 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cornelia Dietrich | -- | 3755 | 2023-04-28 16:14:21 | | | |
| 2 | Jessie Wu | -268 word(s) | 3487 | 2023-05-04 07:09:24 | | | | |
| 3 | Jessie Wu | Meta information modification | 3487 | 2023-05-04 07:14:51 | | | | |
| 4 | Jessie Wu | Meta information modification | 3487 | 2023-05-04 07:15:35 | | | | |
| 5 | Jessie Wu | Meta information modification | 3487 | 2023-05-04 10:05:20 | | |
Video Upload Options
Ferroptosis is a regulated form of cell death characterized by iron-dependency and increased lipid peroxidation. Initially assumed to be selectively induced in tumour cells, there is increasing evidence that ferroptosis plays an important role in pathophysiology and numerous cell types and tissues. Deregulated ferroptosis has been linked to human diseases, such as neurodegenerative diseases, cardio-vascular disorders, and cancer. Along these lines, ferroptosis is a promising pathway to overcome therapy resistance of cancer cells. It is therefore of utmost importance to understand the cellular signalling pathways and the molecular mechanisms underlying ferroptosis regulation including context-specific effects mediated by the neighbouring cells through cell-cell contacts.
1. Introduction
Cell death can be executed in a regulated or accidental manner. Accidental cell death generally occurs after harsh conditions and proceeds uncontrolled. In contrast, regulated cell death (RCD) is executed by defined cellular programmes and - according to its nature - can be regulated by pharmacological or genetic interventions. When RCD occurs in physiological contexts, e.g. embryonic development or maintenance of tissue homeostasis, it is referred to as programmed cell death [1]. It has long been considered that apoptosis is the sole example of programmed and regulated cell death. Today we know that a myriad of different regulated cell death modalities exist including necroptosis, parthanatos, mitochondrial permeability transition (MPT)-mediated necrosis, or ferroptosis, to name a few [2]. Each cell death pathway seems to be unique in its key elements and execution processes although also mixed forms and cross-talk between these forms may exist. In particular, necroptosis, a well-studied cell death mechanism, as well as ferroptosis have attracted broad attention in the scientific community due to their pathophysiological roles in various human diseases, such as cardiovascular disorders, and neuro-degenerative diseases [3]. However, also the therapeutic potential of these cell death forms has been recognized since they represent a promising alternative option to eliminate apoptosis-resistant cancer cells.
Interestingly, ferroptosis can be modulated by specific signalling pathways, and increasing evidence points to an important role of cell-cell contacts in regulating ferroptosis. Before researchers will summarize and discuss the emerging concept on the role of cell-cell contacts in ferroptosis regulation, researchers first need to provide an overview on what is the current knowledge on the molecular mechanisms and characteristics underlying ferroptosis.
2. Ferroptosis: Characterization and the Core Machinery
Ferroptosis is defined by two characteristic hallmarks, which are excessive lipid peroxidation and iron dependency.
2.1. Lipid Peroxidation
Healthy cells maintain an equilibrium between permanently occurring lipid peroxidation and the detoxification processes thereby keeping the overall level of lipid hydroperoxides low. Ferroptosis is triggered by an imbalance between production and removal of lipid hydroperoxides in favour of their production. In most cases described, ferroptosis is induced by inhibition of detoxification processes. However, it can also be triggered by an increase in lipid peroxidation. Typically, lipid peroxidation of esterified fatty acids, i.e. phospholipids of cellular membranes, but not of free fatty acids, is required for ferroptosis [4]. Two enzymes are of main importance for ferroptosis: the acyl CoA-synthetase long chain family member 4 (ACSL4) which preferentially transfers arachidonic acid to acetyl-CoA and which is required for subsequent incorporation of the fatty acid into phospholipids by lysophosphatidylcholine acyltransferase 3 (LPCAT3) [4][5]. At least in some cell types, such as ovarian cancer cells, neurons, and cardiomyocytes, also formation of ether phospholipids appears to regulate ferroptosis [6].
Polyunsaturated fatty acids (PUFAs, > 1 double bond), for instance alpha-linolenic and arachidonic acid, are prone to peroxidation. Monounsaturated fatty acids (MUFAs, one double bond) are much less susceptible to peroxidation, while saturated fatty acids withstand peroxidation. In accordance, ferroptosis can be attenuated by exogenous addition of MUFAs which are incorporated instead of PUFAs by ACSL3 into the phospholipids of the plasma membrane [7].
