Metabolic syndrome (MetS) is a cluster of metabolic risk factors for diabetes, coronary heart disease, non-alcoholic fatty liver disease, and some tumors. It includes insulin resistance, visceral adiposity, hypertension, and dyslipidemia. MetS is primarily linked to lipotoxicity, with ectopic fat deposition from fat storage exhaustion, more than obesity per se. Excessive intake of long-chain saturated fatty acid and sugar closely relates to lipotoxicity and MetS through several pathways, including toll-like receptor 4 activation, peroxisome proliferator-activated receptor-gamma regulation (PPARγ), sphingolipids remodeling, and protein kinase C activation. These mechanisms prompt mitochondrial dysfunction, which plays a key role in disrupting the metabolism of fatty acids and proteins and in developing insulin resistance.
1. Unveiling Lipotoxicity from LCSFA
There is a positive direct association between LCSFA consumption and MetS involving lipotoxicity that was previously explored in a systematic review
[1]. Lipotoxicity is a term first coined by Young Lee and Roger H. Unger in 1994 to describe ẞ-cell abnormalities related to increased plasma levels of fatty acyl-CoA leading to insulin resistance. In their experiment, in obese prediabetic rodents, increased plasma free fatty acids (FFA) and triacylglycerol content in pancreatic islets preceded the loss of glucose-stimulated insulin secretion and correlated with glucose levels. These effects were alleviated by caloric restriction
[2].
In obese conditions, increased plasma FFA is observed following increased LCSFA intake, as well as saturation of fat storage and loss of adipose tissue expandability
[3]. Excess plasma FFA levels prompt a compensatory response to counteract lipid overload. This response includes: (1) triacylglycerol incorporation into fat cells for storage as “lipid droplets” (LD); (2) activation of oxidative programs in mitochondria and peroxisome; and (3) membrane lipid remodeling in cell membranes, with sphingolipids’ generation and functional membrane microdomains’ remodeling, or the “lipid rafts”
[4].
Once fat storage is exhausted and the mitochondrial capacity of fatty acid oxidation is surpassed, the incomplete FA oxidation increases the generation of an intermediate metabolite (acylcarnitine), increases the lipid membrane remodeling through ceramide synthesis, and leads to an ectopic fat accumulation
[4]. Several self-perpetuating mechanisms have been proposed to explain lipid-induced insulin resistance in MetS as listed in
Table 1.
Table 1. Mechanisms proposed to explain insulin resistance induced by lipids.
| Causes |
Consequences |
References |
| TLR4 activation in adipocytes, macrophages, and skeletal cells |
Pro-inflammatory cytokine production (TNF-α, IL-1ẞ, IL-6) |
[5] |
| PKC activation |
↓ insulin-stimulated IRS-1 tyrosine phosphorylation |
[6][7][8] |
| Mitochondrial dysfunction |
↑ oxidative stress and anaplerosis |
[9][10][11] |
| ↓ n3-PUFAs intake |
↑ pro-inflammatory cytokine production and impaired insulin signaling |
[12][13] |
| UPR and JNK activation |
ER stress |
[10][14] |
Experimentally, excessive exogenous LCSFA intermediates the disruption of mitochondrial function, leading to mitochondrial damage and apoptosis. This effect can occur in different ways: mitochondrial membrane depolarization, mPTP opening, and ROS generation
[15]. LCSFA are precursors of ceramides during de novo synthesis, which have been pointed out as central players in lipotoxic mitochondrial dysfunction (see
[16]). Ceramides are key molecules of sphingolipid metabolism, and their generation involves several metabolic pathways, including sphingomyelin hydrolysis by sphingomyelinase and de novo synthesis by ceramide synthase (CerS)-isoforms 1 to 6
[17].
Ceramides interact with oxidized cytochrome c from mitochondria; decrease mitochondria transmembrane potential; increase ROS and oxidative stress; and initiate the mitochondrial outer membrane permeabilization with cytochrome c release, caspase activation, and apoptosis in a dose-dependent manner
[18][19]. These events might be prevented by enhancing LCSFA oxidation through increased carnitine-palmitoyl-transferase 1 expression (which shuttles FA into mitochondria through ẞ-oxidation) or by glutathione activity
[19][20].
