 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chaiyavat Chaiyasut | -- | 4630 | 2023-04-27 11:46:49 | | | |
| 2 | Rita Xu | Meta information modification | 4630 | 2023-04-27 11:53:53 | | |
Video Upload Options
Neuromyelitis optica (NMO) is a rare autoimmune inflammatory disorder affecting the central nervous system (CNS), specifically the optic nerve and the spinal cord, with severe clinical manifestations, including optic neuritis (ON) and transverse myelitis. Initially, NMO was wrongly understood as a condition related to multiple sclerosis (MS), due to a few similar clinical and radiological features, until the discovery of the AQP4 antibody (NMO-IgG/AQP4-ab).NMO was expanded to NMO spectrum disorder (NMOSD) because of its varied clinical phenotypes. NMOSD is characterized by the activation of the complement cascade, granulocyte, eosinophil and lymphocyte infiltration, oligodendrocyte, and astrocyte injury, demyelination, and neuronal loss. The studies on gut microbiota and NMOSD explain the correlation between gut dysbiosis and NMOSD by signifying the abundance of pathogenic bacteria and reduction in commensal organisms, which cause abnormal metabolism and metabolic signals in the pathogenesis of autoimmune diseases such as NMOSD.
1. Introduction
2. Molecular Mechanism of NMOSD
2.1. AQP4 Dependent Pathology
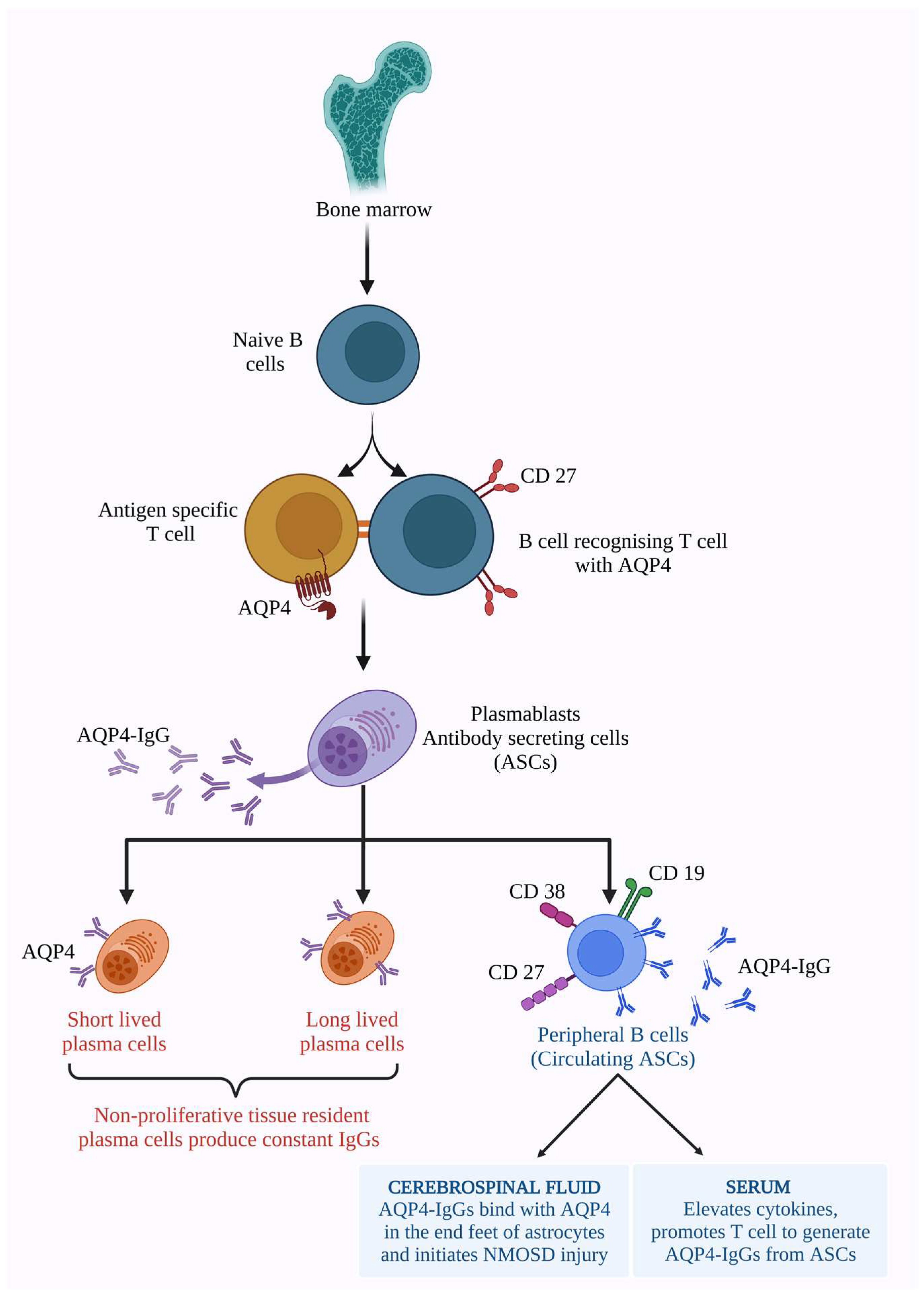
