Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mannix Balanay | -- | 2024 | 2023-04-04 12:44:42 | | | |
| 2 | Rita Xu | -59 word(s) | 1965 | 2023-04-04 13:23:26 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Barredo, P.A.; Balanay, M.P. Molecular Dynamics Simulations of Tau Fibrils and Oligomers. Encyclopedia. Available online: https://encyclopedia.pub/entry/42781 (accessed on 26 July 2026).
Barredo PA, Balanay MP. Molecular Dynamics Simulations of Tau Fibrils and Oligomers. Encyclopedia. Available at: https://encyclopedia.pub/entry/42781. Accessed July 26, 2026.
Barredo, Prechiel A., Mannix P. Balanay. "Molecular Dynamics Simulations of Tau Fibrils and Oligomers" Encyclopedia, https://encyclopedia.pub/entry/42781 (accessed July 26, 2026).
Barredo, P.A., & Balanay, M.P. (2023, April 04). Molecular Dynamics Simulations of Tau Fibrils and Oligomers. In Encyclopedia. https://encyclopedia.pub/entry/42781
Barredo, Prechiel A. and Mannix P. Balanay. "Molecular Dynamics Simulations of Tau Fibrils and Oligomers." Encyclopedia. Web. 04 April, 2023.
Copy Citation
The study of tau protein aggregation and interactions with other molecules or solvents using molecular dynamics simulations (MDs) is of interest to many researchers to propose new mechanism-based therapeutics for neurodegenerative diseases such as Alzheimer’s disease, Pick’s disease, chronic traumatic encephalopathy, and other tauopathies.
Alzheimer’s disease
narrow Pick’s disease
force fields
membrane lipid
1. Introduction
Tau is a microtubule (MT)-associated protein that is essential for microtubule stabilization [1]. In the adult human brain, there are six different isoforms, of which 4R2N (2N4R), the longest, has 441 residues. This full-length human tau has a charged N-terminal region, a proline-rich region (spanning amino acid residues 208-244) followed by microtubule-binding repeats (MTBRs) designated R1 to R4 (R1 aa 244-274; R2 aa 275-305; R3 aa 306-336; and R4 aa 337-368), which bind to axonal microtubules under physiological conditions, and a C-terminal region [2]. The accumulation of abnormal tau aggregates in neurons is an important pathological feature in several neurodegenerative diseases grouped under the term tauopathies. These include Alzheimer’s disease (AD), chronic traumatic encephalopathy (CTE), and frontotemporal dementias (FTD) such as Pick’s disease (PiD), corticobasal degeneration (CBD), etc. [3]. Tau has been found to fibrillate in vitro in the presence of negatively charged co-factors such as heparin, DNA, or RNA [4][5][6]. At least two amino acid sequences in the microtubule-binding domain (R1 to R4) are critical in the aggregation of tau [7]. The hexapeptide segments 275VQIINK280 (PHF6*) and 306VQIVYK311 (PHF6) are located in the MT-binding domain (MTBD) in peptides R2 and R3, respectively [7]. K18 contains the MTBD (R1–R4) and it is located in the C-terminal region [8].
Alzheimer’s disease (AD) is the most common cause of dementia, accounting for 60–80% of all dementia cases. An estimated 6.5 million Americans aged 65 years and older are now living with AD [9]. This number is expected to increase to 13.85 million by 2060. It was Alois Alzheimer who first described the disease using the brain of his patient, Auguste Deter, who died in 1906, five years after she was diagnosed with what is known as presenile dementia [10]. The characteristic pathologies of AD are the accumulation of beta-amyloid (Aβ) plaques outside neurons and neurofibrillary tangles (NFTs), composed of the protein tau, inside neurons in the brain [11][12]. Several investigators have shifted their focus from Aβ to tau as an alternative target of novel therapeutics [13][14][15], as previous studies have found a weak or even nonexistent correlation between β-amyloid plaques and cognitive decline in the symptomatic phase of dementia [16][17][18][19].