In principal, there are two ways of how lipid peroxidation is triggered, i.e. non-enzymatically or enzymatically (Figure 1). Non-enzymatic lipid peroxidation is driven by radicals. Free electrons might emerge from leakage of electrons from the electron transport chain (ETC) in the inner mitochondrial membrane or might be released by enzymatic systems, such as NADPH oxidases (NOX), e.g. NOX4 or NOX2 [8][9][10]. Such electrons can react with oxygen to form superoxide which is converted to hydrogen peroxide by the superoxide dismutase. Under catalysis of ferrous iron (Fe2+), hydrogen peroxide reacts to the hydroxyl radical, a process known as the Fenton reaction. The hydroxyl radical itself, or rather an iron(IV)-oxo (ferryl) intermediate formed within the reaction, are highly reactive and may abstract a hydrogen from the bisallylic site of a PUFA [11][12]. This creates a lipid radical, known as initiation, and this lipid radical will lead to a chain reaction, referred to as propagation. The reaction will be terminated by an antioxidant or when two radicals meet [13]. In addition, fragmentation of the lipid hydroperoxides leads to formation of secondary products, the most important are 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA). 4-HNE is supposed to be the most toxic, MDA appears to be the most mutagenic product of lipid peroxidation, since it can efficiently attack DNA [14].
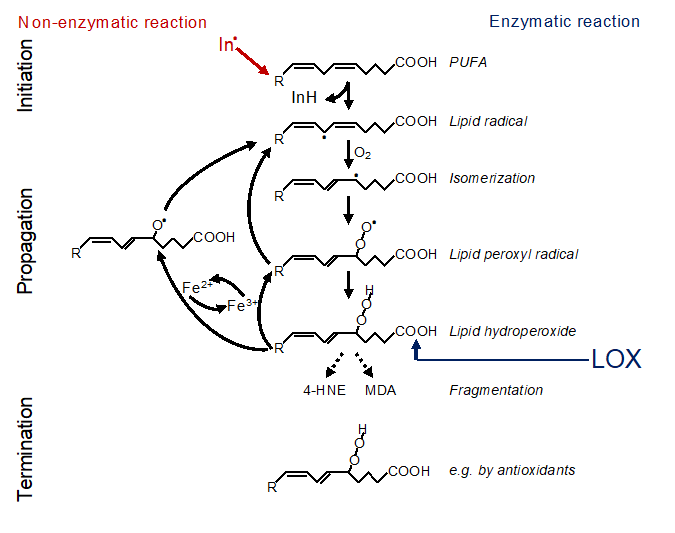
Figure 1. Mechanism of lipid peroxidation. Left: Radical initiation is carried out by a radical (initiator = In.) abstracting a hydrogen from a bisallylic site, i.e. the carbon between two double bonds, of a PUFA thereby creating a carbon-centered lipid radical (L*). Due to the adjacent double bonds, the radical can delocalize its electron system resulting in a resonance-stabilized conformation. In a fast reaction, oxygen is then added, and a lipid peroxyl radical (LOO*) is formed. By abstracting a hydrogen from an adjacent PUFA, the lipid peroxyl radical itself reacts to a lipid hydroperoxide (LOOH) and generates the next carbon-centered radical (L*) thereby propagating the chain reaction. The lipid hydroperoxide (LOOH) is readily decomposed, either by ferrous iron (Fe2+) to the lipid alkoxyl radical (LO*), or by ferric iron (Fe3+) back to the lipid peroxyl radical (LOO*). Both radicals can fuel the chain reaction. The reaction is terminated when two radicals meet each other forming a lipid dimer (L-L) or a peroxide-bridged lipid dimer (LOOL) (not shown), or when a radical meets a radical trapping agent, for instance coenzyme Q10 or alpha-tocopherol (Vitamin E) creating a lipid hydroperoxide (LOOH). Right: Alternatively, lipid peroxidation is carried out in a controlled manner by lipoxygenases (LOX). Although initiating the process at the level of lipid hydroperoxide formation, the lipid hydroperoxide will be decomposed by ferrous or ferric iron to lipid alkoxyl and peroxyl radicals, respectively, and the vicious circle of radical formation will be fueled as described above. Hence, the initial enzymatic reaction is switched in a second step to a radical process [15].
Recently, two groups independently discovered an important role of cytochrome P450 oxidoreductase (POR) in lipid peroxidation although the two molecular mechanisms proposed are rather contradictory [16][17]. While Yan and coworkers demonstrate an elevation of hydrogen peroxide and, in line, inhibition of lipid peroxidation by catalase, Zou and coworkers could not modulate sensitivity towards ferroptosis by expression of catalase and the authors postulate POR-mediated formation of an iron-oxo species [17]. Thus, additional research efforts are required to solve these open questions.
Oxidation of lipids can also be started in a controlled manner by enzymatic processes. The most important enzymes that are capable to initiate lipid oxidation belong to the family of lipoxygenases (LOXs), which dioxygenate PUFAs to the lipid hydroperoxides (Figure 1). Cyclooxygenases are generally not involved in ferroptosis [18]. However, upregulation of COX2 (also known as PTGS2) is frequently observed and can be used as a marker for ferroptosis [19]. A potential role of cytochrome P450 in enzymatic lipid peroxidation during ferroptosis remains to be explored.
