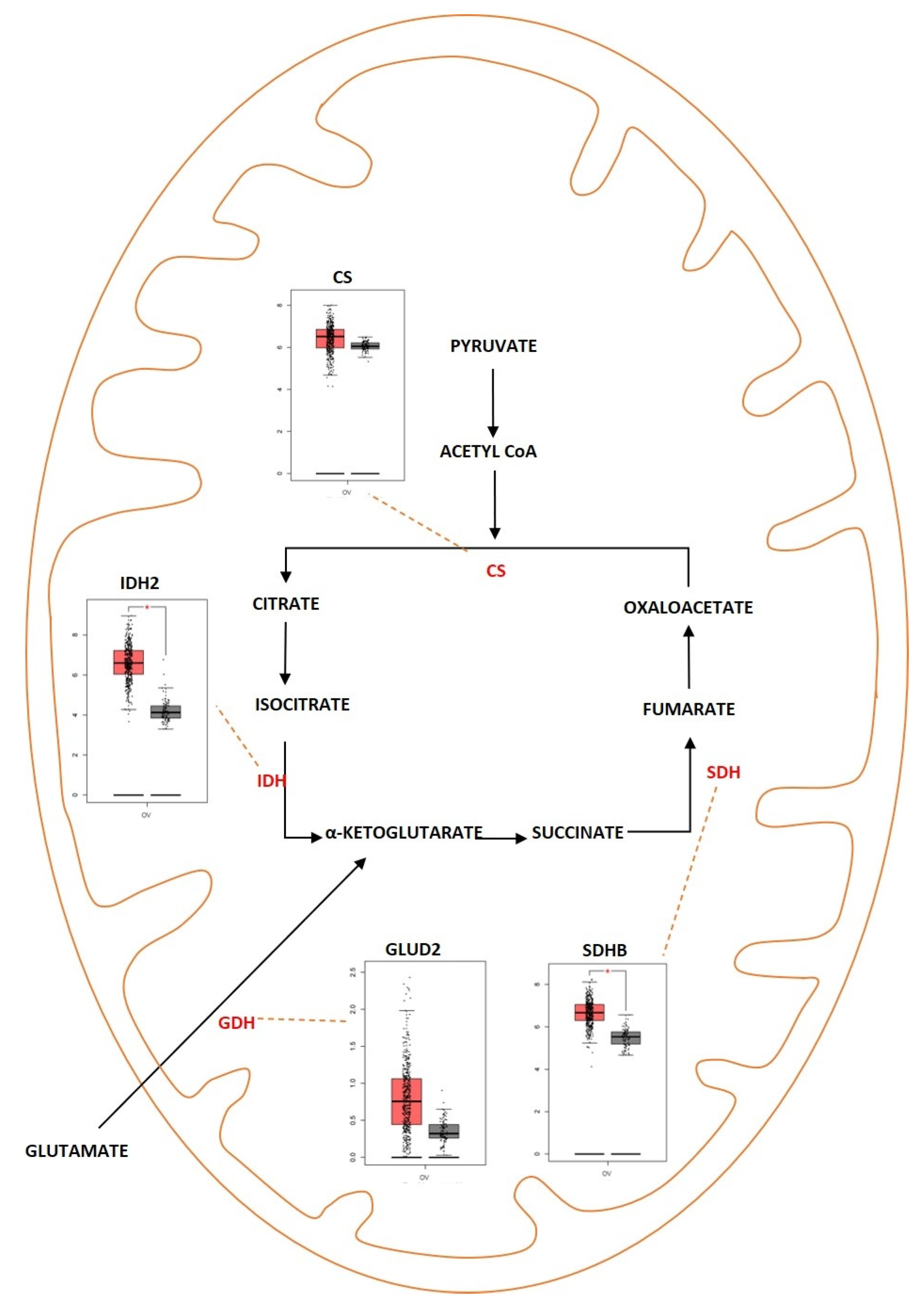
Figure 2. Differentially expressed proteins of the TCA cycle in ovarian cancer. Schematic representation highlighting the differentially regulated genes of the TCA cycle in ovarian cancer. (CS; overexpressed, IDH2; overexpressed, SDHB; overexpressed, GLUD2; overexpressed). Boxplot derived from TGCA expression datasets with tumor (red) and control (gray). * Represents statistical significance of p < 0.05.
Outcome of Somatic Driver Mutations in the TCA Cycle
Oncogenes and TSG were identified to play a critical role in the control and regulation of the TCA cycle. The oncogene c-MYC, which is aberrantly expressed in ovarian cancer, was observed to regulate TCA either directly or indirectly. c-MYC acts indirectly as a master driver of glutamine metabolism through the TCA cycle. c-MYC also controls the conversion of glutamine to glutamate by activating glutaminase 1 (GLS1) through transcriptional suppression of its negative regulator miR-23a/b [62][63]. The c-MYC acts directly on the TCA cycle by interacting with the components of the TCA cycle. For example, c-MYC was determined to co-express with mutant IDH1/2 and increase the state of malignancy in MYC overexpressed cancers [64]. The c-MYC was reported to inactivate SDHA. This resulted in kick-starting a regulatory cascade in cancer cells that led to the activation of H3K4me3 and induced tumor-specific gene expression and the promotion of tumorigenesis. The inhibition of SDH-complex activity led to the accumulation of succinate in cancer cells [65]. KRAS, which is one of the most frequently mutated oncogenes from the RAS family, plays a pivotal role in ovarian cancer metabolism by regulating the TCA cycle. KRAS-driven cancer cells scavenge vital proteins that contain glutamine from the extracellular space and utilize them to fuel the TCA cycle [66]. KRAS-driven cancer cells also scavenge branch chain amino acids such as isoleucine, valine, and leucine and convert them into acetyl-CoA to trigger the TCA cycle [67]. A study conducted by Kerr et al. showed that the copy number variant of KRAS promoted glucose anaplerosis fueling the TCA cycle [68]. Tumor suppressor genes such as TP53 also play a vital role in the regulation of the TCA cycle. Pyruvate metabolism is one of the initiating factors for activating the TCA cycle. TP53 is involved in regulating the pyruvate metabolism. The p53 transcriptionally represses the enzyme pyruvate dehydrogenase kinase-2 (PDK2), which inhibits the activity of pyruvate dehydrogenase (PDH). Using this mechanism, TP53 helps in advancement into the TCA cycle by the conversion of pyruvate into acetyl-CoA [69]. TP53 suppresses the expression of MCT1 (lactate/proton symporter monocarboxylate transporter 1), which reduces the ability of the cells to regenerate NAD+ through the conversion of pyruvate to lactate. Because of this, cells that lack TP53 generate less ATP through oxidative phosphorylation when compared to cells that expresses TP53 [70][71]. The TCA cycle can also be fueled with the help of the amino acid glutamine via α-ketoglutarate-dependent anaplerosis. Glutaminase 2 is a mitochondrial enzyme that is involved in the hydrolysis of glutamine to glutamate. TP53 regulates the process of glutaminolysis by binding to the P53 consensus DNA-binding elements in the promoter region of GLS2, the gene coding for glutaminase 2. Increased expression of GLS2 enhances mitochondrial respiration, ATP generation and the production of glutathione (GSH), an anti-oxidant [72][73]. BRCA1, another important tumor suppressor gene, is involved in regulating metabolism by strongly inhibiting glycolysis while activating the TCA cycle and oxidative phosphorylation. Privat et al. showed that cells expressing BRCA1 were discovered to increase the rate of transcription of SDHC. The enzymes IDH1 in the cytosol and IDH2 in the mitochondria were inversely regulated in BRCA1 positive cells. IDH1 was upregulated whereas IDH2 expression was downregulated. The upregulation of IDH1 could supply NADPH for glutathione reduction to combat against cellular oxidative stress [42][74]. SDHB was also downregulated in BRCA1-silenced cancer cells [75].