The R2 and R3 peptides in the MTBD have different properties than the R1 and R4 peptides, which is probably due to the presence of the β-structure driving hexapeptides PHF6* and PHF6 [7]. The major functions of the microtubule-binding domain are listed in Figure 1 [2]. Paired helical filaments (PHFs-tau) and straight filaments (SFs-tau) form the neurofibrillary tangles (NFTs), one of the pathological hallmarks of AD [3]. The PHF-tau structure from an AD-diseased brain (PDB ID:5O3L) has a total of 11,360 atoms, while SFs-tau (PDB ID:5O3T) has a total of 5,570 atoms. There are a total of ten chains in PHFs-tau, each chain having an equal but opposite pair [3][20], and consisting of eight β-sheets (β1- β8) running along the length of the protofilament and encompassing amino acids 306-378 [3][20]. SFs form neurofibrillary tangles with a width of ∼150 Å in AD. Hybrid filaments of SFs and PHFs have been observed, implying that they have a similar C-shaped subunit but are different in structure [3].

Figure 1. The major functions of MTBD.
Tau proteins may misfold and aggregate, leading to the formation of NFTs in the brain. Studies have shown that microtubule-binding repeats undergo a conformational change when they bind to microtubules, which may affect their tendency to aggregate. In Ding and their colleagues’ study [21], using an all-atom discrete molecular dynamics (MDs) simulation, they observed that both repeats can aggregate into metastable β-sheet-rich dimers according to their respective conformational ensembles and dimerization kinetics, with R2 and R4 being highly comparable in this regard. The β-sheets between chains in R2 aggregates were driven by residues in the PHF6* regions, whereas β-hairpins predominated in the formation of R4 dimers. The greatest propensity for amyloid aggregation was observed in the R3 repeat. In R3, residues in Paired helical filaments the PHF6 regions rapidly self-assembled into intermolecular β-sheets and subsequently stimulated the formation of larger β-sheets by other residues. Both PHF6 in R3 and PHF6* in R2 generated a considerable number of intermolecular interactions and contributed significantly to the early aggregation of tau.
The tau fibril that correlates with Pick’s disease is also called narrow Pick’s filament (NPF). Pick’s disease (PiD) is a rare neurological disorder in which the so-called Pick’s bodies serve as diagnostic markers, similar to neurofibrillary tangles in AD [21]. They consist exclusively of 3R tau isoforms (isoforms comprising repeats R1, R3, and R4 in the MTBD) (Figure 2) [22]. The NPF contains the R1 repeat in the fibril core and does not have the steric zipper PHF6: 306VQIVYK311 or PHF6*: 275VQIINK280 and 373THKLTF378 interaction [21]. As in AD [3], a fuzzy coat consisting of the disordered N- and C-terminal regions of tau surrounds the filament cores and is removed by pronase treatment [23].