There are six isoforms of LOXs in humans: 5-LOX (gene ALOX5), 15-LOX1 (also 12/15-LOX, gene ALOX15), 15-LOX2 (gene ALOX15b), pl12-LOX (gene ALOX12), 12R-LOX (gene ALOX12B), and eLOX3 (gene ALOXE3) [20]. However, the specific role of LOX enzymes in ferroptosis is still a mystery and is highly context specific. Analysis is hampered by the fact that most of the LOX-specific inhibitors are also radical trapping agents and hence unsuitable to discriminate between a LOX-dependent or LOX-independent mechanism [21]. LOX enzymes appear to be required for ferroptosis in some, but not all circumstances and might be dependent on the cellular context and the expression of specific LOX isoforms and / or the ferroptotic stimulus, which can trigger different routes to ferroptosis.
2.2. Iron Dependency
Iron homeostasis in a cell is tightly regulated by import, storage and export (for detailed overview see [22][23][24]). The precise role of iron in ferroptosis has not been elucidated so far [25]. The conclusion that iron is absolutely required for ferroptosis comes from the observation that iron chelators inhibit ferroptosis [8][26][27]. In accordance, decreasing the LIP, e.g. by increased expression of ferritin, suppresses ferroptosis [28]. Vice versa, intracellular increase in LIP, e.g. by lysosomal destabilization or ferritinophagy, supports ferroptosis [29]. In principle, three possibilities can be considered to explain this finding: (i) iron is present as cofactor in the active centres of enzymes, (ii) iron acts as an inducer of lipid peroxidation, (iii) iron functions as a mediator of propagation.
2.3. Detoxification Processes
Three main antioxidant mechanisms regulating ferroptosis have been revealed, (i) the GPX4/GSH, (ii) the FSP1/CoQ10, and (iii) the GCH1/BH4 system. Among the various isoforms, the selenoprotein GPX4 is the only one which - with its cofactor GSH - directly detoxifies lipid hydroperoxides by promoting the reaction to lipid alcohols [30]. The synthesis of GSH is dependent on the availability of cysteine, which is derived from its precursor molecule cystine. Cystine is taken up by specific amino acid transporters, an important one is system xc-. The importance of the GPX4/GSH detoxification mechanism to prevent ferroptosis is demonstrated by the fact, that genetic deletion or pharmacological inhibition of GPX4 as well as downregulation of its cofactor GSH by any means will cause an accumulation of lipid hydroperoxides and results in ferroptosis [8][19][31].
A second important detoxification system, which acts in parallel to GPX4/GSH, is the FSP1/CoQ10 system [32][33]. CoQ10 is well-known for its electron shuttling role in the ETC in the mitochondria, but it also plays an essential role as a lipid radical trapping agent. Mechanistically, the reduced form of CoQ10 (CoQ10H2) can successively trap two electrons thereby preventing radical initiation as well as propagation of lipid peroxidation by scavenging lipid peroxyl radicals. A second function is to regenerate alpha-tocopherol from the tocopheroxyl radical, another important physiological lipid antioxidant [34]. However, genetic deletion or pharmacological inhibition of FSP1 is not sufficient to induce ferroptosis, but it sensitizes to ferroptosis triggered by inhibition of GPX4 or GSH synthesis.
A third independent protective pathway has been identified involving the GTP cyclohydrolase 1 (GCH 1), which is responsible for the synthesis of tetrahydrobiopterin (BH4) [35]. Beside other functions, BH4 acts as a direct radical scavenger [12]. It can interfere with lipid peroxidation at the level of initiation and propagation [36]. Since BH4 is also required for CoQ10 synthesis, it additionally acts by increasing CoQ10 levels. For unknown reasons, BH4 specifically prevents degradation of phospholipids containing two PUFAs. To note, BH4 is a diffusible molecule and therefore able to protect surrounding cells [35]. However, deletion of GCH1 is not sufficient to trigger spontaneously induction of ferroptosis [35].
2.4. Inducers of Ferroptosis
In principle, ferroptosis can be induced by the canonical pathway, which is inhibition of the GPX4/GSH defence pathway, or, non-canonically, by direct stimulation of lipid peroxidation (for detailed overview see [37]). Canonical inducers comprise class I to class III ferroptosis inducers (FIN).
