The Golgi is the central organelle of the secretory pathway and it houses the majority of the glycosylation machinery, which includes glycosylation enzymes and sugar transporters. Correct compartmentalization of the glycosylation machinery is achieved by retrograde vesicular trafficking as the secretory cargo moves forward by cisternal maturation. The vesicular trafficking machinery which includes vesicular coats, small GTPases, tethers and SNAREs, play a major role in coordinating the Golgi trafficking thereby achieving Golgi homeostasis. Glycosylation is a template-independent process, so its fidelity heavily relies on appropriate localization of the glycosylation machinery and Golgi homeostasis. Mutations in the glycosylation enzymes, sugar transporters, Golgi ion channels and several vesicle tethering factors cause congenital disorders of glycosylation (CDG) which encompass a group of multisystem disorders with varying severities.
1. Introduction
Eukaryotic cells have a remarkable feature of intracellular compartments or organelles enclosed by lipid membranes. Compartmental identity is determined by a number of factors, including the membrane lipid and luminal composition as well as by the transmembrane and peripheral proteins associated with the organelle [
1,
2]. The secretory pathway transports cargo not only to the extracellular space for secretion but also between organelles. The endoplasmic reticulum (ER), the Golgi apparatus (GA) and the endolysosomal system are key organelles of this pathway. Proteins of the secretory pathway collectively referred to as cargo are synthesized in the ER by ribosomes. Upon completion of synthesis, the Golgi receives incoming cargo from the ER. Within the lumen of the Golgi, proteins undergo posttranslational modifications and are finally sorted into appropriate carriers targeted to their destination.
The GA is the central hub of the secretory pathway. It is composed of at least four morphologically distinct cisternae named
cis,
medial,
trans, and the
trans-Golgi network (TGN), each having its own identity. The GA functions as a station that processes and packages cargo into appropriate membrane carriers. The cargo arrives at the GA continuously and bidirectionally. Biosynthetic cargo is transported in an anterograde manner from the ER to the Golgi and then to the post Golgi compartments, while retrograde cargo is brought to the Golgi by the endosomal system. The currently accepted cisternal maturation model of Golgi transport describes that the anterograde cargo stays in the Golgi cisternae and is carried forward by the gradual maturation of the
cis-Golgi to the
trans-Golgi while Golgi resident proteins are cycled back to newly formed cisternae [
3,
4,
5,
6]. This way, the identity of each compartment is maintained even as the cisternal maturation goes on.
Lipids and proteins transported by the secretory pathway undergo modifications such as glycosylation within the Golgi. The different known types of glycosylation are classified according to the amino acid residue to which the carbohydrate is attached. In humans, N-glycosylation and O-glycosylation are the two main types. N-glycans are where glycans are attached to Asn in the consensus peptide sequence Asn-X-Ser/Thr, while O-glycans are where glycans are attached to hydroxyl groups of serine or threonine. N-glycosylation is a well-defined process that begins in the ER while O-glycosylation is more varied and mostly begins in the Golgi [
7,
8,
9,
10]. Protein glycosylation includes the addition of N-linked glycans, O-linked glycans, phosphorylated glycans, glycosaminoglycans and glycosylphosphatidylinositol (GPI) anchors to peptide backbones as well as C-mannosylation of tryptophan residues [
11]. The Golgi houses the majority of the glycosylation machinery within its cisternae. The glycosyltransferases, glycosidases together with their accessory machinery, modify and process glycans as they traverse the Golgi. At the TGN, fully processed glycoconjugates are packaged into carrier membranes and transported to appropriate destinations.
The glycosylation of macromolecules is involved in nearly every biological process and its defects give rise to various pathologies [
11,
12]. Glycosylation defects affect multiple organ systems with varying severities, and they fall under the umbrella of congenital disorders of glycosylation (CDG). CDGs are autosomal recessive inherited diseases due to mutations in genes involved in glycosylation [
13,
14]. CDGs are further subdivided into type I (CDG-I) and type II (CDG-II) [
15,
16]. CDG-I is due to defects in the transfer of preassembled dolichol-glycan in the ER (or cytosol). Defects in glycan processing in the Golgi give rise to CDG-II. Formerly, a CDG diagnosis was made only based on the isoelectric focusing of serum transferrin which only detected N-glycosylation defects. Nowadays, glycan mass spectrometry and/or ApoCIII IEF are often used to complement transferrin IEF testing [
17].
