Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ewa Żesławska | -- | 2192 | 2023-04-13 08:51:07 | | | |
| 2 | Jason Zhu | Meta information modification | 2192 | 2023-04-14 03:56:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Żesławska, E.; Tejchman, W.; Kincses, A.; Spengler, G.; Nitek, W.; Żuchowski, G.; Szymańska, E. 5-Arylidenerhodanines as P-gp Modulators. Encyclopedia. Available online: https://encyclopedia.pub/entry/43013 (accessed on 25 June 2026).
Żesławska E, Tejchman W, Kincses A, Spengler G, Nitek W, Żuchowski G, et al. 5-Arylidenerhodanines as P-gp Modulators. Encyclopedia. Available at: https://encyclopedia.pub/entry/43013. Accessed June 25, 2026.
Żesławska, Ewa, Waldemar Tejchman, Annamária Kincses, Gabriella Spengler, Wojciech Nitek, Grzegorz Żuchowski, Ewa Szymańska. "5-Arylidenerhodanines as P-gp Modulators" Encyclopedia, https://encyclopedia.pub/entry/43013 (accessed June 25, 2026).
Żesławska, E., Tejchman, W., Kincses, A., Spengler, G., Nitek, W., Żuchowski, G., & Szymańska, E. (2023, April 13). 5-Arylidenerhodanines as P-gp Modulators. In Encyclopedia. https://encyclopedia.pub/entry/43013
Żesławska, Ewa, et al. "5-Arylidenerhodanines as P-gp Modulators." Encyclopedia. Web. 13 April, 2023.
Copy Citation
Multidrug resistance (MDR) is considered one of the major mechanisms responsible for the failure of numerous anticancer and antiviral chemotherapies. Various strategies to overcome the MDR phenomenon have been developed, and one of the most attractive research directions is focused on the inhibition of MDR transporters, membrane proteins that extrude cytotoxic drugs from living cells.
cancer multidrug resistance
P-glycoprotein
efflux pump inhibition
1. Introduction
Multidrug resistance (MDR) is a severe problem in the treatment of various diseases such as cancer, bacterial, fungal, and parasitic infections. One of the mechanisms of cancer drug resistance is associated with increased drug efflux from cells, mediated by the ATP-binding cassette (ABC) transporters [1]. More than 40 ABC transporters have been identified in humans and classified into seven subfamilies (ABC-A to ABC-G) [2]. Among them, 11 proteins have been reported to be related to MDR, an example of which is P-glycoprotein (P-gp, ABCB1), MDR-associated proteins (MRP, ABCC), and breast cancer resistance protein (BCRP, ABCG2) [3][4]. These proteins and modulation of drug efflux are considered important therapeutic targets in the fight against drug resistance in cancer [5][6][7].
The MDR-ABC transporter most studied in cancer is P-glycoprotein (P-gp, ABCB1). A wide range of compounds with beneficial inhibitory activity toward P-gp have been reported, however, most clinical trials targeting P-gp inhibition have failed, mainly due to the toxicity of the compounds [1][7]. Multiple studies have been performed on P-gp inhibitors, including structure-activity relationship (SAR), crystallization of P-gp in complex with small-molecule compounds, and molecular modeling studies [8][9][10][11]. Most of the collected data suggested that aromatic/hydrophobic interactions could be the key features responsible for binding of the compound to P-gp; however, weak electrostatic interactions (including hydrogen bonding, π-π stacking and cation-π interactions) are also important [8][12].
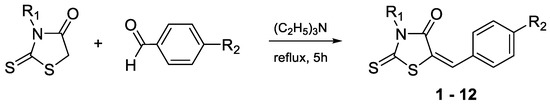
2. Synthesis
The target compounds have been synthesized according to the procedure described previously for compounds 10–12 [13]. The route of the Knoevenagel condensation of rhodanine or rhodanine-3-carboxyalkyl acids with benzaldehyde derivatives is presented in Scheme 1. The rhodanine used for the syntheses was obtained according to the procedure developed by Nencki [14], while the rhodanine-3-carboxyalkyl acids were obtained according to the procedure proposed by Körner [15].