2.5. Inhibitors of Ferroptosis
Ferroptosis is driven by iron-dependent lipid peroxidation, which takes place in phospholipids, and – independent from the initial trigger – is carried out in a second step by a radical chain reaction. In consequence, ferroptosis induction can be counterbalanced by antioxidants, and, in addition, can be pharmacologically attacked at any of these levels. Among the various compounds known to inhibit ferroptosis, the most typical ones need to be mentioned in the following. As already outlined above, iron-chelators, such as deferoxamine and ciclopirox, are powerful inhibitors of ferroptosis [8]. Lipid peroxidation can efficiently be blocked by the radical trapping agents ferrostatin 1 and liproxstatin 1 as well as the endogenous antioxidant alpha-tocopherol (Vitamin E) [8][31][38][39]. Increasing the cellular antioxidative activity by idebenone, a synthetic and soluble analogue of CoQ10, also attenuates ferroptosis [32]. It has further been demonstrated that thiazolidinediones decrease ferroptosis by blocking ACSL4. In general, ferroptosis inhibition could be of relevance in specific pathophysiological settings including neurodegenerative diseases, which may also include ferroptotic cell death. For a more comprehensive description of inhibitors, the reader is referred to some recent overview articles [40][41][42][43].
3. Regulation of Ferroptosis by Cell-Cell Contacts
A role of adherens junctions and E-cadherin in growth control and differentiation
E-cadherin is important to establish epithelial cell differentiation and cell polarity, which is critical for the function of epithelial cell layers. Furthermore, E-cadherin prevents epithelial-mesenchymal transition (EMT), a biological process essentially contributing to the dedifferentiation and carcinogenesis of epithelial cells. During EMT, epithelial cells lose their cell contacts, their characteristic cobblestone morphology, and their polarity to acquire a mesenchymal phenotype supporting migration and metastasis (for detailed description see [44][45][46]). Three core transcription factors are involved in EMT, i.e. SNAI1/SNAI2 (formerly Snail/Slug), TWIST, and ZEB1/2, which repress epithelial markers including E-cadherin transcription. As a consequence, mesenchymal markers, such as fibronectin, vimentin and N-cadherin are upregulated [47].
How do cadherins crosstalk to the Hippo pathway?
E-cadherin regulates the Hippo pathway at multiple levels (Figure 2).
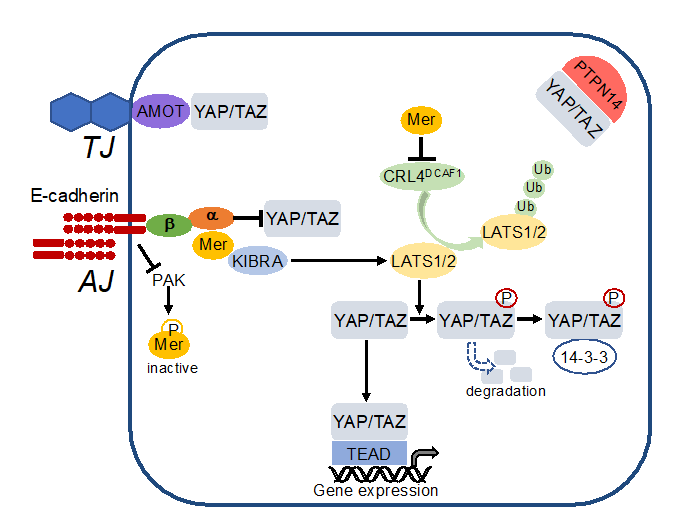
Figure 2. Cell-cell contacts turn on the Hippo pathway. E-cadherin inactivates p21-activated kinase (PAK) thereby blocking phosphorylation and inactivation of merlin (Mer). Active merlin associates with KIBRA, activates LATS1/2 which phosphorylates YAP/TAZ. This leads to cytosolic sequestration and degradation of YAP/TAZ. In addition, merlin inhibits activity of the E3 ubiquitin ligase CRL4DCAF1 thereby blocking degradation of LATS1/2. YAP/TAZ is also directly sequestered to adherens junctions (AJ) by a-catenin (a) which is associated via b-catenin (b) to E-cadherin. Moreover, YAP/TAZ is bound to tight junctions (TJ) via angiomotin (AMOT) and sequestered in the cytoplasm by the protein tyrosine phosphatase nonreceptor (PTPN)14.
4. Cell-Cell Contacts as Regulators of Ferroptosis
Interestingly, recent observations have demonstrated a significant role of cell-cell contacts, in particular adherens junctions, in controlling ferroptosis. This fascinating link was not urgently expected, since it is widely accepted that especially E-cadherin signalling supports apoptosis and, vice versa, EMT protects cancer cells against apoptosis correlating with a poor clinical prognosis [48][49][50].
