Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Weicheng Zhang | -- | 3597 | 2023-03-14 11:37:54 | | | |
| 2 | Jason Zhu | -1 word(s) | 3596 | 2023-03-15 03:13:06 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zhang, H.; Cai, J.; Yu, S.; Sun, B.; Zhang, W. Anticancer Small-Molecule Agents Targeting Eukaryotic Elongation Factor 1A. Encyclopedia. Available online: https://encyclopedia.pub/entry/42173 (accessed on 20 July 2026).
Zhang H, Cai J, Yu S, Sun B, Zhang W. Anticancer Small-Molecule Agents Targeting Eukaryotic Elongation Factor 1A. Encyclopedia. Available at: https://encyclopedia.pub/entry/42173. Accessed July 20, 2026.
Zhang, Han, Jiayou Cai, Siqi Yu, Bin Sun, Weicheng Zhang. "Anticancer Small-Molecule Agents Targeting Eukaryotic Elongation Factor 1A" Encyclopedia, https://encyclopedia.pub/entry/42173 (accessed July 20, 2026).
Zhang, H., Cai, J., Yu, S., Sun, B., & Zhang, W. (2023, March 14). Anticancer Small-Molecule Agents Targeting Eukaryotic Elongation Factor 1A. In Encyclopedia. https://encyclopedia.pub/entry/42173
Zhang, Han, et al. "Anticancer Small-Molecule Agents Targeting Eukaryotic Elongation Factor 1A." Encyclopedia. Web. 14 March, 2023.
Copy Citation
Eukaryotic elongation factor 1A (eEF1A) canonically delivers amino acyl tRNA to the ribosomal A site during the elongation stage of protein biosynthesis. Yet paradoxically, the oncogenic nature of this instrumental protein has long been recognized. Consistently, eEF1A has proven to be targeted by a wide assortment of small molecules with excellent anticancer activity, among which plitidepsin has been granted approval for the treatment of multiple myeloma. Meanwhile, metarrestin is under clinical development for metastatic cancers.
anticancer
eEF1A
mechanism
macrocycle
1. Didemnins and Tamandarins
Didemnins are a family of marine cyclic depsipeptides with strong anticancer, antiviral, and immunosuppressive activities [1]. Since their initial discovery in the early 1980s [2], these macrocycles have become the subject of intense research over the last four decades. Among them, didemnin B (1, Figure 1) and plitidepsin (2) have entered multiple clinical trials. Gratifyingly, plitidepsin was approved in Australia for treating multiple myeloma [3]. With nearly identical architectures but subtly different macrocyclic backbones (highlighted red), tamandarins such as tamandarin A (3) were discovered from a different colony of marine ascidian [4]. As already reviewed in multiple comprehensive monographs [5][6][7][8][9][10][11][12], these compounds will not be reiterated here. Instead, a brief update is presented below on their anticancer mode of action.
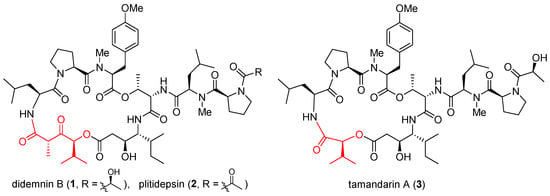
Figure 1. Structures of didemnin B (1), plitidepsin (2), and tamandarin A (3).
Having developed functional signature ontology (FUSION) maps for drug discovery and mechanistic elucidation [13], White et al. revisited the mechanism of 1 and found that it induces rapid and extensive apoptosis in sensitive cancer cell lines through concomitant inhibition of palmitoyl-protein thioesterase 1 (PPT1) and eEF1A1 [14]. Independently, Galmarini et al. showed that eEF1A2 is the specific binding target of 2 with a measured dissociation constant (KD) of 80 nM [15]. Since translation inhibition cannot account per se for the observed antiproliferative effect of 2, it was suspected that this drug impacts non-canonical functions of eEF1A2. Indeed, double-stranded RNA (dsRNA)-dependent protein kinase (PKR) was later identified as a novel binding partner of eEF1A2 [16]. In this case, eEF1A2 interacts directly with PKR to block its pro-apoptotic activity and boost tumor survival. In the presence of 2, however, PKR was disengaged from eEF1A2, thereby regaining its kinase activity to initiate extrinsic apoptosis through activation of MAPK and NF-κB signaling cascades [16]. More recently, Martinez-Leal et al. reported that 2 induces endoplasmic reticulum (ER) stress in HeLa cells by activating the multipronged unfolded protein response (UPR) in a characteristic pattern [17]. Working simultaneously as an ER stress inducer and an autophagy inhibitor, 2 was combined with bortezomib to synergistically block proteasomal degradation and autophagy, thereby exacerbating accumulation of misfolded proteins that originate from plitidepsin-induced oxidative stress. Such elevated proteotoxic stress led to apoptosis both in vitro (in MM1S multiple myeloma cells) and in vivo (in SCID mice xenografted with RPMI-8226 multiple myeloma cells).