CerS expression has different tissue distribution and acyl chain specificity. These properties seem to affect the effect of CerS. For instance, CerS6 activity favors insulin resistance and diet-induced steatohepatitis, while CerS2 activity (C22:0, C24:0, C24:1), mainly expressed in the liver, seems to protect from these conditions
[21]. Thus, the ceramide composition also seems to have a functionally relevant aspect. In a prospective 7-year follow-up trial, baseline plasma levels of saturated fatty acid chain ceramides were positively associated with higher triacylglycerols levels, retroperitoneal and intraperitoneal fat masses, and homeostatic model assessment of insulin resistance (HOMA-IR), while they were negatively associated with high-density lipoprotein (HDL) cholesterol, adiponectin, and subcutaneous fat. Interestingly, unsaturated fatty acid ceramides had the opposite relation regarding visceral and subcutaneous fat and HOMA-IR
[22].
Understanding the factors influencing CerS activity and the ceramide composition is relevant when considering that ceramides can impact health. For instance, in the PREDIMED study, a high ceramide concentration (a sum of C16:0, C22:0, C24:0, and C24:1 ceramides) at baseline was correlated to increased cardiovascular disease risk
[23]. Similarly, baseline circulating Cer16, Cer18, Cer20, and Cer22 were associated to a higher diabetes risk
[24]. In a cross-sectional analysis, individuals with type 2 diabetes (T2D) had higher Cer18:0, Cer20:0, Cer24:1, and total ceramides levels, where elevated Cer18:0 levels were inversely correlated with insulin sensitivity and directly correlated with circulating TNF-α levels
[25].
Bariatric procedures are highly efficient for T2D therapy. In a recent study, patients with obesity and T2M who underwent Roux-en-Y gastric bypass who exhibited low serum ceramide levels at baseline, and those who presented ceramides decrease from the baseline to the second postoperative year, experienced persistent T2D remission 12 years after surgery. Using a linear mixed effect model ceramides inversely predicted T2D remission, independent of changes in body weight. These observations suggest a metabolic contribution of ceramide on insulin sensitization and T2D resolution independent of weight loss
[26]. Indeed, increased ceramide transport in LDL also is found in T2D and does not correlate with obesity severity, but with insulin resistance. Moreover, the infusion of LDL ceramide in a mice model impaired insulin action and glucose homeostasis
[27].
There is a special role for lipid oversupply and ceramide generation in metabolic disturbances. Increased plasma FFA and total muscle ceramides (primarily C18:1, C20:0, C22:0, C24:1, C24:0) are observed in individuals with obesity and T2D, as well as impaired muscle FFA oxidation in obese premenopausal women and individuals with T2D
[28][29][30] Furthermore, there is an inverse relation between visceral adiposity and insulin-stimulated FFA uptake
[30]. Curiously, ceramides do not seem to interfere with whole-body fat oxidation in an individual without T2D, whereas a persistent lipid oversupply results in excessive ceramide muscle accumulation in people with T2D
[28].
Beyond lipotoxicity by sphingolipids, dietary quality and quantity of fat intake are associated with epigenetic regulation of energy and lipid metabolism through DNA methylation
[31][32]. PGC1-α hypermethylation is associated with reduced gene expression and reduced mitochondrial DNA (mtDNA) content. These alterations are increased by exposing cells to free fatty acids
[33][34]. The epigenetic modifications of liver mtDNA have been linked to insulin resistance and the severity of NAFLD
[35][36]. MtDNA alterations precede mitochondrial dysfunction either with increased mtDNA content (not functional) or with decreased content and reduced oxidative phosphorylation
[37]. Studies with NAFLD in mice show that a HFD is associated with a reduced half-life of mitochondrial proteins along with ATP deficiency
[38].
Mitochondrial dysfunction in MetS is also supported by increased plasma levels of long acyl-carnitines (AcylCNs) and free carnitines (CNs) in patients with obesity and T2D. The formers are intermediate FA metabolites that play significant roles in cellular energy metabolism. Increased circulant levels of these molecules suggest incomplete ẞ oxidation of long-chain fatty acids. In T2D, increased medium-chain AcylCNs (C10- to C14) were associated with nuclear factor kappa B (NFkB) pathway activation. Thus, circulant AcylCNs and free CNs are helpful markers of mitochondrial and peroxisomal oxidation function
[39][40].
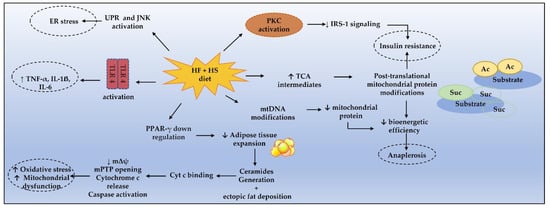
Altogether, these findings support the central role of long-chain ceramides’ generation from dietary LCSFA in lipotoxicity, which along with the mechanisms illustrated in Figure 1, prompts mitochondrial dysfunction and MetS pathogenesis.