Observations from the early 1960s indicated that cells lose sensitivity against cysteine depletion with increased cell-density [51]. Later, it has been shown that mouse embryonic fibroblasts with inducible GPX4 depletion are protected against cell death at high cell densities [18], suggesting a role of cell-cell contacts on ferroptosis sensitivity. However, a specific role of cell-cell contacts in regulating ferroptosis has been shown only recently. Using several murine and human cell lines, researchers demonstrated that cell-cell contacts protect against ferroptosis induced by erastin or the organic tertiary-butyl hydroperoxide (t-BuOOH) [52]. Basal as well as induced lipid peroxidation was strongly reduced in confluent cells. Notably, this protective mechanism occurs independently from contact-inhibition since the cells were seeded below their saturation density thus ensuring cell-cell contact formation but excluding growth inhibition and quiescence at the time of treatment. Interestingly, cell-cell contacts also provide resistance against t-BuOOH-mediated loss of mitochondrial membrane potential and DNA double-strand break formation, but they failed to prevent t-BuOOH-triggered replication blockade or the formation of the oxidative base lesion 8-oxo-dG. These findings indicate that cell-cell contacts confer a broader, selective protection against cellular oxidative stress. Moreover, resistance was also observed in response to treatment with hydrogen peroxide, methyl methanesulfonate or UV-C [52]. Hence, it is reasonable to conclude that protection by cell-cell contacts is more widespread than hitherto expected.
Elegant work by Wu and coworkers identified the Hippo pathway as a crucial mediator of resistance against ferroptosis [53] (Figure 3). Using a colon carcinoma cell line and several genetic gain-of-function as well as loss-of-function analyses, the authors demonstrate that, in confluent cultures, E-cadherin leads to nuclear extrusion of YAP via PAK-merlin-LATS1/2 signalling (see above). Inactivation of YAP results in transcriptional downregulation not only of canonical YAP target genes, but also of the iron-controlling TfR1 (encoded by the TFRC gene) and of ACSL4. A similar protective pathway seems to be induced by N-cadherin in fibroblasts and mesothelioma cells [53]. However, since co-overexpression of TfR1 and ACSL4 could not fully restore ferroptosis in these cells, additional ferroptosis-regulating target genes have to be postulated [53][54]. Moreover, very recently, it has been shown that YAP may regulate ferroptosis via induction of the E3 ubiquitin ligase SKP2, possibly by a positive feedback loop via nonproteolytic polyubiquitination thereby maybe leading to enhanced YAP-TEAD interaction [55]. However, a proteolytic role of SKP2 on an unidentified substrate controlling ferroptosis cannot be formally excluded yet.
It has also been demonstrated that the YAP paralogue TAZ impacts on ferroptosis. In renal cell carcinoma, TAZ stimulates the expression of the epithelial membrane protein 1 (EMP1), which in turn activates p38 MAPK and finally induces the accumulation of NOX4 [9]. In ovarian carcinoma cell lines, TAZ upregulates NOX2 expression via increased induction of ANGPTL4 (angiopoetin-like protein 4) [10]. Although in both publications, ferroptosis was inhibited at high cell density and a regulatory function of TAZ/NOX4 and TAZ/NOX2, respectively, at high cell density is likely, the authors did not demonstrate downregulation of these target genes at cell confluency. Furthermore, the upstream signalling of this effect remains to be elucidated.
These observations raise the interesting question of whether a mesenchymal phenotype of carcinoma cells does also increase ferroptosis sensitivity and, if so, what would be the underlying mechanism. Indeed, recurrent breast cancer cells having acquired a mesenchymal phenotype show upregulation of the receptor for collagen I discoidin domain receptor tyrosine kinase 2 (DDR2), which causes activation of YAP/TAZ and increases ferroptosis susceptibility [56]. The authors also provide evidence that induction of EMT in epithelial breast cancer cells by overexpressing TWIST or SNAI1 leads to similar results [56]. In accordance, a reciprocal association between differentiation and sensitivity to ferroptosis has been shown in a panel of head and neck cancer cells with either epithelial or mesenchymal phenotype [57]. Inducing EMT by different means, such as downregulation of E-cadherin, overexpression of ZEB1, or TGFb1-treatment (which induces SNAI1, TWIST and ZEB1/2), increases the cellular sensitivity to ferroptosis. Vice versa, activation of mesenchymal-epithelial transition by restoring E-cadherin expression, inactivation of ZEB1, or epigenetic reprogramming leads to ferroptosis resistance. A similar correlation was observed in pancreatic cancer cells in response to TGFb1-treatment [58]. In a variety of therapy-resistant cancer cells exhibiting an intrinsic, acquired or induced mesenchymal state, expression of ZEB1 was strongly associated with their susceptibility to ferroptosis triggered by GPX4 inhibition [59]. In contrast, induction of EMT by overexpression of SNAI1 or TWIST did not consistently lead to sensitization [59]. A possible explanation might be provided by the fact that ZEB1 is a lipogenic factor and thereby alters cellular lipid metabolism.