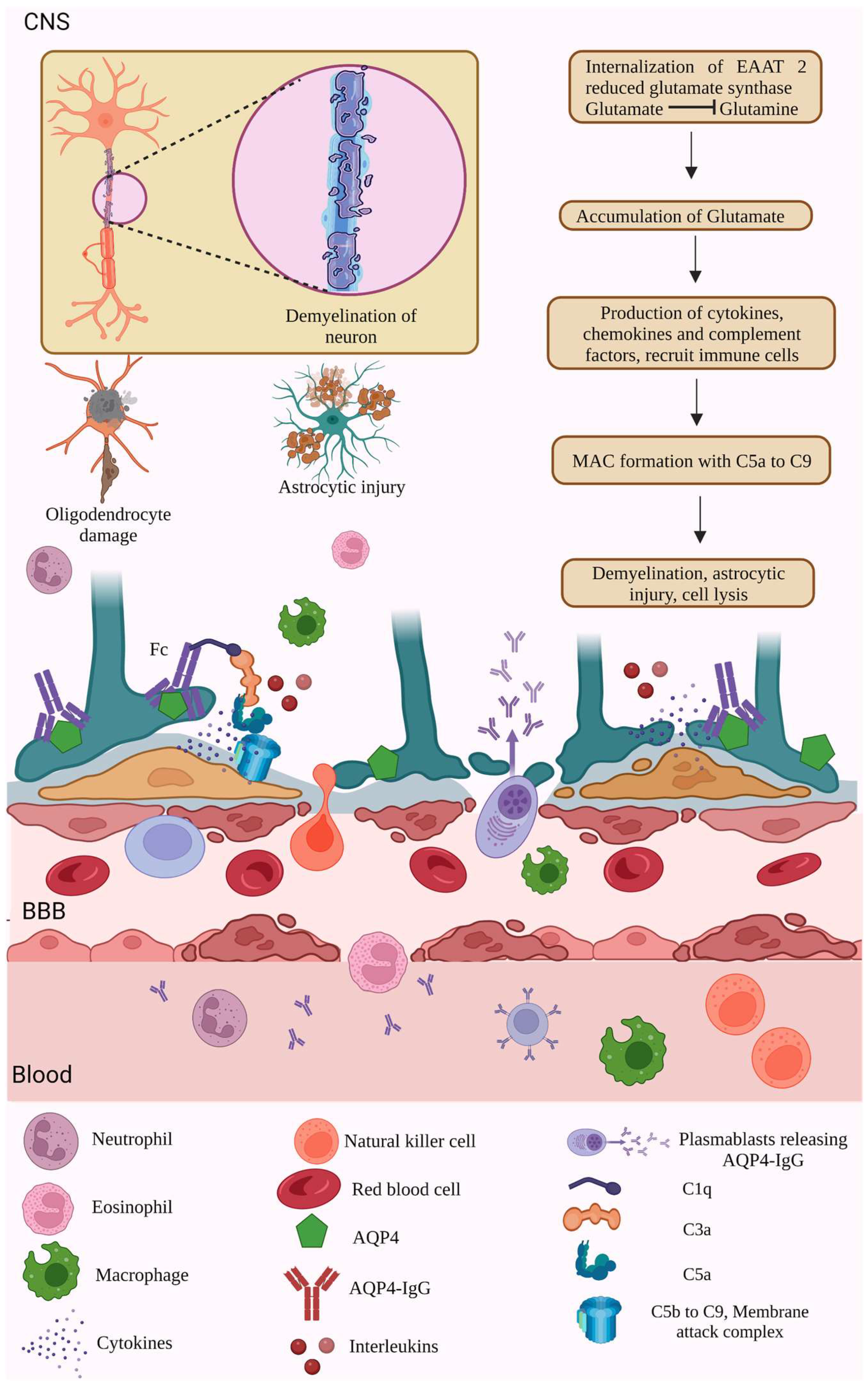
2.2. MOG-IgG-Dependent Pathology
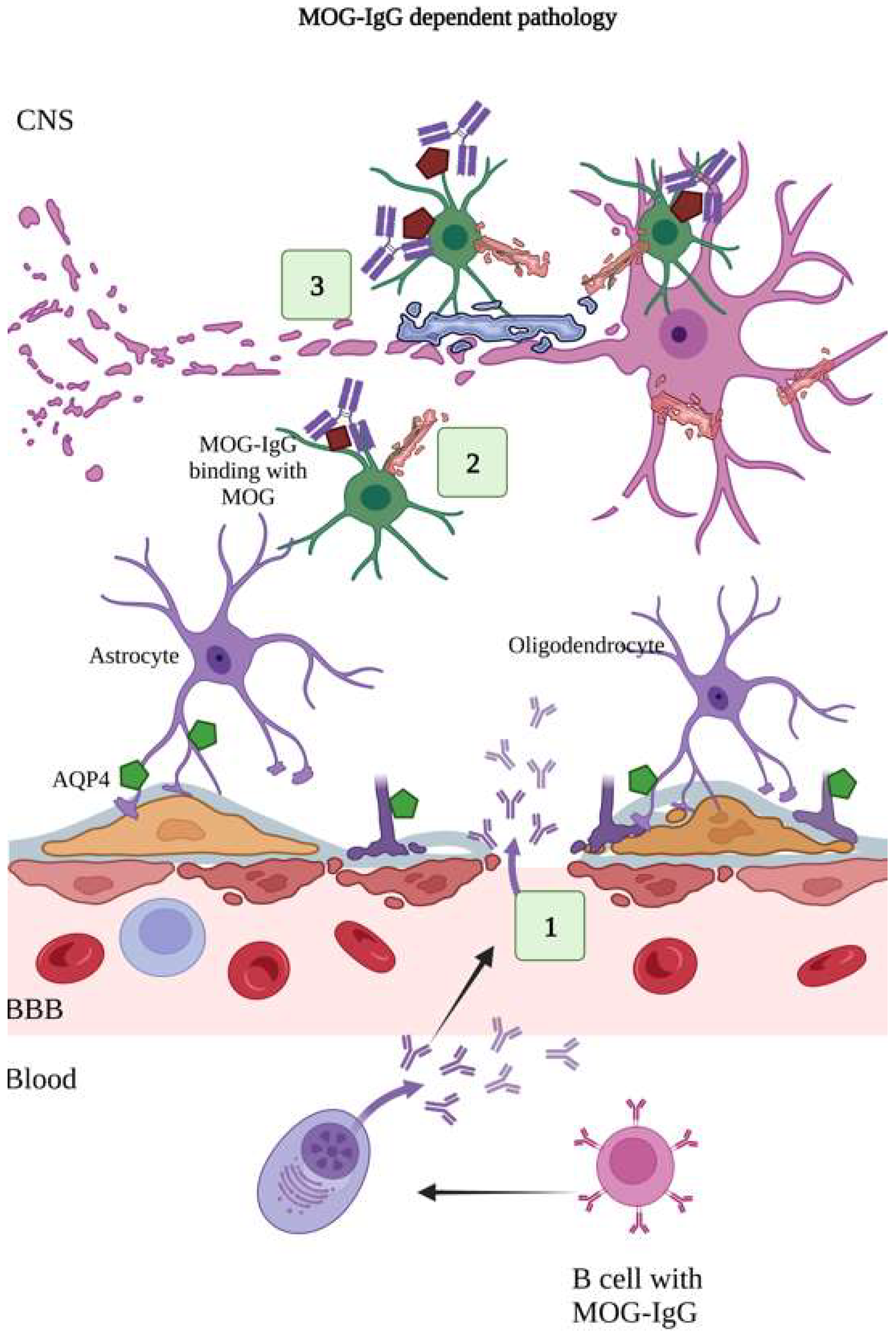
2.3. IL-6 Pathophysiology
2.4. Complement-Mediated Pathology
2.5. Involvement of Gut Microbiome and NMOSD
References
- Anaya, J.M.; Ramirez-Santana, C.; Alzate, M.A.; Molano-Gonzalez, N.; Rojas-Villarraga, A. The Autoimmune Ecology. Front. Immunol. 2016, 7, 139.
- Acosta-Ampudia, Y.; Monsalve, D.M.; Ramírez-Santana, C. Identifying the culprits in neurological autoimmune diseases. J. Transl. Autoimmun. 2019, 2, 100015.
- Pierrot-Deseilligny, C.; Souberbielle, J.C. Vitamin D and multiple sclerosis: An update. Mult. Scler. Relat. Disord. 2017, 14, 35–45.