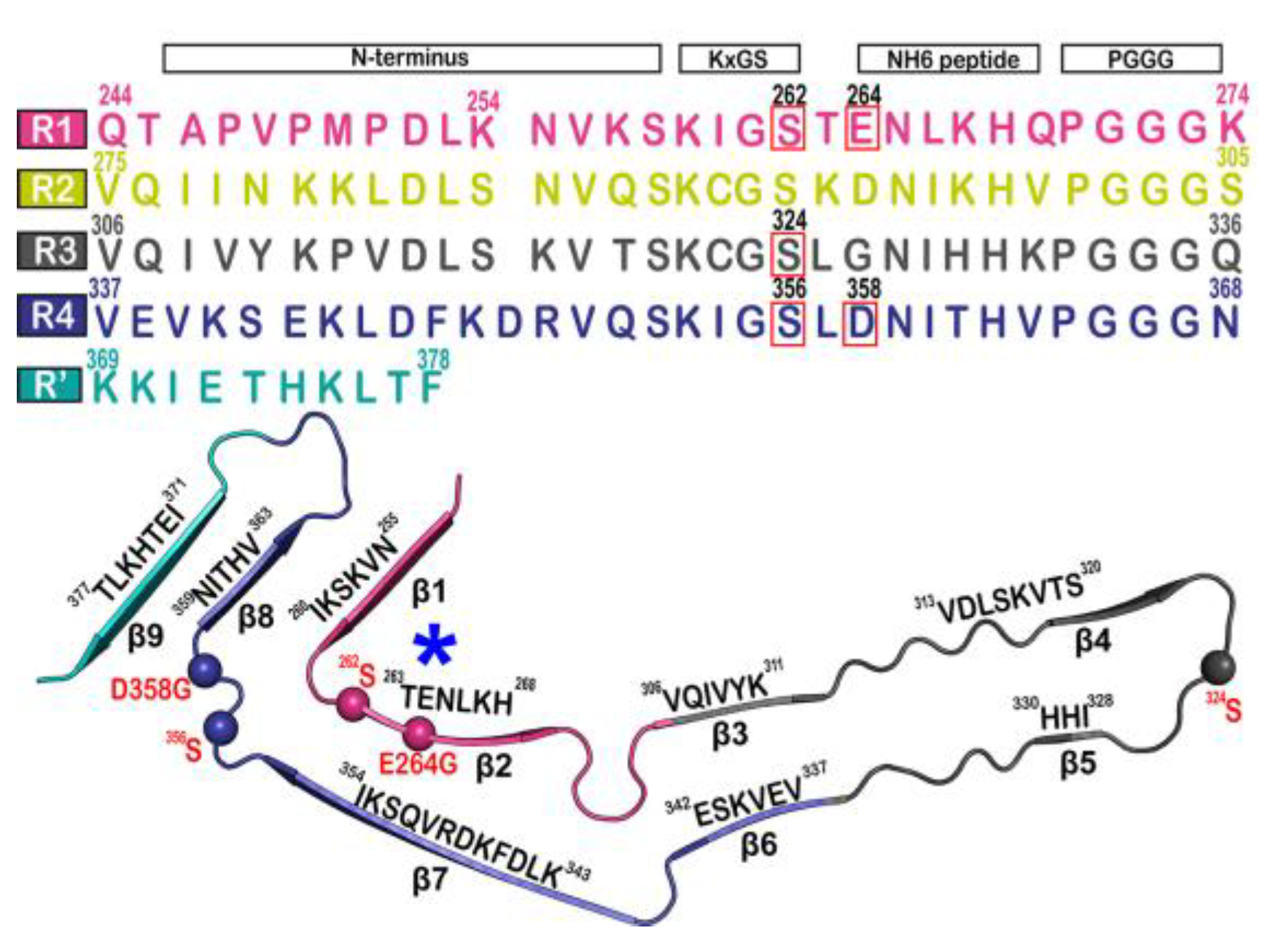
Figure 2. Microtubule-binding repeat region primary sequence, showing color-coded repeats of R1 (pink), R2 (yellow), R3 (gray), R4 (violet), and R’ (teal). The narrow Pick’s filament (NPF) lacks the R2 repeat. The residues of interest are outlined in red. A strand from the NPF cryo-EM structure (PDB-ID: 6GX5), colored according to the repeats of the primary sequence. Residues of interest are outlined in red and shown as spheres. An asterisk indicates the strong curvature of the R1 repeat. The largest tau isoform hTau40 is represented by the residue indices.