3. Amino Acid Metabolism
Proteins are large macromolecules that are present in the living organisms and play a critical role in every function of the body. Proteins consist of multiple sub units of amino acids. Amino acids interact with each other by forming a peptide bond and multiple peptides form complex structures that constitute the protein. Amino acids are molecules that are derivatives of carboxylic acid groups in which the α-hydrogen becomes substituted by an amino group. These molecules play an important part in sustaining life from maintaining the metabolism to act as a catalyst for various biological reactions that maintain the metabolic activities. During catabolism of amino acids, they lose their amino groups and form the “carbon skeleton” α-keto acid form. This form becomes oxidized to form CO2 and H2O. They also play a role in providing three- and four-carbon units that can be converted into glucose by the process of gluconeogenesis. Amino acid metabolism functions by maintaining the amino acid pools producing non-essential amino acids for protein biogenesis and is also involved in conversion of glucose, lipids, and nitrogen-containing metabolites, such as purines and pyrimidines for nucleic acid biosynthesis. Amino acids are required for the activation of important metabolic pathways such as the mTORC pathway and play an important role in detoxification of ammonia by converting it to the non-toxic urea form. In addition, amino acids are required for maintaining the intracellular redox homeostasis (e.g., glutathione, an important antioxidant is metabolized from the amino acids glutamate, cysteine, and glycine) [76]. The complex network of amino acid metabolism is highly interconnected with other important metabolic pathways that include glycolysis, the TCA cycle and fatty acid metabolism [77]. Since amino acid metabolism is involved in almost all the critical functions in maintaining normal cellular homeostasis, dysregulation of amino acid metabolism can be an important mediating factor in different types of cancers. Figure 3 shows the deregulated glutamine metabolic pathway in ovarian cancer. Glutamine is a non-essential α-amino acid that has an amide group that replaces the carboxylic acid group on their carbon structure. It is a highly abundant amino acid found in the body. Even though it is a non-essential amino acid, the majority of tumors require extra-cellular glutamine for their survival. Cancer cells import glutamine with the help of various transporters, which include ASC (alanine/serine/cysteine-preferring), Na+-dependent transporters and the Na+-coupled neutral amino acid transporters. Some members of these transporter families are dysregulated in different types of cancers. ASCT2, a member of ASC transporter family, was determined to play a role in the development of gastric cancer, and inhibition of ASCT2 reduced cancer growth [78]. ASCT2 was significantly upregulated in EOC. ASCT2 was positively correlated with p-mTOR in the development and progression of ovarian cancer [79]. One of the initial steps in glutamine metabolism is the conversion of glutamine into glutamate by the process of glutaminolysis. This process is catalyzed by the enzyme glutaminase (GA). GA is an enzyme coded by two isoforms of GLS and GLS2. The GA encoded by GLS was reported to promote tumor cell growth as it was regulated by oncogenes, and GA encoded by GLS2 was determined to reduce the sensitivity of the cells towards ROS-associated apoptosis with the help of glutathione-dependent antioxidant defense [80]. GLS was highly expressed in cancers and to promote tumorigenesis and cell proliferation. Knocking down GLS was shown to suppress proliferation and promote apoptosis in prostate cancer [81]. The rate of glutaminolysis was associated with invasiveness in ovarian cancer. The higher the invasive rate, the higher the rate of glutaminolysis [82]. Glutamate is further converted into α-ketoglutarate through oxidative deamination by the enzyme glutamate dehydrogenase (GDH). α-ketoglutarate is an important substrate that is a part of the TCA cycle. Higher levels of glutamine were determined to increase the activity of the enzymes GA and GDH by modulating the mTOR/S6 and MAPK pathways in ovarian cancer [83]. GDH has been proposed as an important marker for metastasis in ovarian cancer [84]. Glutamine helps in the synthesis of the reduced form of glutathione (GSH), an anti-oxidant, as it acts as a carbon and nitrogen donor by supplementing glutamate and mediating cysteine uptake [85][86]. GSH has multiple functions. It functions as a reducing agent and an antioxidant. It mediates the metabolism of xenobiotics and also acts as a physiological reservoir for cysteine, and also regulates Ca2+ homeostasis [87]. GSH is also involved in inducing cellular resistance to ionizing radiation and promotes resistance against cytotoxic drugs [88]. Platinum-based chemotherapeutic drugs such as cisplatin and carboplatin are widely used for the treatment of ovarian cancer. GSH provides resistance to cisplatin and carboplatin therapy by different mechanisms, such as by reducing drug uptake and increasing intracellular drug detoxification, increased DNA repair mechanisms, and by suppressing drug-induced oxidative stress in ovarian cancer [89][90]. GSH was determined to have complex and contrasting roles in providing resistance against cisplatin. Decreased levels of reduced GSH and enzymes involved in GSH synthesis were identified to play a key role in cisplatin resistance [91]. Pompella et al. showed that the enzyme gamma-glutamyltransferase 1 (GGT1) expression on the surface of the cell favors the cellular resupply of antioxidant glutathione (GSH) which in turn favors protection against cisplatin by the formation of glycyl-cysteine dipeptide which forms an adduct with cisplatin thereby preventing its entry into the cell and its interaction with the DNA [92][93]. Platinum-resistant ovarian cancer cells showed increased dependency on glutamine metabolism as ASCT2 and GA were significantly upregulated. There is also increased dependency of glutamine utilization through the TCA cycle in platinum-resistant ovarian cancer cells. Knocking down glutaminase sensitized the resistant cells to the platinum-based compounds [94]. Glutamine, as an amide donor, is also involved in de novo synthesis of nucleotides and plays a role in maintaining the nucleotide balance. For example, purines are involved in controlling cell proliferation in ovarian cancer [95]. Myc, an important transcription factor and an oncoprotein that is found overexpressed in ovarian cancer, was shown to regulate glutamine metabolism. Increased expression of Myc was reported to promote glutaminolysis and make it highly dependent on exogenous glutamine for the survival of cell. Cells with c-MYC amplification undergo apoptosis because of glutamine deprivation [63][96]. Overexpressed c-Myc was reported to upregulate GA and promote glutaminolysis [97]. The above studies indicate that glutamine is equally important as glucose for cell survival and proliferation and balancing the cells’ bioenergetic needs. Serine is another important amino acid that helps in ovarian cancer progression and proliferation. Extra-cellular serine is required for the cancer cell progression since serine deprivation affects the tumor growth and cancer cell proliferation [98][99]. Serine is transported into the cell by Na+-dependent transporter ASCT1 (SLC1A4). ASCT1 is upregulated in different cancer types [100][101]. ASCT1 was also expressed in ovarian cancer and its expression was positively associated with the expression of L-type amino acid transporter 1 (LAT1), another amino acid transporter [102]. Serine metabolism is also a key player in promoting ovarian cancer [103]. Serine can be synthesized intracellularly with the help of glucose through a de novo serine synthesis pathway. The pathway is initiated when 3-phosphoglycerate (3-PG), the intermediate metabolite of glycolysis, becomes converted to 3-phosphohydroxypyruvate (3-PH). This reaction is catalyzed by 3-phosphoglycerate dehydrogenation (PHGDH). 