- Rubin, D.B.; Batra, A.; Vaitkevicius, H.; Vodopivec, I. Autoimmune Neurologic Disorders. Am. J. Med. 2018, 131, 226–236.
- Kawachi, I.; Lassmann, H. Neurodegeneration in multiple sclerosis and neuromyelitis optica. J. Neurol. Neurosurg. Psychiatry 2017, 88, 137–145.
- Guo, Y.; Lennon, V.A.; Parisi, J.E.; Popescu, B.; Vasquez, C.; Pittock, S.J.; Howe, C.L.; Lucchinetti, C.F. Spectrum of sub lytic astrocytopathy in neuromyelitis optica. Brain 2022, 145, 1379–1390.
- Carnero Contentti, E.; Correale, J. Neuromyelitis optica spectrum disorders: From pathophysiology to therapeutic strategies. J. Neuroinflamm. 2021, 18, 208.
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin 4 and Neuromyelitis Optica. Lancet Neurol. 2012, 11, 535–544.
- Chan, K.H.; Lee, C.Y. Treatment of Neuromyelitis Optica Spectrum Disorders. Int. J. Mol. Sci. 2021, 22, 8638.
- Wingerchuk, D.M. Neuromyelitis optica: New findings on pathogenesis. Int. Rev. Neurobiol. 2007, 79, 665–688.
- Pereira, W.L.; Reiche, E.M.; Kallaur, A.P.; Kaimen-Maciel, D.R. Epidemiological, clinical, and immunological characteristics of neuromyelitis optica: A review. J. Neurol Sci. 2015, 355, 7–17.
- Kowarik, M.C.; Dzieciatkowska, M.; Wemlinger, S.; Ritchie, A.M.; Hemmer, B.; Owens, G.P.; Bennett, J.L. The cerebrospinal fluid immunoglobulin transcriptome and proteome in neuromyelitis optica reveals central nervous system-specific B cell populations. J. Neuroinflamm. 2015, 12, 19.
- Lucchinetti, C.F.; Guo, Y.; Popescu, B.F.G.; Fujihara, K.; Itoyama, Y.; Misu, T. The Pathology of an Autoimmune Astrocytopathy: Lessons Learned from Neuromyelitis Optica. Brain. Pathol. 2014, 24, 83–97.
- Pittock, S.J.; Lucchinetti, C.F. Neuromyelitis optica and the evolving spectrum of autoimmune aquaporin-4 channelopathies: A decade later. Ann. N. Y. Acad. Sci. 2016, 1366, 20–39.
- Popescu, B.; Lennon, V.A.; Parisi, J.E.; Howe, C.L.; Weigand, S.D.; Cabrera-Gomez, J.A.; Newell, K.; Mandler, R.N.; Pittock, S.J.; Weinshenker, B.G.; et al. Neuromyelitis optica unique area postrema lesions: Nausea, vomiting, and pathogenic implications. Neurology 2011, 76, 1229–1237.
- Katoozi, S.; Rao, S.B.; Skauli, N.; Froehner, S.C.; Ottersen, O.P.; Adams, M.E.; Amiry-Moghaddam, M. Functional specialization of retinal Müller cell endfeet depends on an interplay between two syntrophin isoforms. Mol. Brain 2020, 13, 40.
- Roemer, S.F.; Parisi, J.E.; Lennon, V.A.; Benarroch, E.E.; Lassmann, H.; Bruck, W.; Mandler, R.N.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; et al. Pattern-Specific Loss of Aquaporin-4 Immunoreactivity Distinguishes Neuromyelitis Optica from Multiple Sclerosis. Brain 2007, 130, 1194–1205.
- Whittam, D.; Wilson, M.; Hamid, S.; Keir, G.; Bhojak, M.; Jacob, A. What’s new in neuromyelitis optica? A short review for the clinical neurologist. J. Neurol. 2017, 264, 2330–2344.
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorder. Neurology 2015, 85, 177–189.
- Takano, R.; Misu, T.; Takahashi, T.; Sato, S.; Fujihara, K.; Itoyama, Y. Astrocytic damage is far more severe than demyelination in NMO: A clinical CSF biomarker study. Neurology 2010, 75, 208–216.
- Storoni, M.; Verbeek, M.M.; Illes, Z.; Marignier, R.; Teunissen, C.E.; Grabowska, M.; Confavreux, C.; Plant, G.T.; Petzold, A. Serum GFAP levels in optic neuropathies. J. Neurol. Sci. 2012, 317, 117–122.
- Fang, B.; McKeon, A.; Hinson, S.R.; Kryzer, T.J.; Pittock, S.J.; Aksamit, A.J.; Lennon, V.A. Autoimmune glial fibrillary acidic protein astrocytopathy: A novel meningo encephalomyelitis. JAMA Neurol. 2016, 73, 1297–1307.