2. Cytotrienin A and Ansatrienin B
Cytotrienin A (4, Figure 2) was initially isolated from the culture broth of soil-dwelling Streptomyces sp. RK95-74 [18]; this compound has strong cytotoxicity (IC50 = 7.7 nM) against human leukemia cell line HL-60 [19]. Despite early mechanistic studies [20][21][22], its target remained elusive until Pelletier et al. identified 4 as a translation inhibitor through high-throughput screening [23]. Their finding is that akin to didemnin B, cytotrienin A modulates eEF1A-dependent loading of aa-tRNA to the ribosome, most likely by stabilizing the eEF1A/GTP/aa-tRNA assembly positioned at the ribosomal A site. Thus, without release of eEF1A from the ribosome, translation elongation stops. Further insight into this compound’s mode of action came from a 2015 study led by Taunton, who aimed to seek out the target of a potent antiproliferative ternatin derivative 7 (ternatin-4, cross-refer to the following section) [24]. With the help of photoaffinity labeling, they were able to capture the binding partner with a ternatin-based probe. Interestingly, the photolabeled protein is a ternary complex comprising eEF1A, GTP, and aa-tRNA rather than eEF1A alone. Subsequent competitive-binding experiments noted that the photolabeling efficiency is diminished dose-dependently with the addition of didemnin B or ansatrienin B (5, a close side-chain analogue of cytotrienin A shown in Figure 2). Therefore, it was concluded that ternatin, didemnin B, and cytotrienin A/ansatrienin B may share a binding hot spot on the eEF1A surface, probably located near A399, as indicated by resistance-conferring mutation experiments.
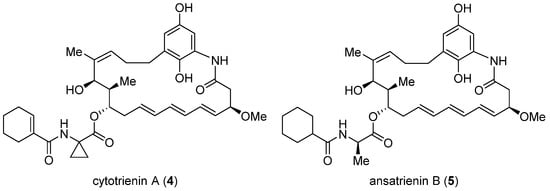
Figure 2. Structures of cytotrienin A (4) and ansatrienin B (5).
3. Ternatin-4
The highly cytotoxic natural product A3, together with several other congeners, was isolated from an Aspergillus fungus [25]. Although its structure was determined to be a partially N-methylated cyclic heptapeptide, the chirality of 7 out of 11 stereo-centers (marked in the structure of A3, Figure 3) remained unknown. The strong structural similarity between A3 and ternatin (6, CAS registry number: 148619-41-4) [26], another natural product with anti-obesity activity [27], inspired Taunton et al. to design ternatin-4 (7) by incorporating the dehydromethyl leucine and pipecolic acid residues of A3 (highlighted red in the structure of 7, Figure 3) into 6 [24]. The resulting hybrid molecule 7 attained more than 10-fold enhancement of potency over the parent compound 6 (IC50 4.6 nM vs. 71 nM against HCT-116 cancer cell line), thus solving all but one stereo-configuration of A3. Wondering the molecular target of ternatins, they developed a bifunctional photoaffinity probe 10. Under UV irradiation, its photolabile diazirine subunit at residue 4 (highlighted red) decomposes into a highly reactive carbene that instantaneously crosslinks to the nearby binding protein. The alkyne at residue 6 (highlighted blue) will then connect to a fluorescent reporter via click cycloaddition, thus tagging the photolabeled target for characterization. In this way, eEF1A was captured and confirmed as the target of ternatins.