A different mechanism is provided by increased expression of metadherin. Metadherin (also known as astrocyte elevated gene 1 or LYRIC) is a tumour associated antigen, which promotes EMT including downregulation of E-cadherin by activating multiple pathways, such as Wnt/b-catenin, MAPK and PI3K/AKT signalling [60]. Interestingly, metadherin also functions as an RNA binding protein leading to downregulation of GPX4 and SLC3A2, the system xc- dimerization partner, and consequently to an elevated vulnerability to ferroptosis inducers [61].
These observations argue against the hypothesis that EMT is per se sufficient to increase ferroptosis susceptibility. Indeed, ferroptosis sensitivity might be counterbalanced by the AKT signalling pathway (Figure 3). In gallbladder cancer cells, downregulation of the histone deacetylase sirtuin 3 leads to typical features of EMT and increased phosphorylation of AKT (at ser473). However, they are protected against ferroptosis by AKT-mediated reduction of ACSL4 [62]. Ferroptosis resistance might also be a result of activating PI3K mutations or loss of PTEN function leading to sustained mTORC1 activity via the PI3K-AKT-mTOR pathway [63]. Sustained mTORC1 activity leads to increased transcriptional activity of the sterol regulatory element-binding protein 1 (SREBP1) resulting in increased expression of the stearoyl CoA desaturase 1 (SCD1) [63]. SCD1 is an enzyme which converts unsaturated fatty acids to MUFAs. Hence, it is likely that MUFAs are incorporated instead of PUFAs into the plasma membrane thereby decreasing the rate of lipid peroxidation in response to ferroptotic triggers [7]. Furthermore, SCD1 induces BH4 synthesis which also contributes to ferroptosis resistance [64].
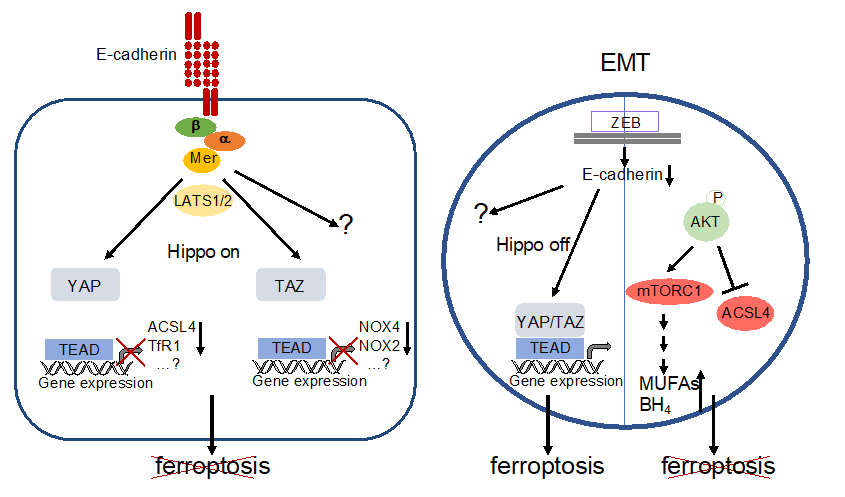
Figure 3. Regulation of ferroptosis by E-cadherin and EMT. Left: E-cadherin activates the Hippo pathway thereby inactivating YAP- and TAZ-activity. This leads directly or indirectly to decreased expression of proteins that are relevant for ferroptosis (note that decreased levels of NOX4 and NOX2 are due to reduced expression of EMP1 and ANGPTL4, resp., see text for details). However, involvement of additional genes and / or signalling pathways is likely. Ferroptosis is blocked. Right: EMT, for instance triggered by activation of ZEB, leads to a loss of E-cadherin expression and inhibition of the Hippo pathway. Although the downstream targets have not been elucidated so far, YAP/TAZ activation supports ferroptosis. Additional pathways remain to be elucidated. However, this can be counterbalanced by phosphorylation of AKT (or, possibly, other mechanism). Activated AKT may lead to a decreased level of ACSL4 as well as increased formation of MUFAs and BH4 via mTORC1 stimulation. Ferroptosis is prevented.
References
- Galluzzi, L.; Kepp, O.; Krautwald, S.; Kroemer, G.; Linkermann, A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol 2014, 35, 24-32, doi:10.1016/j.semcdb.2014.02.006.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018, 25, 486-541, doi:10.1038/s41418-017-0012-4.
- Conrad, M.; Lorenz, S.M.; Proneth, B. Targeting Ferroptosis: New Hope for As-Yet-Incurable Diseases. Trends Mol Med 2021, 27, 113-122, doi:10.1016/j.molmed.2020.08.010.
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 2017, 13, 91-98, doi:10.1038/nchembio.2239.