- Jarius, S.; Aboul-Enein, F.; Waters, P.; Kuenz, B.; Hauser, A.; Berger, T.; Lang, W.; Reindl, M.; Vincent, A.; Kristoferitsch, W. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 2008, 131, 3072–3080.
- Ho, J.D.; Yeh, R.; Sandstrom, A.; Chorny, I.; Harries, W.E.; Robbins, R.A.; Miercke, L.J.; Stroud, R.M. Crystal structure of human aquaporin 4 at 1.8 a and its mechanism of conductance. Proc. Natl. Acad. Sci. USA 2009, 106, 7437–7442.
- Xu, M.; Xiao, M.; Li, S.; Yang, B. Aquaporins in Nervous System. Adv. Exp. Med. Biol. 2017, 969, 81–103.
- Jung, J.S.; Bhat, R.V.; Preston, G.M.; Guggino, W.B.; Baraban, J.M.; Agre, P. Molecular characterization of an aquaporin cDNA from brain: Candidate osmoreceptor and regulator of water balance. Proc. Natl. Acad. Sci. USA 1994, 91, 13052–13056.
- Zhang, S.; Shang, D.; Shi, H.; Teng, W.; Tian, L. Function of Astrocytes in Neuroprotection and Repair after Ischemic Stroke. Eur. Neurol. 2021, 84, 426–434.
- Szu, J.I.; Binder, D.K. The Role of Astrocytic Aquaporin-4 in Synaptic Plasticity and Learning and Memory. Front. Integr. Neurosci. 2016, 24, 10:18.
- Li, L.; Zhang, H.; Verkman, A.S. Greatly attenuated experimental autoimmune encephalomyelitis in aquaporin-4 knockout mice. BMC Neurosci. 2009, 10, 94.
- Netti, V.; Fernández, J.; Melamud, L.; Garcia-Miranda, P.; Di Giusto, G.; Ford, P.; Echevarría, M.; Capurro, C. Aquaporin-4 Removal from the Plasma Membrane of Human Müller Cells by AQP4-IgG from patients with neuromyelitis optica induces changes in cell volume homeostasis: The first step of retinal injury? Mol. Neurobiol. 2021, 58, 5178–5193.
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S. Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477.
- Jarius, S.; Wildemann, B.; Paul, F. Neuromyelitis optica: Clinical features, immunopathogenesis and treatment. Clin. Exp. Immunol. 2014, 176, 149–164.
- Dalakas, M.C. B cells as therapeutic targets in autoimmune neurological disorders. Nat. Clin. Pract. 2008, 4, 557–567.
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmoblasts in neuromyelitis optica. Proc. Nat. Acad. Sci. USA 2011, 108, 3701–3706.
- Phuan, P.W.; Ratelade, J.; Rossi, A.; Tradtrantip, L.; Verkman, A.S. Complement-Dependent Cytotoxicity in Neuromyelitis Optica Requires Aquaporin-4 Protein Assembly in Orthogonal Arrays. J. Biol. Chem. 2012, 287, 13829–13839.
- Elsone, L.; Townsend, T.; Mutch, K.; Das, K.; Boggild, M.; Nurmikko, T.; Jacob, A. Neuropathic pruritis (itch) in neuromyelitis optica. Mult. Scler. 2012, 19, 475–479.
- Wilson, R.; Makuch, M.; Kienzler, A.K.; Varley, J.; Taylor, J.; Woodhall, M.; Palace, J.; Leite, M.I.; Waters, P.; Irani, S.R. Condition-dependent generation of aquaporin-4 antibodies from circulating B cells in neuromyelitis optica. Brain 2018, 141, 1063–1074.
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570.
- Hinson, S.R.; Roemer, S.F.; Lucchinetti, C.F.; Fryer, J.P.; Kryzer, T.J.; Chamberlain, J.L.; Howe, C.L.; Pittock, S.J.; Lennon, V.A. Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down-regulating EAAT2. J. Exp. Med. 2008, 205, 2473–2481.
- Marignier, R.; Nicolle, A.; Watrin, C.; Touret, M.; Cavagna, S.; Varrin-Doyer, M.; Cavillon, G.; Rogemond, V.; Confavreux, C.; Honnorat, J.; et al. Oligodendrocytes are damaged by neuromyelitis optica immunoglobulin G via astrocyte injury. Brain 2010, 133, 2578–2591.
- Howe, C.L.; Kaptzan, T.; Magaña, S.M.; Ayers-Ringler, J.R.; LaFrance-Corey, R.G.; Lucchinetti, C.F. Neuromyelitis optica IgG stimulates an immunological response in rat astrocyte cultures. Glia 2014, 62, 692–708.