Scheme 1. Condensation of rhodanine and rhodanine-3-carboxyalkyl acids with benzaldehyde derivatives.
3. Biological Screening
3.1. The Rhodamine 123 Accumulation Assay
All investigated compounds evaluated for their efflux pump modulating effects in the sensitive parental (PAR) and resistant (MDR) mouse T-lymphoma cells overexpressing ABCB1 using the standard rhodamine 123 functional assay at 2 and 20 μM concentrations. The fluorescence activity ratio (FAR) was calculated based on the obtained fluorescence data and shows a measure of the efflux pump modulating properties under the influence of the investigated compounds. Compounds with FAR values below 1 were considered inactive (Table 1).
Table 1. The effects on rhodamine 123 retention on MDR T-lymphoma cells of investigated compounds.
| Cmpd. | FAR 1 2 μM |
FAR 1 20 μM |
|---|---|---|
| 1 | <1 | 12.704 |
| 2 | <1 | <1 |
| 3 | <1 | <1 |
| 4 | <1 | <1 |
| 5 | <1 | 15.366 |
| 6 | <1 | <1 |
| 7 | <1 | <1 |
| 8 | <1 | <1 |
| 9 | 21.658 | 32.184 |
| 10 | 27.501 | 75.107 |
| 11 | 35.245 | 76.226 |
| 12 | 35.547 | 78.491 |
| Verapamil | nd | 4.380 |
Among structures containing the 5-(4′-phenylbenzylidene) rhodanine moiety (1–4), the modulatory effect was shown only for the derivative 1 at a concentration of 20 μM. An introduction of the carboxyl group to the structure of 1 definitely removed activity (2–4). Similar results were observed for compounds 5–8 that possess the 5-(4′-phenylmethoxybenzylidene) rhodanine moiety.
An opposite effect was shown in the case of the 5-(4′-N, N-diphenylaminobenzylidene)rhodanine series 9–12 that proved to be the most potent P-gp modulators among all compounds tested. All four compounds affected the efflux activity of ABCB1 at both concentrations (Table 1). Furthermore, contrary to other series, the introduction of the carboxyl group into the structure of 9 has caused a significant increase in the FAR coefficient and resulted in compounds 10–12 that at 20 μM concentration showed more than 17 times stronger efflux pump inhibitory effects than the reference inhibitor, verapamil (FAR = 4.38).
3.2. Cytotoxicity and Antiproliferative Assays
The presented compounds were also investigated for their cytotoxic and antiproliferative effects in PAR and MDR mouse T-lymphoma cells, using MTT assay to estimate inhibitory concentration of 50% (IC50) values (Table 2). Most of the compounds under these conditions did not show significant cytotoxic effects against two tested cell lines, except for 3 (IC50 = 13.88 μM and 17.02 μM for PAR and MDR cells, respectively). Antiproliferative activity was observed for all compounds. Interesting results were obtained for 4, which was not cytotoxic, while its antiproliferative activity was more pronounced in the MDR cell line (IC50 = 4.3 μM vs. 8.12 μM), similar to 12 (IC50 = 11.16 μM vs. 15.3 μM).
Table 2. Cytotoxic and antiproliferative effects of the compounds 1–12 on sensitive (PAR) and P-gp overexpressing-resistant (MDR) mouse T-lymphoma cells.