3-PH then obtains an amino group from glutamate and forms 3-phosphoserine (3-PS) with the help of phosphoserine transaminase (PSAT) followed by dephosphorylation through serine phosphatase (PSPH) to produce serine. PHGDH, PSAT and PSPH were overexpressed in various cancers [104][105][106]. PHGDH was significantly upregulated at the protein level in ovarian cancer and involved in invasiveness of the cells as well as provision of resistance to platinum-based drugs [107]. Suppressing PHGDH was reported to inhibit proliferation, migration, and invasion, and increase cellular ROS levels in epithelial ovarian cancer [108]. PSAT1 was overexpressed in EOC. Higher PSAT1 expression indicated poor survival rate in the EOC patients and the expression was associated with increased GSH (reduced glutathione)/GSSG (oxidized glutathione) ratio and reduced ROS levels. This showed that PSAT1 promotes cancer growth by regulating the oxidation–reduction balance [109]. Glycine is an amino acid of lowest molecular weight which carries a hydrogen atom as a side-chain. Glycine is a non-essential amino acid which forms a building block for proteins as well as in different metabolic pathways such as glutathione metabolism and in regulating one-carbon metabolism [110]. Glycine is synthesized when Serine hydroxymethyl transferase (SHMT1 [cytoplasm]; SHMT2 [mitochondria]) catalyzes the transfer of the beta carbon of serine to tetrahydrofolate (THF). During this reaction, glycine is formed along with 5,10-methylene-THF, which is involved in nucleotide biosynthesis. SHMT1 was overexpressed in HGSOC and was necessary for tumor growth and cell migration [111]. SHMT2 was also overexpressed in different cancers and especially in ovarian cancer [112]. c-MYC which is overexpressed in ovarian cancer efficiently targets SHMT [113]. Arginine is a positively charged semi-essential amino acid and it acts as a precursor for amino acids, nitric oxide, polyamines, and creatine. Arginine is taken up into the cells by CAT-1 (SLC7A1), which is a cationic amino acid transporter (CAT). Increased expression of CAT-1 was observed in tumors that are highly L-arginine-dependent [114][115]. CAT-1 is overexpressed in EOC and it is involved in transporting phenylalanine and arginine. The expression of CAT-1 is associated with poorer survival rate in EOC patients and it was shown to promote proliferation and migration in EOC [116]. Arginine is synthesized de novo by the urea cycle. The urea cycle is a detoxification process in which toxic ammonia is converted into non-toxic urea. The substrate carbamoyl phosphate which is produced by the enzyme carbamoyl phosphate synthetase 1 (CPS1) along with ornithine is used to produce citrulline. Then, argininosuccinate synthetase 1 (ASS1), the rate-limiting enzyme involved in arginine synthesis, catalyzes the conversion of citrulline and aspartate into argininosuccinate. The enzyme argininosuccinate lyase (ASL) then cleaves argininosuccinate into arginine and fumarate. There is a relatively strong cross-talk between the enzymes involved in the urea cycle and other metabolic pathways, which promotes tumorigenesis in different cell types [117]. A reduced expression of ASS1 enzyme was observed in different types of cancers such as melanoma and hepatocellular cancers [118][119][120]. Contrastingly, higher expression of ASS1 was observed in primary high-grade and low-grade serous ovarian carcinomas and there was relatively increased expression in recurrent tumors, whereas in non-serous subtypes of ovarian cancer, a decreased expression of ASS1 was reported [121]. Silencing ASS1 through epigenetic modification imparted ovarian cancer cells with resistance to platinum-based drugs resulting in treatment failure and clinical relapse in ovarian cancer [122]. NO is produced by the conversion of arginine catalyzed by the enzyme nitric oxide synthetase (NOS). The ASS and ASL complexes are involved in NO production along with NOS [123]. An isoform of NOS enzyme, NOS1, was upregulated in ovarian cancer. The upregulation of NOS1 promoted proliferation and invasion, and the cells with high NOS1 expression showed resistance to anti-cancer drugs [124]. The branched-chain amino acids (BCAA) such as leucine, isoleucine, and valine are group of essential amino acids that are obtained via dietary intake and scavenged by protein recycling. BCAA is transported into the cells with the help of LAT-1 (SLC7A5), a member of the SLC7 family, which consists of Na+- and pH-independent L-type amino acid transporters (system L/antiporter). LAT-1 is highly expressed in cancers such as breast and lung [125][126]. LAT1 is highly expressed in ovarian cancer. This transporter plays a role in cell proliferation and invasion in ovarian cancer [127]. With the help of certain highly reversible enzymes, the intracellular BCAAs are catabolized to provide nitrogen and carbon groups for various biological needs such as energy production and cell signaling [128]. The BCAA is broken down into the specific forms of branched chain keto acids (BCKA) and the branched chain amino acid transaminase 1 and 2 (BCAT1/2) simultaneously in order to produce glutamine to transfer the nitrogen to α-ketoglutarate (αKG). The enzyme-branched chain alpha-keto acid dehydrogenase complex (BCKDH) metabolizes the BCKAs in order to generate branched chain acyl-CoA (R-CoA), which then becomes further processed into the key intermediates of the TCA cycle (acetyl-CoA or succinyl-CoA (from isoleucine and valine)) [129]. BCAT enzymes comprise two isoforms: BCAT1 (cytosolic) and BCAT2 (mitochondrial). While BCAT2 is expressed in most tissues, BCAT1 is predominantly expressed in the brain, ovary, and placenta [130]. The BCAT1 gene was notably hypomethylated in low-malignant potential (LMP) and HGSOC tumors [131]. BCAT1 was also overexpressed in LMP and HGSOC tumors. The expression of BCAT1 was associated with ovarian cancer progression as it induced cell proliferation, migration, and invasion, and inhibited cell cycle progression [132]. Branched-chain α-keto acid dehydrogenase kinase (BCKDK) acts as a negative regulator of BCAA catabolism by phosphorylating and inactivating BCKDH. A study conducted by Li et al. showed that BCKDK was highly expressed in patients with advanced pathological grade ovarian cancer. This ectopic expression of BCKDK promoted the proliferation and migration of OC cells [133].