2. Glycosylation Enzyme Compartmentalization
Apart from the glycosylation enzymes, the Golgi glycosylation machinery also includes ion channels and sugar transporters that maintain intraluminal pH and bring in cofactors and charged monosaccharides required for glycosylation. The key quality control for glycosylation is the proper cisternal compartmentalization of the glycosylation machinery [
9,
18,
19,
20]. Glycan processing/addition occurs in the sequence in which the enzymes encounter their substrates. As the
cis-Golgi matures, eventually becoming the TGN and carrying cargo forward, the correct compartmentalization of the glycosylation machinery must be maintained within the framework of cisternal maturation. At the TGN, the cargo is sorted into appropriate anterograde carriers targeted to various destinations.
All Golgi glycosylation enzymes are transmembrane (TM) proteins and the majority of them are type II TM proteins. They possess a catalytic C-terminal domain in the lumen, a luminal stem region, a transmembrane domain (TMD) and a short N-terminal cytosolic tail. Evidence suggests that the cytoplasmic tail and TMD of Golgi enzymes, cytosolic adaptor and coat proteins, and membrane lipid composition play a role in the sorting and correct cisternal localization of the glycosylation machinery.
“Kin recognition” was the earliest proposed model for enzyme retention in the Golgi [
24]. Kin oligomerization was first identified in the medial Golgi enzymes N-Acetylglucosaminyltransferase-1 (GlcNAcT1/MGAT1) and Mannosidase-2 (ManII/MAN2A1) [
25]. These enzymes interact with each other via their TMDs and stem regions. Because enzyme oligomers would be too big to enter transport vesicles exiting the Golgi, they are retained in the Golgi. The enzyme α-2,6-sialyltransferase-1 (ST6Gal1) is believed to possess trans Golgi localization signals in its cytoplasmic tail, TMD and stem region because mutations in these regions lead to the secretion of the enzyme [
26,
27]. ST6Gal1 not only forms homodimers but also heteroligomerizes with β-1,4-Galactosyltransferase-1 (B4GALT1) [
28]. All the currently known oligomers of the known Golgi enzymes in various glycosylation pathways have been described by de Graffenried et al. in [
29].
Transmembrane domain-dependent sorting relies on compatibility between the TMD and membrane thickness [
30]. Membrane thickness is believed to gradually increase from the ER to the PM due to different lipid compositions. Phospholipids are enriched in ER and Golgi membranes, while the enrichment of sphingolipids and sterols gradually increases in the TGN and post-Golgi compartments with the highest abundance in the PM. An extensive and detailed comparison of the TM proteins found that ER and Golgi residents have shorter TMDs compared to post-Golgi residents and bulkier residues were distributed closer to the exoplasmic side of the bilayer TMDs [
31]. Simply stated, shorter TMDs reside in thinner membranes. Thus, Golgi enzymes are kept from entering the post-Golgi compartments by the mere incompatibility of their short TMDs in thicker membranes.