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol 2015, 10, 1604-1609, doi:10.1021/acschembio.5b00245.
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603-608, doi:10.1038/s41586-020-2732-8.
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol 2019, 26, 420-432 e429, doi:10.1016/j.chembiol.2018.11.016.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060-1072, doi:10.1016/j.cell.2012.03.042.
- Yang, W.H.; Ding, C.C.; Sun, T.; Rupprecht, G.; Lin, C.C.; Hsu, D.; Chi, J.T. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep 2019, 28, 2501-2508 e2504, doi:10.1016/j.celrep.2019.07.107.
- Yang, W.H.; Huang, Z.; Wu, J.; Ding, C.C.; Murphy, S.K.; Chi, J.T. A TAZ-ANGPTL4-NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol Cancer Res 2020, 18, 79-90, doi:10.1158/1541-7786.MCR-19-0691.
- Cheng, Z.; Li, Y. What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: an update. Chem Rev 2007, 107, 748-766, doi:10.1021/cr040077w.
- Prescott, C.; Bottle, S.E. Biological Relevance of Free Radicals and Nitroxides. Cell Biochem Biophys 2017, 75, 227-240, doi:10.1007/s12013-016-0759-0.
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev 2011, 111, 5944-5972, doi:10.1021/cr200084z.
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014, 2014, 360438, doi:10.1155/2014/360438.
- Spiteller, G. The relation of lipid peroxidation processes with atherogenesis: a new theory on atherogenesis. Mol Nutr Food Res 2005, 49, 999-1013, doi:10.1002/mnfr.200500055.
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol Cell 2021, 81, 355-369 e310, doi:10.1016/j.molcel.2020.11.024.
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol 2020, 16, 302-309, doi:10.1038/s41589-020-0472-6.
- Seiler, A.; Schneider, M.; Forster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Radmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 2008, 8, 237-248, doi:10.1016/j.cmet.2008.07.005.
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317-331, doi:10.1016/j.cell.2013.12.010.
- Kuhn, H.; Banthiya, S.; van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta 2015, 1851, 308-330, doi:10.1016/j.bbalip.2014.10.002.
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent Sci 2018, 4, 387-396, doi:10.1021/acscentsci.7b00589.
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am J Clin Nutr 2017, 106, 1559S-1566S, doi:10.3945/ajcn.117.155804.
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: current questions. Expert Rev Hematol 2017, 10, 65-79, doi:10.1080/17474086.2016.1268047.
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the "Iron Maiden" Cell Death. Cells 2020, 9, doi:10.3390/cells9061505.
- Doll, S.; Conrad, M. Iron and ferroptosis: A still ill-defined liaison. IUBMB Life 2017, 69, 423-434, doi:10.1002/iub.1616.
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864-868, doi:10.1038/nature05859.
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 2008, 15, 234-245, doi:10.1016/j.chembiol.2008.02.010.
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front Aging Neurosci 2016, 8, 308, doi:10.3389/fnagi.2016.00308.
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J 2016, 473, 769-777, doi:10.1042/BJ20150658.
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim Biophys Acta 2013, 1830, 3289-3303, doi:10.1016/j.bbagen.2012.11.020.
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014, 16, 1180-1191, doi:10.1038/ncb3064.
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688-692, doi:10.1038/s41586-019-1705-2.
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693-698, doi:10.1038/s41586-019-1707-0.
- Bentinger, M.; Brismar, K.; Dallner, G. The antioxidant role of coenzyme Q. Mitochondrion 2007, 7 Suppl, S41-50, doi:10.1016/j.mito.2007.02.006.
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Muller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kossl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci 2020, 6, 41-53, doi:10.1021/acscentsci.9b01063.
- Soule, B.P.; Hyodo, F.; Matsumoto, K.; Simone, N.L.; Cook, J.A.; Krishna, M.C.; Mitchell, J.B. The chemistry and biology of nitroxide compounds. Free Radic Biol Med 2007, 42, 1632-1650, doi:10.1016/j.freeradbiomed.2007.02.030.
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830-849, doi:10.1016/j.ccell.2019.04.002.
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 2014, 136, 4551-4556, doi:10.1021/ja411006a.
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent Sci 2017, 3, 232-243, doi:10.1021/acscentsci.7b00028.
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat Chem Biol 2019, 15, 1137-1147, doi:10.1038/s41589-019-0408-1.
- Stockwell, B.R.; Jiang, X. The Chemistry and Biology of Ferroptosis. Cell Chem Biol 2020, 27, 365-375, doi:10.1016/j.chembiol.2020.03.013.
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: process and function. Cell Death Differ 2016, 23, 369-379, doi:10.1038/cdd.2015.158.