| IC50 ± SD (μM) | ||||
|---|---|---|---|---|
| Cmpd. | Cytotoxic Effect | Antiproliferative Effect | ||
| PAR | MDR | PAR | MDR | |
| 1 | 48.28 ± 1.54 | 56.09 ± 1.72 | 35.84 ± 0 | 39.26 ± 1.05 |
| 2 | >100 | >100 | 43.10 ± 0.66 | 55.17 ± 1.89 |
| 3 | 13.88 ± 0.49 | 17.02 ± 1.05 | 28.60 ± 0.72 | 36.75 ± 1.72 |
| 4 | >100 | >100 | 8.12 ± 0.89 | 4.30 ± 0.21 |
| 5 | 45.31 ± 0.94 | 49.57 ± 0.29 | 28.13 ± 0.43 | 50.08 ± 1.44 |
| 6 | >100 | >100 | 36.14 ± 1.49 | 63.48 ± 2.80 |
| 7 | 63.26 ± 2.61 | >100 | 36.12 ± 1.11 | 47.16 ± 0.54 |
| 8 | 47.01 ± 1.99 | 20.99 ± 0.95 | 7.45 ± 0.57 | 8.37 ± 0.61 |
| 9 * | nd | nd | nd | nd |
| 10 | >25 | >25 | 7.31 ± 0.70 | 9.28 ± 0.22 |
| 11 | >25 | >25 | 8.92 ± 1.35 | 42.10 ± 0.73 |
| 12 | >25 | >25 | 15.30 ± 2.24 | 11.16 ± 1.48 |
3.3. Drug Combination Assay
To evaluate the ability of target rhodanines to resensitize MDR cells to the anticancer drug doxorubicin, interactions between doxorubicin and compounds showing efficient cytotoxic and antiproliferative activities (1, 3, 5, and 8) were evaluated using a checkerboard combination assay (Table 3). The level of interactions was expressed by the drug combination index (CI) value, calculated on the basis of IC50 data for individual drug or two-drug combinations, and is defined as additivity for the CI value close to 1, synergy for CI < 1, and antagonism CI > 1. Among the compounds tested, 1, 3, and 5 displayed an antagonistic effect toward doxorubicin, while interactions between 8 and doxorubicin can be described as nearly additive.
Table 3. Drug combination assay results on MDR cells.
| Cmpd. | Ratio 1 | CI | Interaction |
|---|---|---|---|
| 1 | 69.6:1 | 1.50 (18) | Antagonism |
| 3 | 11.6:1 | 2.33 (83) | Antagonism |
| 5 | 34.8:1 | 1.60 (18) | Antagonism |
| 8 | 27.8:1 | 0.92 (8) | Nearly additive |
4. X-ray Studies of Compounds 3, 7, and 11
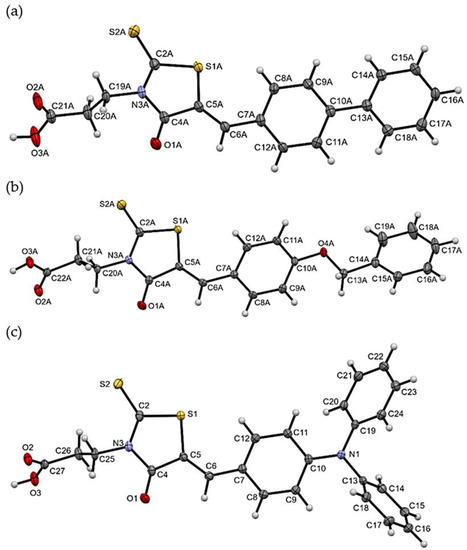
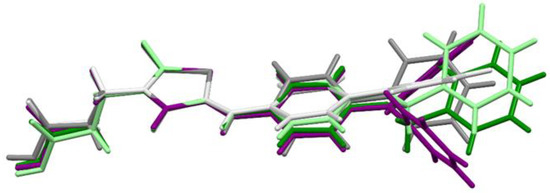
The molecular geometries in the crystals of the selected compounds (3, 7, and 11) are presented in Figure 1. Compounds 3 and 7 crystallize with two molecules (labelled A and B) in the asymmetric unit. For all compounds, the isomer Z is observed. The 5-bezylidenerhodanine fragment of 7 and 11 is almost planar (Figure 2), while 3 shows a greater deviation from planarity. The biggest differences for the compounds analyzed are observed in the mutual arrangement of aromatic rings. Only for 11 the aromatic rings are arranged almost perpendicular to each other.
Figure 1. The molecular geometries of (a) 3 (molecule A), (b) 7 (molecule A), and (c) 11, with the atom-numbering schemes. Displacement ellipsoids are drawn at the 50% probability level.