- Zhang, H.; Verkman, A.S. Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J. Clin. Investig. 2013, 123, 2306–2316.
- Tradtrantip, L.; Yao, X.; Su, T.; Smith, A.J.; Verkman, A.S. Bystander mechanism for complement-initiated early oligodendrocyte injury in neuromyelitis optica. Acta. Neuropathol. 2017, 134, 35–44.
- Duan, T.; Smith, A.J.; Verkman, A.S. Complement-dependent bystander injury to neurons in AQP4-IgG seropositive neuromyelitis optica. J. Neuroinflamm. 2018, 15, 294.
- Luppe, S.; Robertson, N.P. MOG-IgG in neuromyelitis optica. J. Neurol. 2014, 261, 640–642.
- John, T.G.; Bernard, C.A.C. The structure and function of myelin oligodendrocyte glycoprotein. J. Neurochem. 2002, 72, 1–9.
- Mader, S.; Gredler, V.; Schanda, K.; Rostasy, K.; Dujmovic, I.; Pfaller, K.; Lutterotti, A.; Jarius, S.; Di Pauli, F.; Kuenz, B.; et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J. Neuroinflamm. 2011, 8, 184.
- Höftberger, R.; Guo, Y.; Flanagan, E.P.; Lopez-Chiriboga, A.S.; Endmayr, V.; Hochmeister, S.; Joldic, D.; Pittock, S.J.; Tillema, J.M.; Gorman, M.; et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. 2020, 139, 875–892.
- De Lott, L.B.; Bennett, J.L.; Costello, F. The Changing Landscape of Optic Neuritis: A Narrative Review. J. Neurol. 2022, 269, 111–124.
- Petrikowski, L.; Reinehr, S.; Haupeltshofer, S.; Deppe, L.; Graz, F.; Kleiter, I.; Dick, H.B.; Gold, R.; Faissner, S.; Joachim, S.C. Progressive Retinal and Optic Nerve Damage in a Mouse Model of Spontaneous Opticospinal Encephalomyelitis. Front. Immunol. 2022, 12, 759389.
- Tanaka, S.; Hashimoto, B.; Izaki, S.; Oji, S.; Fukaura, H.; Nomura, K. Clinical and Immunological Differences between Mog Associated Disease and Anti Aqp4 Antibody-Positive Neuromyelitis Optica Spectrum Disorders: Blood-Brain Barrier Breakdown and Peripheral Plasmablasts. Mult. Scler. Rela. Disord. 2020, 41, 102005.
- Ataie-Kachoie, P.; Pourgholami, M.H.; Morris, D.L. Inhibition of the IL-6 signaling pathway: A strategy to combat chronic inflammatory diseases and cancer. Cytokine Growth Factor Rev. 2013, 24, 163–173.
- Van Wagoner, N.J.; Benveniste, E.N. Interleukin-6 expression and regulation in astrocytes. J. Neuroimmunol. 1999, 100, 124–139.
- Fujihara, K.; Bennett, J.L.; de Seze, J.; Haramura, M.; Kleiter, I.; Weinshenker, B.G.; Kang, D.; Mughal, T.; Yamamura, T. Interleukin-6 in neuromyelitis optica spectrum disorder pathophysiology. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e841.
- Wang, Y.; Zhang, J.; Chang, H.; Wang, H.; Xu, W.; Cong, H.; Zhang, X.; Liu, J.; Yin, L. NMO-IgG Induce Interleukin-6 Release via Activation of the NF-κB Signaling Pathway in Astrocytes. Neuroscience 2022, 496, 96–104.
- Wang, H.; Wang, K.; Zhong, X.; Dai, Y.; Qiu, W.; Wu, A.; Hu, X. Notable increased cerebrospinal fluid levels of soluble interleukin-6 receptors in neuromyelitis optica. Neuroimmunomodulation 2012, 19, 304–308.
- Du, L.; Chang, H.; Xu, W.; Wei, Y.; Wang, Y.; Yin, L.; Zhang, X. Effect of NMO-IgG on the interleukin-6 cascade in astrocytes via activation of the JAK/STAT3 signaling pathway. Life Sci. 2020, 258, 118217.
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6: Designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412.
- Uzawa, A.; Mori, M.; Ito, M.; Uchida, T.; Hayakawa, S.; Masuda, S.; Kuwabara, S. Markedly increased CSF interleukin-6 levels in neuromyelitis optica, but not in multiple sclerosis. J. Neurol. 2009, 256, 2082–2084.