The four most commonly used software packages for molecular dynamics simulations of tau–peptide–lipid membrane complexes such as the tau–lipid bilayer, are GROMACS [24], AMBER [25], CHARMM [26], and NAMD [27]. GROMACS, for example, is a versatile package for performing molecular dynamics, i.e., simulating Newton’s equations of motion for systems with hundreds to millions of particles [24]. The wide availability of experimentally determined protein structures from the Protein Data Bank (PDB), which can be used directly in whole-atom simulations (AA) or as templates in coarse-grained (CG) MDs, and the use of HPC (High-Performance Computing) [28][29], have contributed much to new relevant biological insights, especially in the study of protein membrane systems. The improvement of GPUs (Graphics Processing Units) hardware and better accessibility of software packages have generated tremendous interest in using GPUs for scientific computations [30]. The Fast Multipole Method (FMM) was recently integrated into GROMACS as an alternative to Particle Mesh Ewald (PME), as a first step toward exascale [31]. This highly efficient GPU (GPU-FMM) has the ability to enable efficient and scalable biomolecular protein–membrane complex simulations on future exascale supercomputers [31]. In large-scale calculations, the most time-consuming computational steps are often those involving nonbonding interactions, especially electrostatic interactions due to their long-range nature and the complexity of the Coulomb potential. The PME algorithm is the usual method for dealing with such interactions. However, the PME algorithm still requires significant computational resources, and in systems with a large number of charged particles, electrostatic interactions can become a bottleneck in the simulation. Recent advances in using FMM in combination with GPU hardware have shown that it is possible to solve such problems and enable faster and more efficient simulations.
2. Multiscale Molecular Dynamics Simulations of Tau Filaments (NPF and PHF/SF) in a Solution and in the Surface of a Lipid Bilayer
A series of simulations has focused on elucidating the structures of stable tau fibrils and oligomers. Narrow Pick’s filament (NPFPS) phosphorylated at three experimentally verified sites in the MTBD (S262, S324, and S356) and is used to study the effects of phosphorylation on the local conformation [22]. In addition, NPFGG, a double-mutant fibril system with mutations E264G and D358G, was selected to understand the role of E264 and D358 on the local conformations and to study the influence of the salt bridges that they form throughout the fibril architecture. The fibril systems were studied together with a wild-type fibril, NPFWT (as a control), using a 1 μs-long conventional molecular dynamic. Calculations were performed using CHARMM36m FF with the TIP3P water model. The fibril was constructed with water and 150 mM NaCl to neutralize the charges and replicate the in vivo environment. To keep the temperature at 310 K, the velocities are assigned according to the Maxwell distribution. Their results show that the phosphorylated (NPFPS) and mutant (NPFGG) systems were found to diverge during simulation, indicating major changes in the fibril architecture compared to NPFWT. The largest divergences in backbone conformation from the wild-type fibril system were observed during phosphorylation and mutation (Figure 3) (although a 1 μs-long simulation is not sufficient to capture all fibril conformations). For the production run, assuming a constrained structure, the last 700 ns of the trajectory are used for further analysis [22].