Figure 2. The overlap of the rhodanine rings of 3 (gray molecule A, light gray molecule B), 7 (green molecule A, light green molecule B), and 11 (purple).
The packing of molecules in the crystals is dominated by intermolecular O-H···O hydrogen bonds between two carboxylic groups, which leads to the formation of dimers. The dimers are joined to each other by weak C-H···O and, in the case of 3 and 7, also by C-H···S interactions.
5. Molecular Modeling
The large and hydrophobic transmembrane interior of P-gp is reported to contain a common binding site for both substrates and small-molecule inhibitors [11]. Competitive inhibitor/substrate binding to the drug-binding cavity is one of the mechanisms suggested for the inhibition of rhodamine 123 efflux, observed in the accumulation assay described above. In an attempt to determine putative binding modes for the described amphiphilic molecules, one of the most active efflux pump modulators, 11, was docked into the large ABCB1 drug-binding pocket using the induced-fit docking (IFD) protocol implemented in the Schrödinger Suite [16] to mimic binding to a flexible protein. The likely binding interactions between hP-gp and 11 were carefully examined and compared with the results of similar docking performed for verapamil, the known first-generation P-gp inhibitor.
Several X-ray structures of murine P-gp crystallized in complex with substrates or inhibitors are known [17][18][19][20], with the protein adopting an inward-facing nucleotide-free conformation that occurs at the initial stage of the ATP-dependent transport cycle [21]. However, so far no such structure of fully human inhibitor-bound ABCB1 is available. On the other hand, a dynamic growth of cryo-EM technology enabled the determination with high-resolution structures of the human ABCB1 transporter in complex with inhibitory antibody fragments and bound to small-molecule substrates or inhibitors [22][23][24]. Most of these structures represent an ‘occluded’ conformation of P-gp, characterized by bent transmembrane helices TM4 and TM10 that form a cytoplasmic gate at the entrance of the drug-binding pocket [22], as well as a reduced distance between two nucleotide-binding domains (NBD), compared to an inward-facing conformation.
Taking into account the above, in the docking studies two following protein structures were used: (1) the homology model of the human P-gp (hP-gp) in an inward-facing conformation, constructed as previously reported [25]; (2) the recently published cryo-EM structure of encequidar bound to the hP-gp (PDB code 7O9W) [24] that represents an occluded conformation of the transporter. The docking score, extra precision glide score, and induced-fit docking score values were calculated and used to rank the obtained ligand-protein complexes. Additionally, the molecular mechanics/generalized Born surface area (MM/GBSA) approach was applied to estimate the free energy of binding of 11 and verapamil to individual conformations of hP-gp.
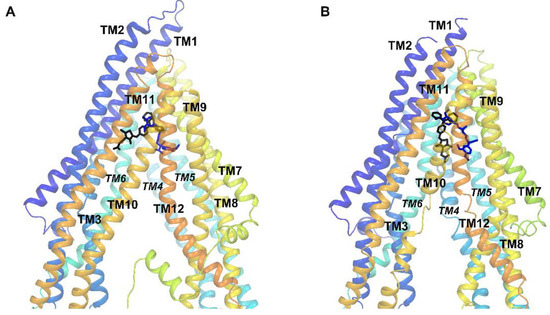
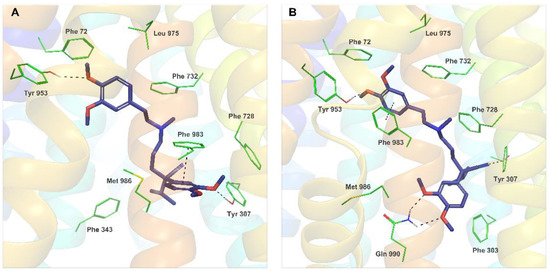
The binding sites of 11 and verapamil in hP-gp predicted by IFD were located in the upper part of the internal binding cavity (Figure 3A,B), similarly to other P-gp inhibitors seen in X-ray or cryo-EM complexes [22][23][24]. For verapamil, the binding modes found in docking to the homology model and to the cryo-EM structure were similar and followed the protein-ligand interaction pattern reported for verapamil by other authors (Figure 4A,B) [26][27][28]. The top-ranked docking poses of verapamil in both protein structures formed hydrogen bonds with Tyr307 and Tyr953 as well as aromatic π-π interactions with Phe983. The calculated free energy of binding (ΔG) of verapamil to hP-gp was −87.19 kcal/mol and −102.10 kcal/mol for the homology model and structure 7O9W, respectively.