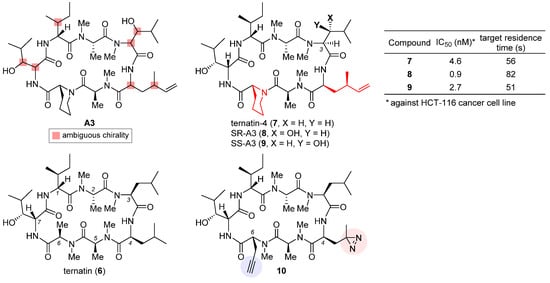
Figure 3. Structures of natural products A3 and ternatin (6), and synthetic analogues ternatin-4 (7), SR-A3 (8), SS-A3 (9), and photoaffinity probe 10. The inset table compares anticancer activity and target residence time of 7–9.
Based on the verified structure of ternatin-4 (7), a β-hydroxyl group was introduced at its residue 3 to obtain two epimers of A3, namely SR-A3 (8) and SS-A3 (9). An improved second-generation total synthesis allowed quick access to both compounds [28]. Hence, the identity of natural product A3 was established as SR-A3 (8). Of special note, despite a minor difference in the side chain of residue 3, 8 was found to display a more prolonged duration of growth inhibition action than 7 and 9. Single-molecule fluorescence resonance energy transfer (smFRET) imaging corroborated this observation, while further quantification through in vitro chase experiments confirmed enhanced drug–target residence time (table inset in Figure 3) and rebinding kinetics of 8. Finally, preclinical evaluation of 8 vis-a-vis 7 was carried out in an aggressive Myc-driven mouse lymphoma model. Compared with its des-hydroxyl variant ternatin-4 (7), SR-A3 (8) significantly reduced tumor burden while extending the survival of the treated Eμ-Myc mice [28]. More recently, with the help of smFRET imaging and cryogenic electron microscopy (cryo-EM), Taunton and collaborators demonstrated that in spite of sharing a common eEF1A-binding site, ternatin-4 (7) and didemnin B exhibit differential inhibition dynamics in that the former traps the eEF1A/GDP/aa-tRNA ternary complex on the ribosome in a more reversible fashion than does the latter [29]. Their in-depth mechanistic investigation also revealed that by trapping eEF1A at the ribosomal A site, ternatin-4 induces ubiquitination and degradation of eEF1A on stalled ribosomes through a previously unknown surveillance pathway for translation quality control [30].
4. Nannocystin A
Nannocystin A (11, Figure 4) is a 21-membered cyclic depsipeptide isolated independently by Brönstrup et al. [31] and Hoepfner et al. [32] from the myxobacteria of the Nannocystis genus. Brönstrup et al. found that 11 is a strong inducer of apoptosis, as such inhibiting the growth of multiple cancer cell lines with low nanomolar IC50 values [31]. Meanwhile, another team led by Hoepfner pinned down its target through a combination of genetic and chemoproteomic approaches [32]. In brief, initial haploinsufficiency profiling and mutagenesis experiments implied eEF1A as the most likely target. To verify direct binding, they set up affinity chromatography with the semisynthetic probe 12 (Figure 4). During elution, this immobilized nannocystin sequestered eEF1A1 and eEF1A2 out of the 3644 proteins comprising the HCT-116 cell lysates. Moreover, it competed with unbound nannocystin A (11) and didemnin B for binding to eEF1A. Hence, eEF1A was determined to be the target of nannocystin A [32].
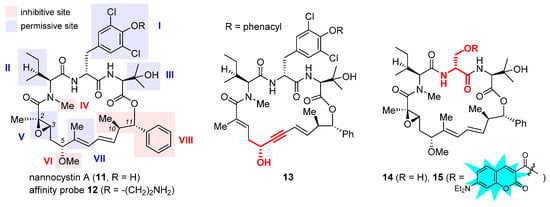
Figure 4. Structures of naturally occurring nannocystin A (11), semisynthetic affinity probe 12, as well as synthetic macrocyclic propargylic alcohol 13, serine-containing intermediate 14, and coumarin-tagged fluorescent probe 15. The SAR illustrated in the structure of 11 consists of inhibitive (red) and permissive (blue) sites: the former must be reserved for high activity, whereas the latter can tolerate moderate change.