Figure 1. Mechanisms enrolling metabolic dysfunction in MetS. Cyt c, cytochrome c; ER, endoplasmic reticulum; HF, high fat; HS, high sugar; IRS-1, IL-1ẞ, interleukin 1ẞ; IL-6, interleukin 6; insulin receptor substrate 1; JNK, c-Jun N-terminal kinase; mPTP, mitochondrial permeability transition pore; mΔψ, mitochondrial transmembrane potential; PKC, protein kinase C; PPAR-γ, peroxisome proliferator-activated receptor γ; TLR4, toll-like receptor 4; UPR, unfolded protein response, TNF-α, tumor necrosis factor α.
2. Protein Modifications from Fructose- and Sugar-Sweetened Foods
Beyond a high-fat diet (HFD), a high-sugar diet also is associated with MetS
[41]. Hepatic epigenetic and post-translational mitochondrial proteins’ modifications (PTM) have been described following long-term sugar intake, including mitochondrial DNA hypomethylation with protein hyperacetylation and/or hypo-succinylation (when combined with HFD). These modifications can rise from an accumulation of acetyl-coenzyme A, as an intermediate of the TCA, leading to disrupted glucose, lipid, and protein metabolism
[11][42].
Fructose metabolism induces the activation of adenosine monophosphate deaminase, leading to uric acid generation and mitochondrial oxidative stress through distinct pathways. These include the activation of nicotinamide adenine dinucleotide phosphate oxidase subunit NOX4, aconitase inhibition (TCA cycle enzyme), and citrate overload
[43]. L6 myotubes exposed to high fructose concentration were shown to induce mitochondrial dysfunction due to reduced mitochondrial enzyme activity, decreased mitochondrial membrane potential and mitochondrial electron transport chain, and disrupted energy metabolism. In turn, these events lead to increased ROS, reactive nitrogen species, and apoptosis
[44].
It is suggested that the impact of sugars on MetS risk may be related to its caloric component. A United States survey with teenagers correlated increased sugar ingestion with progressive MetS risk, despite body mass index (BMI) values, physical activity, and total energy intake, mainly when consumed above 70 g/day
[45]. Conversely, a meta-analysis showed an association of fructose consumption to MetS only when this was consumed as extra energy in hypercaloric diets (>+21% to 35% extra energy)
[46]. In a large cohort study, the total carbohydrate intake showed an association with mortality where the lower mortality risk ranged between 50 and 55% of total energy intake
[47].
The dietary source of sugar is an important issue concerning MetS risk, with sugar-sweetened beverages (SSB) conferring a higher risk and yogurt and fruits conferring a lower risk
[48]. Together with foods with added sugar, SSB consumption is often a source of high sugar/fructose intake in children, adolescents, and adults and has been linked to insulin resistance and MetS
[45][48]. In the Framingham Offspring Cohort, the frequency of SSB consumption correlated to plasma Cer16:0, Cer22:0, and Cer24:0. In individuals with prediabetes or T2D, plasma Cer24:0 correlated to more recurrent SSB ingestion
[49]. In addition, a meta-analysis showed that subjects consuming more than 1–2 serving of SSB/day exhibited a 26% and 20% higher risk of developing T2D and MetS, respectively
[50]. Curiously, artificially sweetened beverages (diet or non-carbohydrate low calorie foods) have shown a linear dose–response relationship in MetS risk
[51].
In the KNHANES survey (2007–2014), a higher carbohydrate intake (≥ 74.2% of energy intake) correlated to MetS risk in women irrespective of dietary lipid composition
[52]. Nevertheless, the long-term association of high fat + high sugar diet (HFSD) seems to be the most deleterious combination, exacerbating every isolated nutrient overload toxicity and culminating in mitochondrial inefficiency, reduced fatty acid utilization, and tissue lipid overload. Indeed, the HFSD diet has been used as an effective experimental model to induce MetS in rats
[53].
Overall, a high-sugar diet prompts hepatic epigenetics and PTM, which are related to impaired glucose, lipid, and protein metabolism along with mitochondrial dysfunction. Therefore, sugar intake should be discouraged in individuals aiming to maintain a healthy status or to manage MetS. Artificially sweetened beverages (with no sugar) also have shown to be positively associated to MetS, but the mechanisms enrolled remain poorly elucidated.
 +1 credit
+1 credit