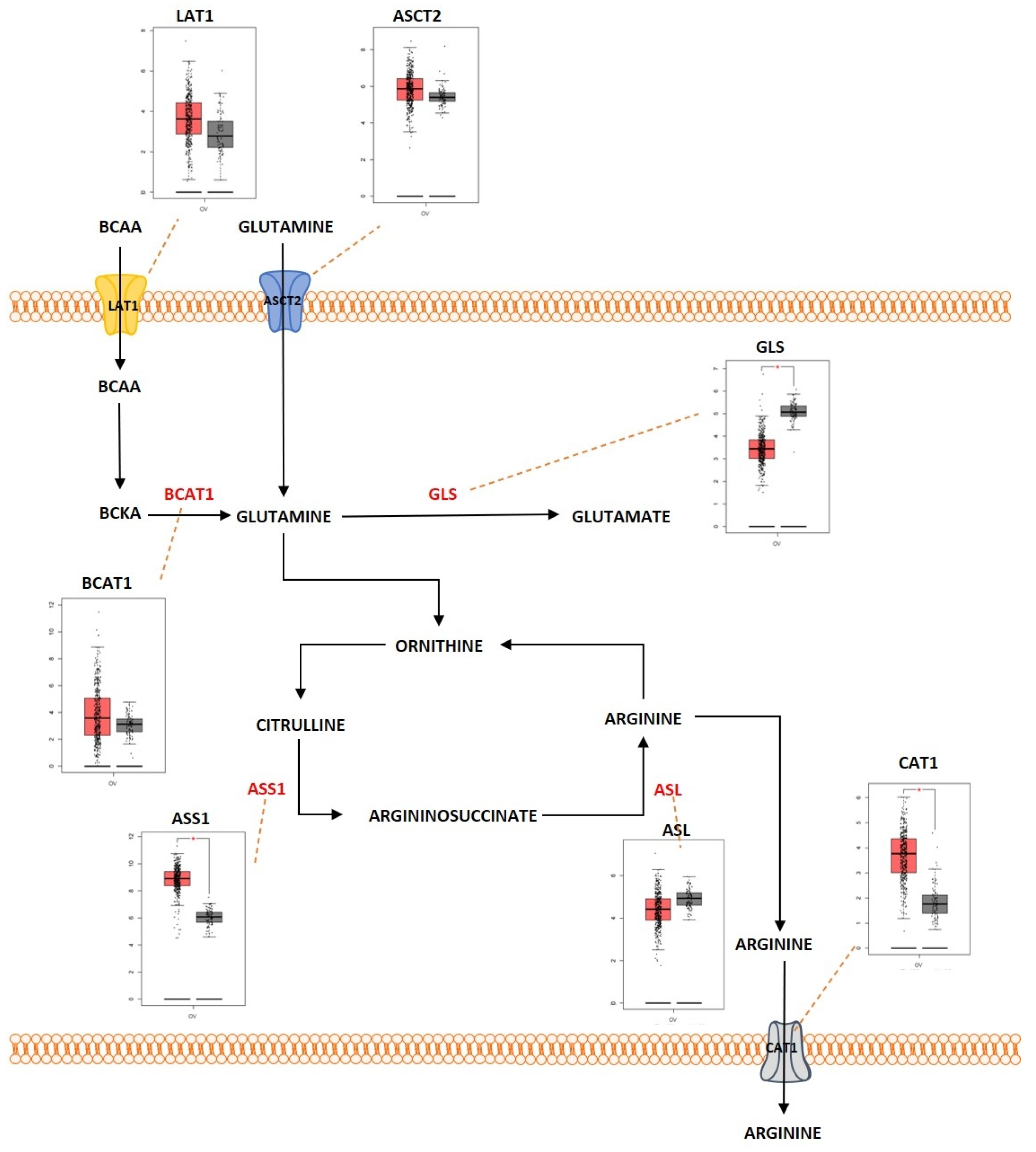
Figure 3. Differentially expressed proteins of the glutamine metabolic pathway in ovarian cancer. Schematic representation of the glutamine metabolic pathway in ovarian cancer that highlights the differentially regulated genes (LAT1; overexpressed, ASCT2; overexpressed, BCAT1; overexpressed, GLS; overexpressed, ASS1; overexpressed, ASL; underexpressed; CAT1; overexpressed). Boxplot derived from TGCA ex-pression datasets with tumor (red) and control (gray). * Represents statistical significance of p < 0.05.
References
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218.
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102.
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899.
- Teng, Y.; Zhang, Y.; Qu, K.; Yang, X.; Fu, J.; Chen, W.; Li, X. MicroRNA-29B (mir-29b) regulates the Warburg effect in ovarian cancer by targeting AKT2 and AKT3. Oncotarget 2015, 6, 40799–40814.
- Okamoto, T.; Mandai, M.; Matsumura, N.; Yamaguchi, K.; Kondoh, H.; Amano, Y.; Baba, T.; Hamanishi, J.; Abiko, K.; Kosaka, K.; et al. Hepatocyte nuclear factor-1beta (HNF-1beta) promotes glucose uptake and glycolytic activity in ovarian clear cell carcinoma. Mol. Carcinog. 2015, 54, 35–49.
- Caneba, C.A.; Yang, L.; Baddour, J.; Curtis, R.; Win, J.; Hartig, S.; Marini, J.; Nagrath, D. Nitric oxide is a positive regulator of the Warburg effect in ovarian cancer cells. Cell Death Dis. 2014, 5, e1302.
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662.
- Ma, Y.; Wang, W.; Idowu, M.O.; Oh, U.; Wang, X.Y.; Temkin, S.M.; Fang, X. Ovarian Cancer Relies on Glucose Transporter 1 to Fuel Glycolysis and Growth: Anti-Tumor Activity of BAY-876. Cancers 2018, 11, 33.
- Wang, Y.; Yun, Y.; Wu, B.; Wen, L.; Wen, M.; Yang, H.; Zhao, L.; Liu, W.; Huang, S.; Wen, N.; et al. FOXM1 promotes reprogramming of glucose metabolism in epithelial ovarian cancer cells via activation of GLUT1 and HK2 transcription. Oncotarget 2016, 7, 47985–47997.
- Fan, J.Y.; Yang, Y.; Xie, J.Y.; Lu, Y.L.; Shi, K.; Huang, Y.Q. MicroRNA-144 mediates metabolic shift in ovarian cancer cells by directly targeting Glut1. Tumour Biol. 2016, 37, 6855–6860.