The firsthand structure–activity relationship of nannocystins was derived from isolated and semisynthetic nannocystins [31][32], which indicates that modification at the tyrosine phenol moiety (subdomain I in the structure of 11, Figure 4) or the side chain of N-Me-L-isoleucine (subdomain II) is tolerated. To obtain comprehensive SAR, nevertheless, total synthesis is a must. Thus far, seven routes have been reported for the total syntheses of nannocystin A (11) or its 2E-alkene surrogate nannocystin Ax (16, structure shown in Table 1) [33][34][35][36][37][38][39][40], each involving a distinct macrocyclization reaction as the key strategic step [41]. With the dual purpose of (1) total synthesis and (2) SAR validation concerning the binding role of the polyketide C5-C7 region, Fürstner et al. devised a motif-oriented strategy so that the macrocyclic propargylic alcohol 13 underwent post-macrocyclization elaboration [42] into an array of novel analogues besides nannocystin Ax (16) [39]. It was found that the 5R-methoxy ether (subdomain VI), instead of the neighboring C6-C7 (E)-alkene (subdomain VII), must be reserved for high activity. As an illustration, Table 1 compares the anticancer activity of four pairs of nannocystin derivatives with or without methylation at the C5-OH group (R = Me or H). Clearly, removal of this moiety causes a drastic reduction in potency (16 vs. 17, 18 vs. 19, 20 vs. 21, 22 vs. 23); on the other hand, changing the C6-methyl (16) to a fluorine (18) or hydrogen (20) atom, or even curtailing the (Z) alkene to an alkyne (22), has insignificant impact on activity.
Following a total synthesis of nannocystin A [35] via Heck macrocyclization [43][44], researchers prepared a diversity of non-natural nannocystins modified at different sites. The findings demonstrated that (1) the (2R, 3S)-epoxide (subdomain V) may be substituted for a 2E-alkene without compromising activity [45], (2) the side chain of β-OH-L-valine (subdomain III) is tolerant of minor change [45], and (3) the polyketide C9-C10 segment including its entire (10R, 11S) stereo-chemistry (subdomain VIII) is a key determinant of potency [46][47]. In parallel, He et al. synthesized more variants via Heck macrocyclization too and observed that removal of the N-methyl moiety (subdomain IV) incurred a dramatic loss of activity [48]. Taken together, the SAR of nannocystins is illustrated in the structure of 11.
Aside from target elucidation and SAR profiling, the exact mechanism of nannocystins is a subject of enduring interest [49][50][51] given the poorly understood role of eEF1A in tumorigenesis. Chen et al. showed that the antimetastatic effect of nannocystin Ax in lung cancer cells is attributable to its interference with the TGF-β/Smad signaling pathway [49]. Their result is in step with another study uncovering the promigratory ability of eEF1A2 to promote lung adenocarcinoma metastasis [52]. The additional finding that the regulation of TGFβI (TGF β receptor I) by nannocystin Ax occurs at the transcriptional rather than the (post-)translational level implied the presence of an alternative mechanism independent of eEF1A inhibition [49]. Therefore, similar to the case of plitidepsin [16], the possibility that nannocystins impact certain protumorigenic pathway(s) mediated by eEF1A cannot be ruled out. Recently, researchers designed a serine-incorporating nannocystin 14 (Figure 4) to leverage a post-macrocyclization diversification strategy for efficient side-chain variation [51]. Thus obtained SAR concurred with the general trend depicted in Figure 4 and further informed the development of a coumarin-conjugated fluorescent probe 15. With good permeability into the cancer cells, this probe was localized to the ER, as visualized by confocal fluorescence microscopy, which implies that nannocystins act on eEF1A predominantly at the ER-bound ribosome.