Figure 3. Position of the top-ranked docking poses of 11 (black) and verapamil (blue) inside the hP-gp drug-binding pocket: (A) the homology model and (B) the cryo-EM structure 7O9W.
Figure 4. Binding modes and molecular interactions observed for the best docking poses of verapamil (reference poses). (A) Verapamil docked to the hP-gp homology model (XP GScore −9.387, IFD score −2651.60); (B) Verapamil docked to the cryo-EM structure 7O9W (XP GScore −10.974, IFD score −2243.60).
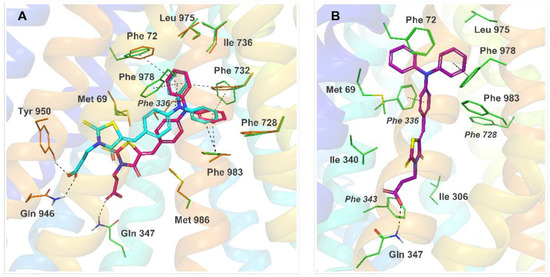
The IFD results for 11 revealed that the predominant number of observed top-ranked poses of this compound docked to the hP-gp homology model represented one out of two variants—pose Ia or Ib, illustrated in Figure 5A. In both variants, the triphenylamine moiety of 11 was trapped inside the hydrophobic pocket built by residues of TM1, TM6, TM7, and TM12 and formed multiple π-π stacking contacts with some of the surrounding phenylalanines. The carboxylic group of 11 anchored the molecule by hydrogen bonding with a polar residue, mainly Gln946 (pose Ia) or Gln347 (pose Ib). Substantial binding interactions observed for both variants of pose I resulted in high docking scores and free binding energy: −87.96 kcal/mol and −68.07 kcal/mol for the best docking poses Ia and Ib, respectively.
Figure 5. Binding modes and molecular interactions for 11 bound to hP-gp, predicted by IFD. (A) Two top-ranked docking poses of 11 in the homology model of hP-gp (pose Ia in cyan, pose Ib in pink); (B) the top-ranked docking pose of 11 in 7O9W (pose II, purple). Dashed lines represent hydrogen bonds and π-π interactions.
During IFD docking of 11 to the cryo-EM structure 7O9W, a docking pose II was observed predominantly among the high-scored results (ΔG = −92.06 kcal/mol for the best docking score, Figure 5B). Interestingly, despite a different, occluded conformation of the protein in 7O9W, caused by kinked transmembrane helices TM4 and TM10, the binding mode observed for pose II resembled the interaction pattern of Ib—the triphenylamine moiety of 11 was located inside the phenylalanine region of TM1, TM6, TM7 and TM12, and involved in multiple π-π interactions, while the carboxylic end of the ligand formed the hydrogen bond with Gln347.
It should be noted that all of the discussed binding modes of 11 predicted by IFD were consistent, both in terms of the observed ligand-protein interactions and free energy of binding. Analysis of the interaction pattern for 11 showed a clear overlap with the binding mode seen in the present IFD results for verapamil and was highly in accordance with the experimental mutagenesis data on the verapamil binding site [29][30], suggesting a similar mechanism of inhibition of the rhodamine 123 efflux for both compounds.
Multiple in silico studies were performed for structurally diverse compounds that act as P-gp substrates and inhibitors. In general, P-gp inhibitors tend to be highly lipophilic molecules with a higher log P value and a higher molecular weight compared to P-gp substrates. The main impact on interactions between the inhibitor and the P-gp binding site usually comes from a large number of hydrophobic and van der Waals contacts formed by the aryl, alkyl, and other lipophilic groups of the ligand [8][10][12][22][31]. Functional groups such as carbonyl, ether, or a tertiary amine are also often present in the structure of P-gp inhibitors, allowing hydrogen bond formation or ionic interactions with the protein.