- Siu, M.K.Y.; Jiang, Y.X.; Wang, J.J.; Leung, T.H.Y.; Han, C.Y.; Tsang, B.K.; Cheung, A.N.Y.; Ngan, H.Y.S.; Chan, K.K.L. Hexokinase 2 Regulates Ovarian Cancer Cell Migration, Invasion and Stemness via FAK/ERK1/2/MMP9/NANOG/SOX9 Signaling Cascades. Cancers 2019, 11, 813.
- Lu, J.; Wang, L.; Chen, W.; Wang, Y.; Zhen, S.; Chen, H.; Cheng, J.; Zhou, Y.; Li, X.; Zhao, L. miR-603 targeted hexokinase-2 to inhibit the malignancy of ovarian cancer cells. Arch. Biochem. Biophys. 2019, 661, 1–9.
- Lu, J.; Zhen, S.; Tuo, X.; Chang, S.; Yang, X.; Zhou, Y.; Chen, W.; Zhao, L.; Li, X. Downregulation of DNMT3A Attenuates the Warburg Effect, Proliferation, and Invasion via Promoting the Inhibition of miR-603 on HK2 in Ovarian Cancer. Technol. Cancer Res. Treat. 2022, 21, 15330338221110668.
- Koukourakis, M.I.; Kontomanolis, E.; Giatromanolaki, A.; Sivridis, E.; Liberis, V. Serum and tissue LDH levels in patients with breast/gynaecological cancer and benign diseases. Gynecol. Obstet. Investig. 2009, 67, 162–168.
- Qiu, H.; Jackson, A.L.; Kilgore, J.E.; Zhong, Y.; Chan, L.L.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. JQ1 suppresses tumor growth through downregulating LDHA in ovarian cancer. Oncotarget 2015, 6, 6915–6930.
- Donadeu, F.X.; Schauer, S.N. Differential miRNA expression between equine ovulatory and anovulatory follicles. Domest. Anim. Endocrinol. 2013, 45, 122–125.
- Han, R.L.; Wang, F.P.; Zhang, P.A.; Zhou, X.Y.; Li, Y. miR-383 inhibits ovarian cancer cell proliferation, invasion and aerobic glycolysis by targeting LDHA. Neoplasma 2017, 64, 244–252.
- Xintaropoulou, C.; Ward, C.; Wise, A.; Marston, H.; Turnbull, A.; Langdon, S.P. A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget 2015, 6, 25677–25695.
- Holloway, R.W.; Marignani, P.A. Targeting mTOR and Glycolysis in HER2-Positive Breast Cancer. Cancers 2021, 13, 2922.
- Choi, J.; Jung, W.H.; Koo, J.S. Metabolism-related proteins are differentially expressed according to the molecular subtype of invasive breast cancer defined by surrogate immunohistochemistry. Pathobiology 2013, 80, 41–52.
- Choi, J.; Kim, E.S.; Koo, J.S. Expression of Pentose Phosphate Pathway-Related Proteins in Breast Cancer. Dis. Markers 2018, 2018, 9369358.
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701.
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800.
- Wu, Y.; Deng, Y.; Zhu, J.; Duan, Y.; Weng, W.; Wu, X. Pim1 promotes cell proliferation and regulates glycolysis via interaction with MYC in ovarian cancer. Onco Targets Ther. 2018, 11, 6647–6656.
- Racker, E.; Resnick, R.J.; Feldman, R. Glycolysis and methylaminoisobutyrate uptake in rat-1 cells transfected with ras or myc oncogenes. Proc. Natl. Acad. Sci. USA 1985, 82, 3535–3538.
- Sasaki, H.; Shitara, M.; Yokota, K.; Hikosaka, Y.; Moriyama, S.; Yano, M.; Fujii, Y. Overexpression of GLUT1 correlates with Kras mutations in lung carcinomas. Mol. Med. Rep. 2012, 5, 599–602.
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559.
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670.
- Liang, C.; Qin, Y.; Zhang, B.; Ji, S.; Shi, S.; Xu, W.; Liu, J.; Xiang, J.; Liang, D.; Hu, Q.; et al. ARF6, induced by mutant Kras, promotes proliferation and Warburg effect in pancreatic cancer. Cancer Lett. 2017, 388, 303–311.
- Connell, L.C.; Boucher, T.M.; Chou, J.F.; Capanu, M.; Maldonado, S.; Kemeny, N.E. Relevance of CEA and LDH in relation to KRAS status in patients with unresectable colorectal liver metastases. J. Surg. Oncol. 2017, 115, 480–487.
- Sun, H.; Wang, H.; Wang, X.; Aoki, Y.; Wang, X.; Yang, Y.; Cheng, X.; Wang, Z.; Wang, X. Aurora-A/SOX8/FOXK1 signaling axis promotes chemoresistance via suppression of cell senescence and induction of glucose metabolism in ovarian cancer organoids and cells. Theranostics 2020, 10, 6928–6945.
- Nguyen, T.T.T.; Shang, E.; Shu, C.; Kim, S.; Mela, A.; Humala, N.; Mahajan, A.; Yang, H.W.; Akman, H.O.; Quinzii, C.M.; et al. Aurora kinase A inhibition reverses the Warburg effect and elicits unique metabolic vulnerabilities in glioblastoma. Nat. Commun. 2021, 12, 5203.
- Nguyen, T.T.T.; Shang, E.; Westhoff, M.A.; Karpel-Massler, G.; Siegelin, M.D. Therapeutic Drug-Induced Metabolic Reprogramming in Glioblastoma. Cells 2022, 11, 2956.
- Cheng, A.; Zhang, P.; Wang, B.; Yang, D.; Duan, X.; Jiang, Y.; Xu, T.; Jiang, Y.; Shi, J.; Ding, C.; et al. Aurora-A mediated phosphorylation of LDHB promotes glycolysis and tumor progression by relieving the substrate-inhibition effect. Nat. Commun. 2019, 10, 5566.
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120.
- Ohnishi, T.; Kusuyama, J.; Bandow, K.; Matsuguchi, T. Glut1 expression is increased by p53 reduction to switch metabolism to glycolysis during osteoblast differentiation. Biochem. J. 2020, 477, 1795–1811.
- Bustamante, E.; Pedersen, P.L. High aerobic glycolysis of rat hepatoma cells in culture: Role of mitochondrial hexokinase. Proc. Natl. Acad. Sci. USA 1977, 74, 3735–3739.
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185.
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633.
- Chiyoda, T.; Hart, P.C.; Eckert, M.A.; McGregor, S.M.; Lastra, R.R.; Hamamoto, R.; Nakamura, Y.; Yamada, S.D.; Olopade, O.I.; Lengyel, E.; et al. Loss of BRCA1 in the Cells of Origin of Ovarian Cancer Induces Glycolysis: A Window of Opportunity for Ovarian Cancer Chemoprevention. Cancer Prev. Res. 2017, 10, 255–266.
- van der Groep, P.; Bouter, A.; Menko, F.H.; van der Wall, E.; van Diest, P.J. High frequency of HIF-1alpha overexpression in BRCA1 related breast cancer. Breast Cancer Res. Treat. 2008, 111, 475–480.