- Uzawa, A.; Mori, M.; Arai, K.; Sato, Y.; Hayakawa, S.; Masuda, S.; Taniguchi, J.; Kuwabara, S. Cytokine and Chemokine Profiles in Neuromyelitis Optica: Significance of Interleukin-6. Mult. Scler. 2010, 16, 1443–1452.
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740.
- Thoman, M.E.; McKarns, S.C. Metabolomic Profiling in Neuromyelitis Optica Spectrum Disorder Biomarker Discovery. Metabolites 2020, 10, 374.
- Pache, F.; Ringelstein, M.; Aktas, O.; Kleiter, I.; Jarius, S.; Siebert, N.; Bellmann-Strobl, J.; Paul, F.; Ruprecht, K. C3 and C4 complement levels in AQP4-IgG-positive NMOSD and in MOGAD. J. Neuroimmunol. 2021, 360, 577699.
- Maciak, K.; Pietrasik, S.; Dziedzic, A.; Redlicka, J.; Saluk-Bijak, J.; Bijak, M.; Włodarczyk, T.; Miller, E. Th17-Related Cytokines as Potential Discriminatory Markers between Neuromyelitis Optica (Devic’s Disease) and Multiple Sclerosis—A Review. Int. J. Mol. Sci. 2021, 22, 8946.
- Uzawa, A.; Mori, M.; Kuwabara, S. Cytokines and chemokines in neuromyelitis optica: Pathogenetic and therapeutic implications. Brain Pathol. 2014, 24, 67–73.
- Hou, M.M.; Li, Y.F.; He, L.L.; Li, X.Q.; Zhang, Y.; Zhang, S.X.; Li, X.Y. Proportions of Th17 Cells and Th17-Related Cytokines in Neuromyelitis Optica Spectrum Disorders Patients: A Meta-Analysis. Int. Immunopharmacol. 2019, 75, 105793.
- Nytrova, P.; Potlukova, E.; Kemlink, D.; Woodhall, M.; Horakova, D.; Waters, P.; Havrdova, E.; Zivorova, D.; Vincent, A.; Trendelenburg, M. Complement activation in patients with neuromyelitis optica. J. Neuroimmunol. 2014, 274, 185–191.
- Asavapanumas, N.; Tradtrantip, L.; Verkman, A.S. Targeting the complement system in neuromyelitis optica spectrum disorder. Expert. Opin. Biol. Ther. 2021, 21, 1073–1086.
- Soltys, J.; Liu, Y.; Ritchie, A.; Wemlinger, S.; Schaller, K.; Schumann, H.; Owens, G.P.; Bennett, J.L. Membrane assembly of aquaporin-4 autoantibodies regulates classical complement activation in neuromyelitis optica. J. Clin. Investig. 2019, 129, 2000–2013.
- Yao, X.; Verkman, A.S. Complement regulator CD59 prevents peripheral organ injury in rats made seropositive for neuromyelitis optica immunoglobulin G. Acta. Neuropathol. Commun. 2017, 5, 57.
- Kalluri, S.R.; Srivastava, R.; Kenet, S.; Tanti, G.K.; Dornmair, K.; Bennett, J.L.; Misgeld, T.; Hemmer, B.; Wyss, M.T.; Herwerth, M. P2R Inhibitors Prevent Antibody-Mediated Complement Activation in an Animal Model of Neuromyelitis Optica: P2R Inhibitors Prevent Autoantibody Injury. Neurotherapeutics 2022, 19, 1603–1616.
- Pittock, S.J.; Lennon, V.A.; McKeon, A.; Mandrekar, J.; Weinshenker, B.G.; Lucchinetti, C.F.; O’Toole, O.; Wingerchuk, D.M. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: An open-label pilot study. Lancet Neurol. 2013, 12, 554–562.
- Vincent, T.; Saikali, P.; Cayrol, R.; Roth, A.D.; Bar-Or, A.; Prat, A.; Antel, J.P. Functional consequences of neuromyelitis optica-IgG astrocyte interactions on blood-brain barrier permeability and granulocyte recruitment. J. Immunol. 2008, 181, 5730–5737.
- Bennett, J.L.; Lam, C.; Kalluri, S.R.; Saikali, P.; Bautista, K.; Dupree, C.; Glogowska, M.; Case, D.; Antel, J.P.; Owens, G.P.; et al. Intrathecal pathogenic antiaquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol. 2009, 66, 617–629.
- Klos, A.; Tenner, A.J.; Johswich, K.O.; Ager, R.R.; Reis, E.S.; Kohl, J. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009, 46, 2753–2766.