6. Lipophilicity
In search of answers to the observed results of different influences of the carboxyl group on bioactivity, researchers have decided to estimate selected physicochemical properties, namely lipophilicity (log P) and solubility (log S). The obtained results of predicted measures [32] showed comparable solubility of all investigated compounds, but their lipophilicity is more diverse. For two series, 1–4 and 5–8, the introduction of the carboxyl group affects the values of log P more significantly (increase from 3.69 to 4.67 and from 3.78 to 4.15), while for the third series 9–12, such an impact of the carboxyl group is not observed (values of log P in the range of 5.19 to 5.33). The lipophilicity of 9 is so high that the introduction of the carboxyalkyl chain does not play a significant role in changing it.
References
- Nobili, S.; Landini, I.; Giglioni, B.; Mini, E. Pharmacological strategies for overcoming multidrug resistance. Curr. Drug Targets 2006, 7, 861–879.
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166.
- Park, S.H.; Park, C.J.; Kim, D.Y.; Lee, B.R.; Kim, Y.J.; Cho, Y.U.; Jang, S. MRP1 and P-glycoprotein expression assays would be useful in the additional detection of treatment non-responders in CML patients without ABL1 mutation. Leuk. Res. 2015, 39, 1109–1116.
- Slot, A.J.; Molinski, S.V.; Cole, S.P. Mammalian multidrug-resistance proteins (MRPs). Essays Biochem. 2011, 50, 179–207.
- Gatti, L.; Beretta, G.L.; Cossa, G.; Zunino, F.; Perego, P. ABC transporters as potential targets for modulation of drug resistance. Mini Rev. Med. Chem. 2009, 9, 1102–1112.
- Szakacs, G.; Hall, M.D.; Gottesman, M.M.; Boumendjel, A.; Kachadourian, R.; Day, B.J.; Baubichon-Cortay, H.; Di Pietro, A. Targeting the Achilles heel of multidrug-resistant cancer by exploiting the fitness cost of resistance. Chem. Rev. 2014, 114, 5753–5774.
- Lai, J.-I.; Tseng, Y.-J.; Chen, M.-H.; Huang, C.-Y.F.; Chang, P.M.-H. Clinical perspective of FDA approved drugs with P-glycoprotein inhibition activities for potential cancer therapeutics. Front. Oncol. 2020, 10, 561936.
- Seelig, A. P-glycoprotein: One mechanism, many tasks and the consequences for pharmacotherapy of cancers. Front. Oncol. 2020, 10, 576559.
- Leopoldo, M.; Nardulli, P.; Contino, M.; Leonetti, F.; Luurtsema, G.; Colabufo, N.A. An updated patent review on P-glycoprotein inhibitors (2011–2018). Expert Opin. Ther. Pat. 2019, 29, 455–461.
- Dong, J.; Qin, Z.; Zhang, W.-D.; Cheng, G.; Yehuda, A.G.; Ashby, C.R.; Chen, Z.-S.; Cheng, X.-D.; Qin, J.-J. Medicinal chemistry strategies to discover P-glycoprotein inhibitors: An update. Drug Resist. Updat. 2020, 49, 100681.
- Chufan, E.E.; Sim, H.M.; Ambudkar, S.V. Molecular basis of the polyspecificity of P-glycoprotein (ABCB1): Recent biochemical and structural studies. Adv. Cancer Res. 2015, 125, 71–96.
- Mollazadeh, S.; Sahebkar, A.; Hadizadeh, F.; Behravan, J.; Arabzadeh, S. Structural and functional aspects of P-glycoprotein and its inhibitors. Life Sci. 2018, 214, 118–123.
- Tejchman, W.; Korona-Glowniak, I.; Malm, A.; Zylewski, M.; Suder, P. Antibacterial properties of 5-substituted derivatives of rhodanine-3-carboxyalkyl acids. Med. Chem. Res. 2017, 26, 1316–1324.