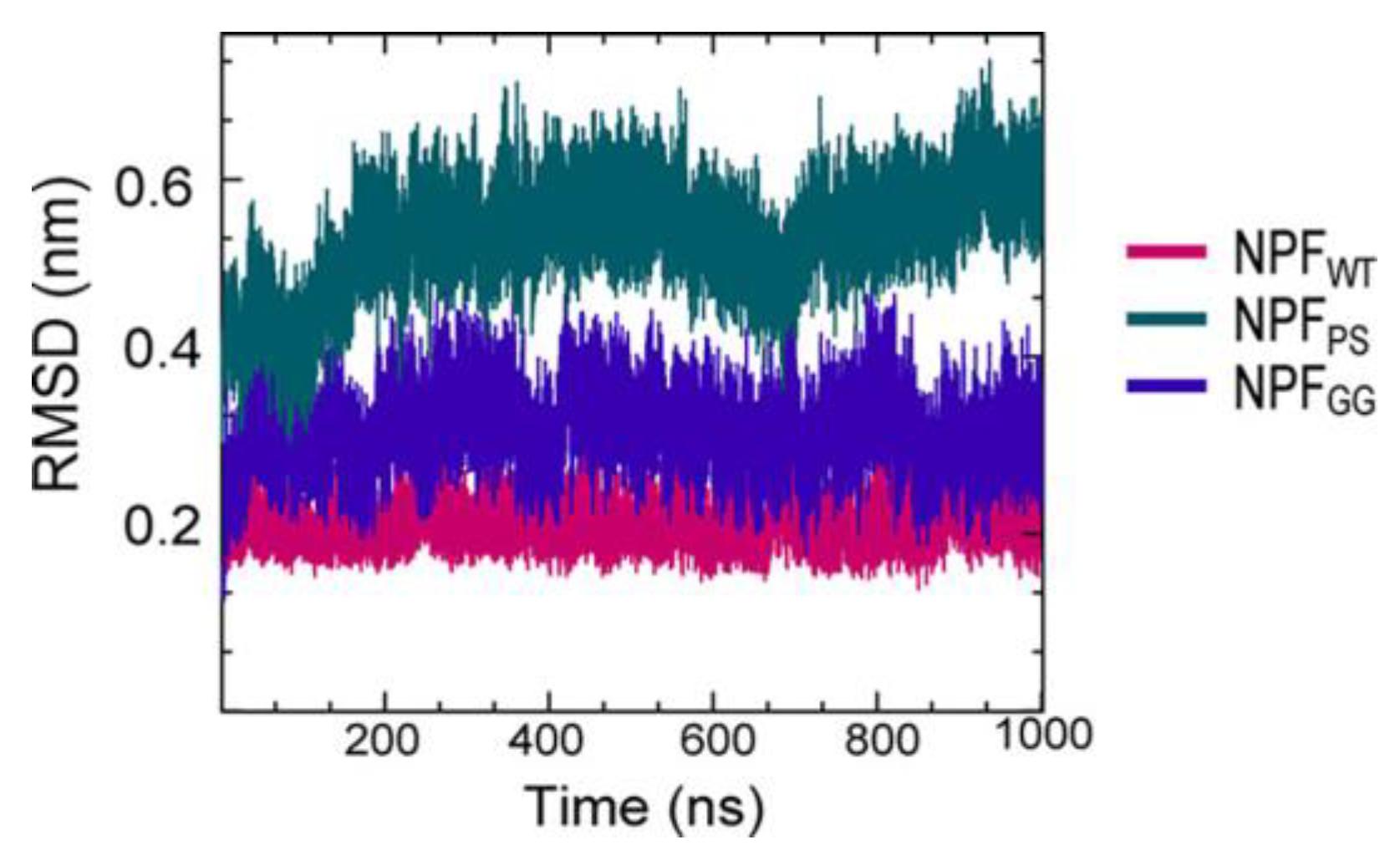
Figure 3. The fibril systems’ root mean square deviation (RMSD) from 1 μs simulations.
The stability of the C-shaped structural motifs of the tau protein was studied by Nussinov and et al. [32]. They used an all-atom explicit solvent MD calculation with the CHARMM FF and the TIP3P water model. They also included ~150 mM NaCl to represent the physiological ion concentration. All systems were heated from 0 to 300 K and held at equilibrium for 4 ns, except for K18, which was heated to 310 K. Their simulations showed that only the third and fourth repeat domains (R3-R4) retain the C-shaped conformation after optimization, while the first and second repeat domains (R1-R2) adopted a linear shape (Figure 4). The β2-β3 and β6-β7 angles of PHF34 remained relatively the same during the simulations, but the β6-β7 angles of PHF13 and PHF23 increased, indicating that the structure tends to elongate. PHF23 adopted a V-shaped conformation, whereas a U-shape was preferred for PHF13. The stability resulted from the interaction between the C-terminal residues and the N-terminus of the adjacent chain. Of all the PHFs studied, PHF34 exhibited the strongest intra-chain interactions.