Golgi enzymes can cycle between the Golgi cisternae [
32,
33] as well as between the Golgi and ER [
34], and the majority of recycling occurs in COPI-coated vesicles. Yeast α-1,6-mannosyltransferase (Mnn9p) is one such example [
35,
36]. This
cis-Golgi enzyme is retrieved in COPI vesicles at the TGN [
35]. A few other glycosylation enzyme complexes such as the Man-Pol-I (Mann9p and Van1p) and Man-Pol-II (Mnn9p, Hoc1p, Anp1, Mnn10p, and Mnn11p), in yeast, cycle through the ER in COPI vesicles. Selective sorting into retrograde COPI-coated carriers either occurs via direct interaction between the cytosolic tails or via an adaptor. Sorting signals—KKXX or RXR on ER residents [
37] or φ(K/R)XLX(K/R) on some
cis-Golgi enzymes—directly interacting with COPI subunits have been identified on the cytoplasmic tails of a subset of enzymes [
38,
39,
40]. Vps74 is a COPI interacting protein that was identified as a protein essential for the proper localization of a subset of glycosylation enzymes in yeast [
41]. Vps74 recognizes the ([F/L][L/I/V]XX[R/K]) peptide motif on the cytoplasmic tails of a subset of glycosyltransferases [
42]. GOLPH3 and GOLPH3L are human homologs of Vps74. They have an affinity for PI4P enriched in the trans-Golgi and recognize membrane-proximal polybasic residues on the cytoplasmic tails of a subset of Golgi enzymes, including ST6Gal1 and N-acetylgalactosaminyltransferase-2 (GALNT2) [
33,
43]. About half the number of enzymes involved in glycosphingolipid synthesis have an LXX[R/K] consensus motif recognized by GOLPH3 on their cytosolic tails [
44].
These mechanisms of enzyme compartmentalization within the Golgi are not mutually exclusive.
3. Membrane Trafficking Machinery
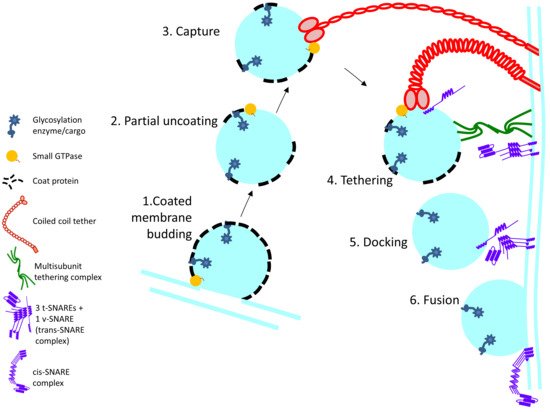
The membrane trafficking machinery moves the cargo in membrane-enclosed carriers to various destinations within the cell or releases secretory cargo at the plasma membrane. Every compartment in the secretory pathway is equipped with its own set of molecular players for transport. This machinery can be broadly categorized based on its function as budding, tethering and fusion machinery (
Figure 1). The events of budding and fusion are controlled by a set of GTPases that modulate the activities of the trafficking machinery. Coat proteins together with cargo adaptors bind to the cargo, recruit them into specialized domains and bud these membrane domains off in the form of small vesicles or tubules [
45,
46]. COPI, COPII and Clathrin are the three known coat proteins. COPI is involved in the intra-Golgi and Golgi to ER retrograde trafficking, COPII is involved in the ER to Golgi anterograde trafficking and Clathrin is involved in TGN-plasma membrane bidirectional transport. Arf1 and Sar1 are small GTPases that are involved in vesicle formation. Membrane localized active Arf1-GTP recruits COPI and clathrin components while Sar1-GTP recruits the COPII components [
47]. Membrane curvature is triggered by dimerization of Arf1-GTP [
48]. Fission is thought to be driven by the interaction between the GTPases’ amphipathic helices and membrane lipids [
48]. The membrane-enclosed cargo carrier is then tethered to its specific target membrane. Alignment of the fusion machinery on the two opposing membranes results in membrane fusion and the delivery of cargo to its destination. The function and intricate interactions between all the factors at play in cellular transport are yet to be fully understood.
Figure 1. Schematic outline of the steps involved in Golgi enzyme recycling: 1. Membrane budding at the donor membrane is initiated by recruitment of coat proteins which directly or indirectly interact with cargo (glycosylation enzyme) 2. and 3. After budding, the coat protein partially falls off and the vesicle is captured by long coiled-coil tethers. 4. Among other interactions, MTCs interact with the SNAREs on the vesicle and target membrane, thereby facilitating SNARE alignment 5. and 6. Priming of the SNAREs and formation of the trans-SNARE complex leads to vesicle fusion at the target membrane and cargo delivery.
 +1 credit
+1 credit