- Privat, M.; Radosevic-Robin, N.; Aubel, C.; Cayre, A.; Penault-Llorca, F.; Marceau, G.; Sapin, V.; Bignon, Y.J.; Morvan, D. BRCA1 induces major energetic metabolism reprogramming in breast cancer cells. PLoS ONE 2014, 9, e102438.
- Cheng, J.C.; McBrayer, S.K.; Coarfa, C.; Dalva-Aydemir, S.; Gunaratne, P.H.; Carpten, J.D.; Keats, J.K.; Rosen, S.T.; Shanmugam, M. Expression and phosphorylation of the AS160_v2 splice variant supports GLUT4 activation and the Warburg effect in multiple myeloma. Cancer Metab. 2013, 1, 14.
- Morani, F.; Phadngam, S.; Follo, C.; Titone, R.; Aimaretti, G.; Galetto, A.; Alabiso, O.; Isidoro, C. PTEN regulates plasma membrane expression of glucose transporter 1 and glucose uptake in thyroid cancer cells. J. Mol. Endocrinol. 2014, 53, 247–258.
- Phadngam, S.; Castiglioni, A.; Ferraresi, A.; Morani, F.; Follo, C.; Isidoro, C. PTEN dephosphorylates AKT to prevent the expression of GLUT1 on plasmamembrane and to limit glucose consumption in cancer cells. Oncotarget 2016, 7, 84999–85020.
- Chen, J.Q.; Russo, J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim. Biophys. Acta 2012, 1826, 370–384.
- Yang, C.; Ko, B.; Hensley, C.T.; Jiang, L.; Wasti, A.T.; Kim, J.; Sudderth, J.; Calvaruso, M.A.; Lumata, L.; Mitsche, M.; et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 2014, 56, 414–424.
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728.
- Migita, T.; Narita, T.; Nomura, K.; Miyagi, E.; Inazuka, F.; Matsuura, M.; Ushijima, M.; Mashima, T.; Seimiya, H.; Satoh, Y.; et al. ATP citrate lyase: Activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008, 68, 8547–8554.
- Cai, Y.; Wang, J.; Zhang, L.; Wu, D.; Yu, D.; Tian, X.; Liu, J.; Jiang, X.; Shen, Y.; Zhang, L.; et al. Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med. Oncol. 2015, 32, 391.
- Bergaggio, E.; Piva, R. Wild-Type IDH Enzymes as Actionable Targets for Cancer Therapy. Cancers 2019, 11, 56.
- Sonego, M.; Baldassarre, G. A new role for IDH1 in the control of ovarian cancer cells metabolism and senescence. Ann. Transl. Med. 2020, 8, 780.
- Dahl, E.S.; Buj, R.; Leon, K.E.; Newell, J.M.; Imamura, Y.; Bitler, B.G.; Snyder, N.W.; Aird, K.M. Targeting IDH1 as a Prosenescent Therapy in High-grade Serous Ovarian Cancer. Mol. Cancer Res. 2019, 17, 1710–1720.
- Lee, S.Y.; Kwon, J.; Woo, J.H.; Kim, K.H.; Lee, K.A. Bcl2l10 mediates the proliferation, invasion and migration of ovarian cancer cells. Int. J. Oncol. 2020, 56, 618–629.
- Lee, S.Y.; Kwon, J.; Lee, K.A. Bcl2l10 induces metabolic alterations in ovarian cancer cells by regulating the TCA cycle enzymes SDHD and IDH1. Oncol. Rep. 2021, 45, 47.
- Gaude, E.; Frezza, C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014, 2, 10.
- Huang, S.; Millar, A.H. Succinate dehydrogenase: The complex roles of a simple enzyme. Curr. Opin. Plant Biol. 2013, 16, 344–349.
- Xia, L.; Zhang, H.; Wang, X.; Zhang, X.; Nie, K. The Role of Succinic Acid Metabolism in Ovarian Cancer. Front. Oncol. 2021, 11, 769196.
- Chen, L.; Liu, T.; Zhang, S.; Zhou, J.; Wang, Y.; Di, W. Succinate dehydrogenase subunit B inhibits the AMPK-HIF-1alpha pathway in human ovarian cancer in vitro. J. Ovarian Res. 2014, 7, 115.
- Aspuria, P.P.; Lunt, S.Y.; Varemo, L.; Vergnes, L.; Gozo, M.; Beach, J.A.; Salumbides, B.; Reue, K.; Wiedemeyer, W.R.; Nielsen, J.; et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014, 2, 21.
- Chen, L.; Liu, T.; Zhou, J.; Wang, Y.; Wang, X.; Di, W.; Zhang, S. Citrate synthase expression affects tumor phenotype and drug resistance in human ovarian carcinoma. PLoS ONE 2014, 9, e115708.
- Gao, C.; Shen, Y.; Jin, F.; Miao, Y.; Qiu, X. Cancer Stem Cells in Small Cell Lung Cancer Cell Line H446: Higher Dependency on Oxidative Phosphorylation and Mitochondrial Substrate-Level Phosphorylation than Non-Stem Cancer Cells. PLoS ONE 2016, 11, e0154576.
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787.
- Odia, Y.; Orr, B.A.; Bell, W.R.; Eberhart, C.G.; Rodriguez, F.J. cMYC expression in infiltrating gliomas: Associations with IDH1 mutations, clinicopathologic features and outcome. J. Neurooncol. 2013, 115, 249–259.
- Li, S.T.; Huang, D.; Shen, S.; Cai, Y.; Xing, S.; Wu, G.; Jiang, Z.; Hao, Y.; Yuan, M.; Wang, N.; et al. Myc-mediated SDHA acetylation triggers epigenetic regulation of gene expression and tumorigenesis. Nat. Metab. 2020, 2, 256–269.
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553.
- Mayers, J.R.; Wu, C.; Clish, C.B.; Kraft, P.; Torrence, M.E.; Fiske, B.P.; Yuan, C.; Bao, Y.; Townsend, M.K.; Tworoger, S.S.; et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med. 2014, 20, 1193–1198.
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, C.P. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016, 531, 110–113.
- Contractor, T.; Harris, C.R. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012, 72, 560–567.
- Boidot, R.; Vegran, F.; Meulle, A.; Le Breton, A.; Dessy, C.; Sonveaux, P.; Lizard-Nacol, S.; Feron, O. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res. 2012, 72, 939–948.
- Ma, W.; Sung, H.J.; Park, J.Y.; Matoba, S.; Hwang, P.M. A pivotal role for p53: Balancing aerobic respiration and glycolysis. J. Bioenerg. Biomembr. 2007, 39, 243–246.
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460.
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466.
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941.
- Salem, A.F.; Howell, A.; Sartini, M.; Sotgia, F.; Lisanti, M.P. Downregulation of stromal BRCA1 drives breast cancer tumor growth via upregulation of HIF-1alpha, autophagy and ketone body production. Cell Cycle 2012, 11, 4167–4173.