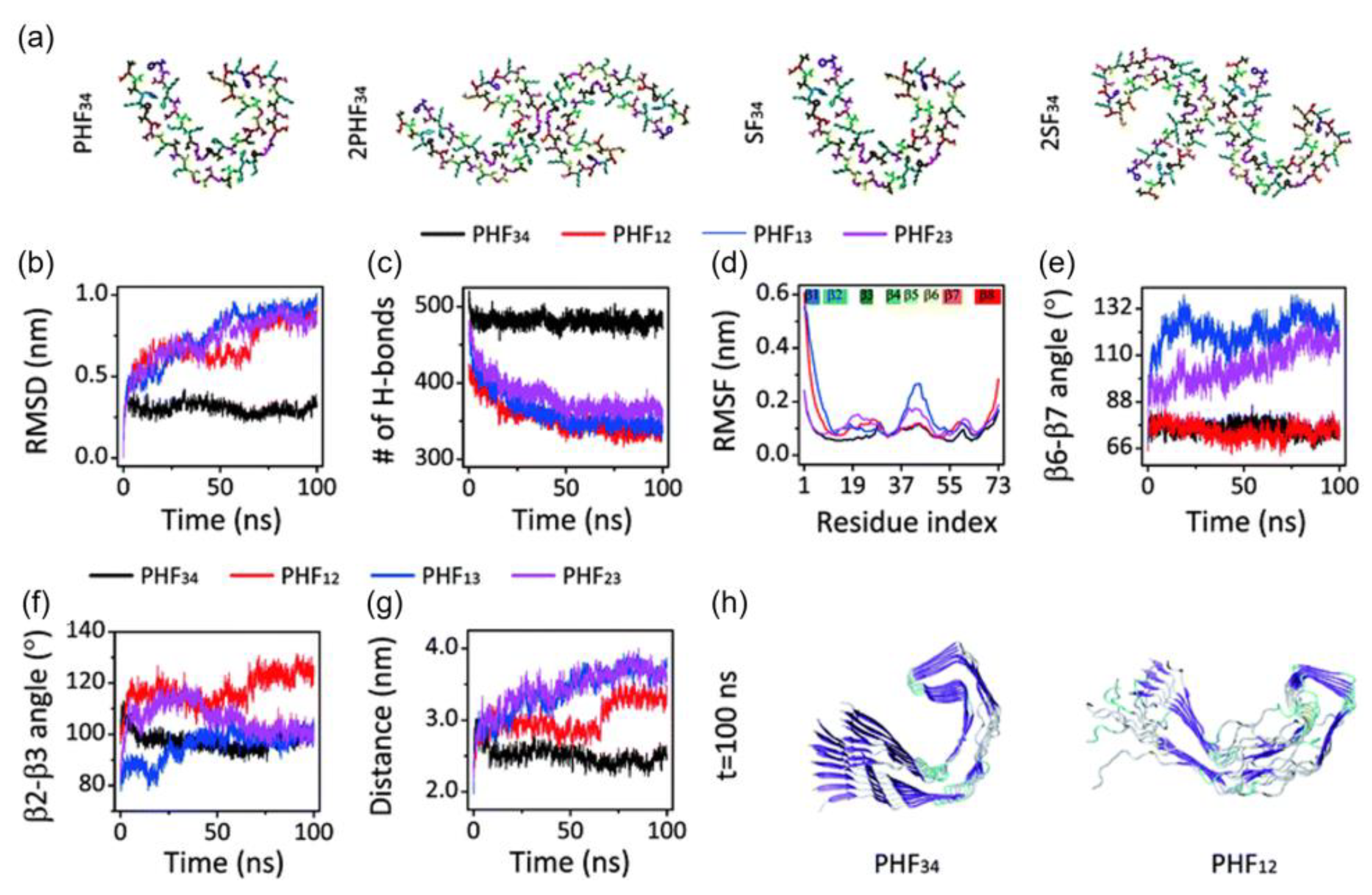
Figure 4. (a) Initial structure of one strand of protofilaments and two strands of filaments of the R3-R4 combination. The RMSD (b), H-bond, (c) and RMSF (d) trajectories for the four PHF protofilaments. All protofilament’s time evolution of β6-β7 (e) and β2-3 (f) angles. (g) The PHF34 Q351-I371; PHF12 Q288-I308; and PHF13 and PHF23 T319-V339 distances. (h) PHF34 and PHF12 snapshots (t = 100 ns).