- Thangaleela, S.; Sivamaruthi, B.S.; Kesika, P.; Bharathi, M.; Chaiyasut, C. Role of the Gut-Brain Axis, Gut Microbial Composition, Diet, and Probiotic Intervention in Parkinson’s Disease. Microorganisms 2022, 10, 1544.
- Cui, C.; Ruan, Y.; Qiu, W. Potential role of the gut microbiota in neuromyelitis optica spectrum disorder: Implication for intervention. J. Clin. Neurosci. 2020, 82, 193–199.
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498.
- Varrin-Doyer, M.; Spencer, C.M.; Schulze-Topphoff, U.; Nelson, P.A.; Stroud, R.M.; Cree, B.A.; Zamvil, S.S. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann. Neurol. 2012, 72, 53–64.
- Zamvil, S.S.; Spencer, C.M.; Baranzini, S.E.; Cree, B.A.C. The Gut Microbiome in Neuromyelitis Optica. Neurotherapeutics 2018, 15, 92–101.
- Cree, B.A.; Spencer, C.M.; Varrin-Doyer, M.; Baranzini, S.E.; Zamvil, S.S. Gut microbiome analysis in neuromyelitis optica reveals overabundance of Clostridium perfringens. Ann. Neurol. 2016, 80, 443–447.
- Zhang, J.; Xu, Y.F.; Wu, L.; Li, H.F.; Wu, Z.Y. Characteristic of gut microbiota in southeastern Chinese patients with neuromyelitis optica spectrum disorders. Mult. Scler. Relat. Disor. 2020, 44, 102217.
- Jarius, S.; Wandinger, K.P.; Platzer, S.; Wildemann, B. Homology between Klebsiella pneumoniae and human aquaporin-4: No evidence for cross-reactivity in neuromyelitis optica. A study on 114 patients. J. Neurol. 2011, 258, 929–931.
- Gong, J.; Qiu, W.; Zeng, Q.; Liu, X.; Sun, X.; Li, H.; Yang, Y.; Wu, A.; Bao, J.; Wang, Y.; et al. Lack of short-chain fatty acids and overgrowth of opportunistic pathogens define dysbiosis of neuromyelitis optica spectrum disorders: A Chinese pilot study. Mult. Scler. 2019, 25, 1316–1325.
- Katz, S.I.; Zhu, Y.; Ntranos, A.; Clemente, J.C.; Cekanaviciute, E.; Brandstadter, R.; Crabtree-Hartman, E.; Singh, S.; Bencosme, Y.; Debelius, J.; et al. Disease-modifying therapies alter gut microbial composition in MS. Neurol. Neuroimmunol. Neuroinflamm. 2018, 6, e517.
- Pandit, L.; Cox, L.M.; Malli, C.; D’Cunha, A.; Rooney, T.; Lokhande, H.; Willocq, V.; Saxena, S.; Chitnis, T. Clostridium bolteae is elevated in neuromyelitis optica spectrum disorder in India and shares sequence similarity with AQP4. Neurol. Neuroimmunol. Neuroinflamm. 2020, 8, e907.
- Cheng, X.; Zhou, L.; Li, Z.; Shen, S.; Zhao, Y.; Liu, C.; Zhong, X.; Chang, Y.; Kermode, A.G.; Qiu, W. Gut Microbiome and Bile Acid Metabolism Induced the Activation of CXCR5+ CD4+ T Follicular Helper Cells to Participate in Neuromyelitis Optica Spectrum Disorder Recurrence. Front. Immunol. 2022, 13, 827865.
- Okumura, R.; Takeda, K. Maintenance of intestinal homeostasis by mucosal barriers. Inflamm. Regen. 2018, 38, 5.
- Nagpal, R.; Yadav, H. Bacterial Translocation from the Gut to the Distant Organs: An Overview. Ann. Nutr. Metab. 2017, 71, 11–16.
- Wang, Y.; Zhu, M.; Liu, C.; Han, J.; Lang, W.; Gao, Y.; Lu, C.; Wang, S.; Hou, S.; Zheng, N. Blood brain barrier permeability could be a biomarker to predict severity of neuromyelitis optica spectrum disorders: A retrospective analysis. Front. Neurol. 2018, 9, 648.
- Schmitz, H.; Barmeyer, C.; Fromm, M.; Runkel, N.; Foss, H.D.; Bentzel, C.J.; Riecken, E.O.; Schulzke, J.D. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology 1999, 116, 301–309.
- Sterlin, D.; Larsen, M.; Fadlallah, J.; Parizot, C.; Vignes, M.; Autaa, G.; Dorgham, K.; Juste, C.; Lepage, P.; Aboab, J.; et al. Perturbed microbiota/immune homeostasis in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e997.