- Wei, Z.; Liu, X.; Cheng, C.; Yu, W.; Yi, P. Metabolism of Amino Acids in Cancer. Front. Cell Dev. Biol. 2020, 8, 603837.
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell. Mol. Life Sci. 2016, 73, 377–392.
- Lu, J.; Chen, M.; Tao, Z.; Gao, S.; Li, Y.; Cao, Y.; Lu, C.; Zou, X. Effects of targeting SLC1A5 on inhibiting gastric cancer growth and tumor development in vitro and in vivo. Oncotarget 2017, 8, 76458–76467.
- Guo, H.; Xu, Y.; Wang, F.; Shen, Z.; Tuo, X.; Qian, H.; Wang, H.; Wang, K. Clinical associations between ASCT2 and pmTOR in the pathogenesis and prognosis of epithelial ovarian cancer. Oncol. Rep. 2018, 40, 3725–3733.
- Mates, J.M.; Segura, J.A.; Martin-Rufian, M.; Campos-Sandoval, J.A.; Alonso, F.J.; Marquez, J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr. Mol. Med. 2013, 13, 514–534.
- Zhang, J.; Mao, S.; Guo, Y.; Wu, Y.; Yao, X.; Huang, Y. Inhibition of GLS suppresses proliferation and promotes apoptosis in prostate cancer. Biosci. Rep. 2019, 39, BSR20181826.
- Erickson, J.W.; Cerione, R.A. Glutaminase: A hot spot for regulation of cancer cell metabolism? Oncotarget 2010, 1, 734–740.
- Yuan, L.; Sheng, X.; Willson, A.K.; Roque, D.R.; Stine, J.E.; Guo, H.; Jones, H.M.; Zhou, C.; Bae-Jump, V.L. Glutamine promotes ovarian cancer cell proliferation through the mTOR/S6 pathway. Endocr. Relat. Cancer 2015, 22, 577–591.
- Fan, S.; Wang, Y.; Zhang, Z.; Lu, J.; Wu, Z.; Shan, Q.; Sun, C.; Wu, D.; Li, M.; Sheng, N.; et al. High expression of glutamate-ammonia ligase is associated with unfavorable prognosis in patients with ovarian cancer. J. Cell. Biochem. 2018, 119, 6008–6015.
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): Cystine supplier and beyond. Amino Acids 2012, 42, 231–246.
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492.
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181.
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913.
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074.
- Stewart, D.J. Mechanisms of resistance to cisplatin and carboplatin. Crit. Rev. Oncol. Hematol. 2007, 63, 12–31.
- Criscuolo, D.; Avolio, R.; Parri, M.; Romano, S.; Chiarugi, P.; Matassa, D.S.; Esposito, F. Decreased Levels of GSH Are Associated with Platinum Resistance in High-Grade Serous Ovarian Cancer. Antioxidants 2022, 11, 1544.
- Pompella, A.; Corti, A.; Visvikis, A. Redox Mechanisms in Cisplatin Resistance of Cancer Cells: The Twofold Role of Gamma-Glutamyltransferase 1 (GGT1). Front. Oncol. 2022, 12, 920316.
- Pompella, A.; De Tata, V.; Paolicchi, A.; Zunino, F. Expression of gamma-glutamyltransferase in cancer cells and its significance in drug resistance. Biochem. Pharmacol. 2006, 71, 231–238.
- Hudson, C.D.; Savadelis, A.; Nagaraj, A.B.; Joseph, P.; Avril, S.; DiFeo, A.; Avril, N. Altered glutamine metabolism in platinum resistant ovarian cancer. Oncotarget 2016, 7, 41637–41649.
- Liu, J.; Hong, S.; Yang, J.; Zhang, X.; Wang, Y.; Wang, H.; Peng, J.; Hong, L. Targeting purine metabolism in ovarian cancer. J. Ovarian Res. 2022, 15, 93.
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 2012, 22, 631–644.
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765.
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258.
- Maddocks, O.D.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546.
- Pollari, S.; Kakonen, S.M.; Edgren, H.; Wolf, M.; Kohonen, P.; Sara, H.; Guise, T.; Nees, M.; Kallioniemi, O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat. 2011, 125, 421–430.
- Riscal, R.; Schrepfer, E.; Arena, G.; Cisse, M.Y.; Bellvert, F.; Heuillet, M.; Rambow, F.; Bonneil, E.; Sabourdy, F.; Vincent, C.; et al. Chromatin-Bound MDM2 Regulates Serine Metabolism and Redox Homeostasis Independently of p53. Mol. Cell 2016, 62, 890–902.
- Kaira, K.; Nakamura, K.; Hirakawa, T.; Imai, H.; Tominaga, H.; Oriuchi, N.; Nagamori, S.; Kanai, Y.; Tsukamoto, N.; Oyama, T.; et al. Prognostic significance of L-type amino acid transporter 1 (LAT1) expression in patients with ovarian tumors. Am. J. Transl. Res. 2015, 7, 1161–1171.
- Van Nyen, T.; Planque, M.; van Wagensveld, L.; Duarte, J.A.G.; Zaal, E.A.; Talebi, A.; Rossi, M.; Korner, P.R.; Rizzotto, L.; Moens, S.; et al. Serine metabolism remodeling after platinum-based chemotherapy identifies vulnerabilities in a subgroup of resistant ovarian cancers. Nat. Commun. 2022, 13, 4578.
- Antonov, A.; Agostini, M.; Morello, M.; Minieri, M.; Melino, G.; Amelio, I. Bioinformatics analysis of the serine and glycine pathway in cancer cells. Oncotarget 2014, 5, 11004–11013.
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350.
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874.
- Bi, F.; An, Y.; Sun, T.; You, Y.; Yang, Q. PHGDH Is Upregulated at Translational Level and Implicated in Platin-Resistant in Ovarian Cancer Cells. Front. Oncol. 2021, 11, 643129.
- Zhang, X.; Sun, M.; Jiao, Y.; Lin, B.; Yang, Q. PHGDH Inhibitor CBR-5884 Inhibits Epithelial Ovarian Cancer Progression via ROS/Wnt/beta-Catenin Pathway and Plays a Synergistic Role with PARP Inhibitor Olaparib. Oxid. Med. Cell. Longev. 2022, 2022, 9029544.
- Zhang, Y.; Li, J.; Dong, X.; Meng, D.; Zhi, X.; Yuan, L.; Yao, L. PSAT1 Regulated Oxidation-Reduction Balance Affects the Growth and Prognosis of Epithelial Ovarian Cancer. Onco Targets Ther. 2020, 13, 5443–5453.
- Alves, A.; Bassot, A.; Bulteau, A.L.; Pirola, L.; Morio, B. Glycine Metabolism and Its Alterations in Obesity and Metabolic Diseases. Nutrients 2019, 11, 1356.