- Nencki, M. Ueber die Einwirkung der Monochloressigsäure auf Sulfocyansäure und ihre Salze. J. Prakt. Chem. Chem. 1877, 16, 1–17.
- Körner, H. Über einige Derivate der Dithiocarbamino-essigsäure. Ber. Dtsch. Chem. Ges. 1908, 41, 1901–1905.
- Schrödinger Release 2021-4: LigPrep; Epik; Protein Preparation Wizard. Macromodel; Glide; Prime; MM-GBSA; Schrödinger, LLC: New York, NY, USA, 2021.
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722.
- Li, J.; Jaimes, K.F.; Aller, S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46.
- Szewczyk, P.; Tao, H.; McGrath, A.P.; Villaluz, M.; Rees, S.D.; Lee, S.C.; Doshi, R.; Urbatsch, I.L.; Zhang, Q.; Chang, G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Cryst. D Biol. Cryst. 2015, 71, 732–741.
- Nicklisch, S.C.; Rees, S.D.; McGrath, A.P.; Gökirmak, T.; Bonito, L.T.; Vermeer, L.M.; Cregger, C.; Loewen, G.; Sandin, S.; Chang, G.; et al. Global marine pollutants inhibit P-glycoprotein: Environmental levels, inhibitory effects, and cocrystal structure. Sci. Adv. 2016, 2, e1600001.
- Lusvarghi, S.; Robey, R.; Gottesman, M.; Ambudkar, S. Multidrug transporters: Recent insights from cryo-electron microscopy-derived atomic structures and animal models. F1000Research 2020, 9, 17.
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756.
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253.
- Urgaonkar, S.; Nosol, K.; Said, A.M.; Nasief, N.N.; Bu, Y.; Locher, K.P.; Lau, J.Y.N.; Smolinski, M.P. Discovery and characterization of potent dual P-glycoprotein and CYP3A4 inhibitors: Design, synthesis, cryo-EM analysis, and biological evaluations. J. Med. Chem. 2022, 65, 191–216.
- Kaczor, A.; Nove, M.; Kincses, A.; Spengler, G.; Szymanska, E.; Latacz, G.; Handzlik, J. Search for ABCB1 modulators among 2-amine-5-arylideneimidazolones as a new perspective to overcome cancer multidrug resistance. Molecules 2020, 25, 2258.
- Pimthon, J.; Dechaanontasup, R.; Ratanapiphop, C.; Phromprasert, C. Homology modeling and substrate binding studies of human P-glycoprotein. Pharm. Sci. Asia 2017, 44, 96–107.
- Syed, S.B.; Arya, H.; Fu, I.H.; Yeh, T.K.; Periyasamy, L.; Hsieh, H.P.; Coumar, M.S. Targeting P-glycoprotein: Investigation of piperine analogs for overcoming drug resistance in cancer. Sci. Rep. 2017, 7, 7972.
- Prajapati, R.; Singh, U.; Patil, A.; Khomane, K.S.; Bagul, P.; Bansal, A.K.; Sangamwar, A.T. In silico model for P-glycoprotein substrate prediction: Insights from molecular dynamics and in vitro studies. J. Comput. Aided. Mol. Des. 2013, 27, 347–363.
- Loo, T.W.; Clarke, D.M. Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane. J. Biol. Chem. 2000, 275, 39272–39278.
- Loo, T.W.; Clarke, D.M. Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. J. Biol. Chem. 2001, 276, 14972–14979.
- Wang, R.B.; Kuo, C.L.; Lien, L.L.; Lien, E.J. Structure-activity relationship: Analyses of p-glycoprotein substrates and inhibitors. J. Clin. Pharm. Ther. 2003, 28, 203–228.
- Tetko, I.V.; Tanchuk, V.Y. Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program. J. Chem. Inf. Comput. Sci. 2002, 42, 1136–1145.
More
Information
Subjects:
Chemistry, Medicinal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
618
Revisions:
2 times
(View History)
Update Date:
14 Apr 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No