Bhargava et al. studied the tau protein on lipid bilayers by CG MD and AA MD simulations [33]. The tau-SF structure (PDB ID:5O3T) was modeled with CG using Martini representations [34] to model the lipids and proteins. Water and 0.15 M KCl were added when calculating AA MD. The Nosé–Hoover thermostat was used to maintain the temperature at 310 K. CHARMM-GUI was used to generate the initial configurations for the fibrils and lipids. The simulations show that the tau proteins interact differently with the zwitterionic compared to the charged lipid membranes. The negatively charged POPG lipid membranes increase the binding tendency of tau fibrils. Fourteen systems consisting of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine (POPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylglycerol (POPG), and cholesterol (CHOL) in seven different compositions were simulated. Throughout the study, the symmetric composition of lipids in the upper and lower leaflets of the bilayer was used. In the case of the pure POPC system, 255 POPC molecules were randomly placed. Using the clustering algorithm, it was shown that the tau fibrils have different modes of interaction with the lipids. They found that the binding of the tau fibrils to the lipid bilayers coincides with the loss of β-sheet zones over the tau filament, which breaks the lipid bilayers. In another CG model, multiple atoms in proteins and lipids are approximated as a single bead and four water molecules are treated as a single particle (known as one bead 4:1 mapping) [29]. The beads can be distinguished by their polarity or hydrophilicity [29][35].
References
- Gustke, N.; Trinczek, B.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Domains of tau protein and interactions with microtubules. Biochemistry 1994, 33, 9511–9522.
- Limorenko, G.; Lashuel, H.A. Revisiting the grammar of Tau aggregation and pathology formation: How new insights from brain pathology are shaping how we study and target Tauopathies. Chem. Soc. Rev. 2022, 51, 513–565.
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190.
- Xu, S.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Characterization of tau fibrillization in vitro. Alzheimers. Dement. 2010, 6, 110–117.
- Wilson, D.M.; Binder, L.I. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer’s disease. Am. J. Pathol. 1997, 150, 2181–2195.
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553.
- von Bergen, M.; Barghorn, S.; Li, L.; Marx, A.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. Mutations of Tau Protein in Frontotemporal Dementia Promote Aggregation of Paired Helical Filaments by Enhancing Local β-Structure. J. Biol. Chem. 2001, 276, 48165–48174.
- Ait-Bouziad, N.; Lv, G.; Mahul-Mellier, A.-L.; Xiao, S.; Zorludemir, G.; Eliezer, D.; Walz, T.; Lashuel, H.A. Discovery and characterization of stable and toxic Tau/phospholipid oligomeric complexes. Nat. Commun. 2017, 8, 1678.
- Rajan, K.B.; Weuve, J.; Barnes, L.L.; McAninch, E.A.; Wilson, R.S.; Evans, D.A. Population estimate of people with clinical Alzheimer’s disease and mild cognitive impairment in the United States (2020–2060). Alzheimers. Dement. 2021, 17, 1966–1975.
- Stelzmann, R.A.; Norman Schnitzlein, H.; Reed Murtagh, F. An english translation of alzheimer’s 1907 paper, “über eine eigenartige erkankung der hirnrinde”. Clin. Anat. 1995, 8, 429–431.
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158.