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273-285, doi:10.1016/j.cell.2017.09.021.
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843-850, doi:10.1038/nature03319.
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 2019, 20, 69-84, doi:10.1038/s41580-018-0080-4.
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461-1473, doi:10.1038/onc.2016.304.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014, 15, 178-196, doi:10.1038/nrm3758.
- Wong, S.H.M.; Fang, C.M.; Chuah, L.H.; Leong, C.O.; Ngai, S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit Rev Oncol Hematol 2018, 121, 11-22, doi:10.1016/j.critrevonc.2017.11.010.
- Na, T.Y.; Schecterson, L.; Mendonsa, A.M.; Gumbiner, B.M. The functional activity of E-cadherin controls tumor cell metastasis at multiple steps. Proc Natl Acad Sci U S A 2020, 117, 5931-5937, doi:10.1073/pnas.1918167117.
- Lu, M.; Marsters, S.; Ye, X.; Luis, E.; Gonzalez, L.; Ashkenazi, A. E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Mol Cell 2014, 54, 987-998, doi:10.1016/j.molcel.2014.04.029.
- Eagle, H. The Sustained Growth of Human and Animal Cells in a Protein-Free Environment. Proc Natl Acad Sci U S A 1960, 46, 427-432, doi:10.1073/pnas.46.4.427.
- Wenz, C.; Faust, D.; Linz, B.; Turmann, C.; Nikolova, T.; Dietrich, C. Cell-cell contacts protect against t-BuOOH-induced cellular damage and ferroptosis in vitro. Arch Toxicol 2019, 93, 1265-1279, doi:10.1007/s00204-019-02413-w.
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, 402-406, doi:10.1038/s41586-019-1426-6.
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Publisher Correction: Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, E20, doi:10.1038/s41586-019-1480-0.
- Yang, W.H.; Lin, C.C.; Wu, J.; Chao, P.Y.; Chen, K.; Chen, P.H.; Chi, J.T. The Hippo Pathway Effector YAP Promotes Ferroptosis via the E3 Ligase SKP2. Mol Cancer Res 2021, doi:10.1158/1541-7786.MCR-20-0534.
- Lin, C.C.; Yang, W.H.; Lin, Y.T.; Tang, X.; Chen, P.H.; Ding, C.C.; Qu, D.C.; Alvarez, J.V.; Chi, J.T. DDR2 upregulation confers ferroptosis susceptibility of recurrent breast tumors through the Hippo pathway. Oncogene 2021, 40, 2018-2034, doi:10.1038/s41388-021-01676-x.
- Lee, J.; You, J.H.; Kim, M.S.; Roh, J.L. Epigenetic reprogramming of epithelial-mesenchymal transition promotes ferroptosis of head and neck cancer. Redox Biol 2020, 37, 101697, doi:10.1016/j.redox.2020.101697.
- Daher, B.; Parks, S.K.; Durivault, J.; Cormerais, Y.; Baidarjad, H.; Tambutte, E.; Pouyssegur, J.; Vucetic, M. Genetic Ablation of the Cystine Transporter xCT in PDAC Cells Inhibits mTORC1, Growth, Survival, and Tumor Formation via Nutrient and Oxidative Stresses. Cancer Res 2019, 79, 3877-3890, doi:10.1158/0008-5472.CAN-18-3855.
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453-457, doi:10.1038/nature23007.
- Dhiman, G.; Srivastava, N.; Goyal, M.; Rakha, E.; Lothion-Roy, J.; Mongan, N.P.; Miftakhova, R.R.; Khaiboullina, S.F.; Rizvanov, A.A.; Baranwal, M. Metadherin: A Therapeutic Target in Multiple Cancers. Front Oncol 2019, 9, 349, doi:10.3389/fonc.2019.00349.
- Bi, J.; Yang, S.; Li, L.; Dai, Q.; Borcherding, N.; Wagner, B.A.; Buettner, G.R.; Spitz, D.R.; Leslie, K.K.; Zhang, J.; et al. Metadherin enhances vulnerability of cancer cells to ferroptosis. Cell Death Dis 2019, 10, 682, doi:10.1038/s41419-019-1897-2.
- Liu, L.; Li, Y.; Cao, D.; Qiu, S.; Li, Y.; Jiang, C.; Bian, R.; Yang, Y.; Li, L.; Li, X.; et al. SIRT3 inhibits gallbladder cancer by induction of AKT-dependent ferroptosis and blockade of epithelial-mesenchymal transition. Cancer Lett 2021, 510, 93-104, doi:10.1016/j.canlet.2021.04.007.
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci U S A 2020, 117, 31189-31197, doi:10.1073/pnas.2017152117.
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res 2019, 79, 5355-5366, doi:10.1158/0008-5472.CAN-19-0369.