- Gupta, R.; Yang, Q.; Dogra, S.K.; Wajapeyee, N. Serine hydroxymethyl transferase 1 stimulates pro-oncogenic cytokine expression through sialic acid to promote ovarian cancer tumor growth and progression. Oncogene 2017, 36, 4014–4024.
- Zeng, Y.; Zhang, J.; Xu, M.; Chen, F.; Zi, R.; Yue, J.; Zhang, Y.; Chen, N.; Chin, Y.E. Roles of Mitochondrial Serine Hydroxymethyltransferase 2 (SHMT2) in Human Carcinogenesis. J. Cancer 2021, 12, 5888–5894.
- Nilsson, L.M.; Forshell, T.Z.; Rimpi, S.; Kreutzer, C.; Pretsch, W.; Bornkamm, G.W.; Nilsson, J.A. Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet. 2012, 8, e1002573.
- Abdelmagid, S.A.; Rickard, J.A.; McDonald, W.J.; Thomas, L.N.; Too, C.K. CAT-1-mediated arginine uptake and regulation of nitric oxide synthases for the survival of human breast cancer cell lines. J. Cell. Biochem. 2011, 112, 1084–1092.
- Kishikawa, T.; Otsuka, M.; Tan, P.S.; Ohno, M.; Sun, X.; Yoshikawa, T.; Shibata, C.; Takata, A.; Kojima, K.; Takehana, K.; et al. Decreased miR122 in hepatocellular carcinoma leads to chemoresistance with increased arginine. Oncotarget 2015, 6, 8339–8352.
- You, S.; Zhu, X.; Yang, Y.; Du, X.; Song, K.; Zheng, Q.; Zeng, P.; Yao, Q. SLC7A1 Overexpression Is Involved in Energy Metabolism Reprogramming to Induce Tumor Progression in Epithelial Ovarian Cancer and Is Associated with Immune-Infiltrating Cells. J. Oncol. 2022, 2022, 5864826.
- Keshet, R.; Szlosarek, P.; Carracedo, A.; Erez, A. Rewiring urea cycle metabolism in cancer to support anabolism. Nat. Rev. Cancer 2018, 18, 634–645.
- Delage, B.; Fennell, D.A.; Nicholson, L.; McNeish, I.; Lemoine, N.R.; Crook, T.; Szlosarek, P.W. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int. J. Cancer 2010, 126, 2762–2772.
- Dillon, B.J.; Prieto, V.G.; Curley, S.A.; Ensor, C.M.; Holtsberg, F.W.; Bomalaski, J.S.; Clark, M.A. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: A method for identifying cancers sensitive to arginine deprivation. Cancer 2004, 100, 826–833.
- Feun, L.G.; Marini, A.; Walker, G.; Elgart, G.; Moffat, F.; Rodgers, S.E.; Wu, C.J.; You, M.; Wangpaichitr, M.; Kuo, M.T.; et al. Negative argininosuccinate synthetase expression in melanoma tumours may predict clinical benefit from arginine-depleting therapy with pegylated arginine deiminase. Br. J. Cancer 2012, 106, 1481–1485.
- Cheon, D.J.; Walts, A.E.; Beach, J.A.; Lester, J.; Bomalaski, J.S.; Walsh, C.S.; Ruprecht Wiedemeyer, W.; Karlan, B.Y.; Orsulic, S. Differential expression of argininosuccinate synthetase in serous and non-serous ovarian carcinomas. J. Pathol. Clin. Res. 2015, 1, 41–53.
- Nicholson, L.J.; Smith, P.R.; Hiller, L.; Szlosarek, P.W.; Kimberley, C.; Sehouli, J.; Koensgen, D.; Mustea, A.; Schmid, P.; Crook, T. Epigenetic silencing of argininosuccinate synthetase confers resistance to platinum-induced cell death but collateral sensitivity to arginine auxotrophy in ovarian cancer. Int. J. Cancer 2009, 125, 1454–1463.
- Erez, A.; Nagamani, S.C.; Shchelochkov, O.A.; Premkumar, M.H.; Campeau, P.M.; Chen, Y.; Garg, H.K.; Li, L.; Mian, A.; Bertin, T.K.; et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat. Med. 2011, 17, 1619–1626.
- Zou, Z.; Li, X.; Sun, Y.; Li, L.; Zhang, Q.; Zhu, L.; Zhong, Z.; Wang, M.; Wang, Q.; Liu, Z.; et al. NOS1 expression promotes proliferation and invasion and enhances chemoresistance in ovarian cancer. Oncol. Lett. 2020, 19, 2989–2995.
- Hafliger, P.; Charles, R.P. The L-Type Amino Acid Transporter LAT1-An Emerging Target in Cancer. Int. J. Mol. Sci. 2019, 20, 2428.
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci. 2018, 43, 752–789.
- Kaji, M.; Kabir-Salmani, M.; Anzai, N.; Jin, C.J.; Akimoto, Y.; Horita, A.; Sakamoto, A.; Kanai, Y.; Sakurai, H.; Iwashita, M. Properties of L-type amino acid transporter 1 in epidermal ovarian cancer. Int. J. Gynecol. Cancer 2010, 20, 329–336.
- Sivanand, S.; Vander Heiden, M.G. Emerging Roles for Branched-Chain Amino Acid Metabolism in Cancer. Cancer Cell 2020, 37, 147–156.
- Harper, A.E.; Miller, R.H.; Block, K.P. Branched-chain amino acid metabolism. Annu. Rev. Nutr. 1984, 4, 409–454.
- Hall, T.R.; Wallin, R.; Reinhart, G.D.; Hutson, S.M. Branched chain aminotransferase isoenzymes. Purification and characterization of the rat brain isoenzyme. J. Biol. Chem. 1993, 268, 3092–3098.
- Keita, M.; Wang, Z.Q.; Pelletier, J.F.; Bachvarova, M.; Plante, M.; Gregoire, J.; Renaud, M.C.; Mes-Masson, A.M.; Paquet, E.R.; Bachvarov, D. Global methylation profiling in serous ovarian cancer is indicative for distinct aberrant DNA methylation signatures associated with tumor aggressiveness and disease progression. Gynecol. Oncol. 2013, 128, 356–363.
- Wang, Z.Q.; Faddaoui, A.; Bachvarova, M.; Plante, M.; Gregoire, J.; Renaud, M.C.; Sebastianelli, A.; Guillemette, C.; Gobeil, S.; Macdonald, E.; et al. BCAT1 expression associates with ovarian cancer progression: Possible implications in altered disease metabolism. Oncotarget 2015, 6, 31522–31543.
- Li, H.; Yu, D.; Li, L.; Xiao, J.; Zhu, Y.; Liu, Y.; Mou, L.; Tian, Y.; Chen, L.; Zhu, F.; et al. BCKDK Promotes Ovarian Cancer Proliferation and Migration by Activating the MEK/ERK Signaling Pathway. J. Oncol. 2022, 2022, 3691635.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
759
Revisions:
3 times
(View History)
Update Date:
08 May 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No