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s Disease. In Protein Aggregation and Fibrillogenesis in Cerebral and Systemic Amyloid Disease; Harris, J.R., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 329–352.
- Drubin, D.G.; Kirschner, M.W. Tau protein function in living cells. J. Cell Biol. 1986, 103, 2739–2746.
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639.
- Bierer, L.M.; Hof, P.R.; Purohit, D.P.; Carlin, L.; Schmeidler, J.; Davis, K.L.; Perl, D.P. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch. Neurol. 1995, 52, 81–88.
- Buchhave, P.; Minthon, L.; Zetterberg, H.; Wallin, A.K.; Blennow, K.; Hansson, O. Cerebrospinal fluid levels of β-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch. Gen. Psychiatry 2012, 69, 98–106.
- Giacobini, E.; Gold, G. Alzheimer disease therapy--moving from amyloid-β to tau. Nat. Rev. Neurol. 2013, 9, 677–686.
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739.
- Selkoe, D.J. Treatments for Alzheimer’s disease emerge. Science 2021, 373, 624–626.
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Cryst. D Biol. Cryst. 1998, 54, 1078–1084.
- He, H.; Liu, Y.; Sun, Y.; Ding, F. Misfolding and Self-Assembly Dynamics of Microtubule-Binding Repeats of the Alzheimer-Related Protein Tau. J. Chem. Inf. Model. 2021, 61, 2916–2925.
- Chung, D.-E.C.; Roemer, S.; Petrucelli, L.; Dickson, D.W. Cellular and pathological heterogeneity of primary tauopathies. Mol. Neurodegener. 2021, 16, 57.
- Sahayaraj, A.E.; Viswanathan, R.; Pinhero, F.; Abdul Vahid, A.; Vijayan, V. Sequence-Dependent Conformational Properties of PGGG Motif in Tau Repeats: Insights from Molecular Dynamics Simulations of Narrow Pick Filament. ACS Chem. Neurosci. 2023, 14, 136–147.
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140.
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25.
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210.
- Brooks, B.R.; Brooks III, C.L.; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614.
- Nelson, M.T.; Humphrey, W.; Gursoy, A.; Dalke, A.; Kalé, L.V.; Skeel, R.D.; Schulten, K. NAMD: A Parallel, Object-Oriented Molecular Dynamics Program. Int. J. Supercomput. Appl. High Perform. Comput. 1996, 10, 251–268.
- Sinha, S.; Tam, B.; Wang, S.M. Applications of Molecular Dynamics Simulation in Protein Study. Membranes 2022, 12, 844.
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47.
- Lee, C.A.; Gasster, S.D.; Plaza, A.; Chang, C.I.; Huang, B. Recent Developments in High Performance Computing for Remote Sensing: A Review. IEEE J. Sel. Top. Appl. Earth Obs. Remote Sens. 2011, 4, 508–527.
- Kohnke, B.; Kutzner, C.; Grubmüller, H. A GPU-Accelerated Fast Multipole Method for GROMACS: Performance and Accuracy. J. Chem. Theory Comput. 2020, 16, 6938–6949.
- Li, X.; Dong, X.; Wei, G.; Margittai, M.; Nussinov, R.; Ma, B. The distinct structural preferences of tau protein repeat domains. Chem. Commun. 2018, 54, 5700–5703.
- Chowdhury, U.D.; Paul, A.; Bhargava, B.L. The effect of lipid composition on the dynamics of tau fibrils. Proteins 2022, 90, 2103–2115.
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824.
- Nguyen, P.H.; Ramamoorthy, A.; Sahoo, B.R.; Zheng, J.; Faller, P.; Straub, J.E.; Dominguez, L.; Shea, J.-E.; Dokholyan, N.V.; De Simone, A.; et al. Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer’s Disease, Parkinson’s Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis. Chem. Rev. 2021, 121, 2545–2647.
More
Information
Subjects:
Medical Informatics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
04 Apr 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No