5. Metarrestin
Perinucleolar compartment (PNC) is a heritable multicomponent dynamic subnuclear organelle located at the periphery of the nucleolus of eukaryotic cells and uniquely associated with metastatic cancer cells [53]. Huang et al. found that PNC prevalence, defined as the percentage of non-apoptotic and non-mitotic cells harboring at least one PNC, is a pan-cancer prognostic marker positively correlated with metastatic capacity [54][55]. Subsequent screening of clinically approved anticancer drugs led to the observation that some of these drugs are capable of reducing PNC prevalence via specific on-target inhibition in lieu of promiscuous toxicity [56]. Having confirmed the existence of mechanism-specific PNC disassemblers with clinical efficacy, the proof-of-concept study supported taking PNC prevalence reduction as a phenotypic screening marker to discover antimetastatic drugs. To this end, a metastatic prostate cancer cell line PC3M with a PNC prevalence of 75% to 85% was engineered to stably express green fluorescent protein (GFP)-fused polypyrimidine-tract-binding protein (PTB) [57]. PTB is an essential PNC marker routinely measured by immunohistochemistry, which is unfortunately inconvenient for automated screening. But now with the self-reporting fluorescent PC3M-GFP-PTB cell line at hand, they were able to establish an image-based high-throughput, high-content assay (HCA) primed for spotting compounds able to reduce PNC prevalence by 50% [58]. Aiming at antimetastasis, the initial hits underwent secondary assays to select for invasion inhibition while excluding those acting via apoptosis induction, DNA intercalation, general cytotoxicity, or cell-cycle arrest [59]. By means of this multistage screening protocol, two leads were eventually identified out of 140,800 structurally diverse compounds from the NIH Molecular Libraries Small Molecule Repository (MLSMR) due to their outstanding PNC-disassembling efficiency and low cytotoxicity, thus setting the stage for the ensuing medicinal chemistry campaign [60].
After a preliminary exploration, pyrrolopyrimidine 24 (Figure 5) was favored over the other lead (structure not shown) for systematic optimization. Robust synthetic methods were next developed to access a broad variety of analogues evaluated for PNC disassembly and drug-like properties as well. The SAR trends are summarized in the structure of 24 (Figure 5). Specifically, (1) the N-3 substitution at the subdomain I prefers a linear alkyl chain bearing a hydroxy, ether, or amine, and conformational constraint with a cyclohexyl ring gives rise to the highest potency; (2) the N-7 position at the subdomain II tolerates a benzyl, phenethyl, or 4-methoxylphenyl group, but the presence of an alkyl substituent diminishes the potency significantly; (3) the unsubstituted C5 and C6 phenyl rings at the subdomain III are indispensable for high potency. While deducing the above trends, multi-round optimization finally yielded metarrestin (25), which possesses a superior selectivity window between PNC reduction and cell viability compared with the classic anticancer drugs doxorubicin and camptothecin (table inset in Figure 5) [60]. The in vitro performance of 25 was smoothly translated into in vivo efficacy in three mouse models of human cancer, where it suppressed metastatic invasion with concomitant reduction in PNC prevalence in the cancer cells of primary and metastasized tumors, offering a remarkable survival advantage to the treated animals [59]. After an in-depth evaluation of its pharmacokinetics [61][62] and safety [63], this drug has been advanced into a phase I clinical trial for the treatment of metastatic solid tumors [64].
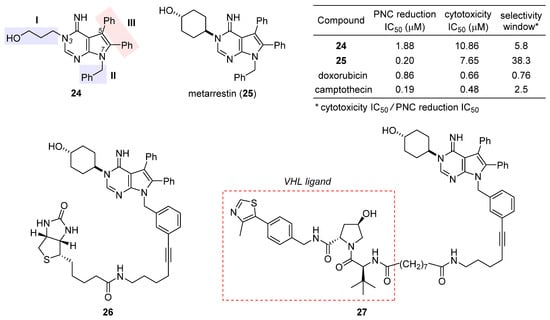
Figure 5. Structures of the initial hit 24, final drug candidate metarrestin (25), biotin-labeled affinity probe 26, and metarrestin-based PROTAC 27. The inset table compares the selectivity window between PNC reduction and cytotoxicity of 24, 25, doxorubicin, and camptothecin.
The excellent antimetastatic capability of 25 prompted Huang et al. to investigate its mechanism, with the primary conclusion that the drug disrupts PNC assembly by blocking RNA polymerase I transcription [59]. Further seeking the binding target of 25, they designed a biotin-conjugated probe 26 (Figure 5) that is likewise efficacious in disassembling PNC. Affinity purification with 26 combined with competition experiments using untagged metarrestin identified eEF1A2 as the binding target. The metarrestin–eEF1A2 interaction was confirmed by cellular thermal shift assay. Subsequent experiments along this line of research showed that (1) eEF1A2 enhances PNC assembly and metastatic progression; and (2) eEF1A2, at least in part, mediates the PNC-elimination function of metarrestin [59]. Whereas further details await elucidation, it was believed that metarrestin interferes with certain non-translational functions of eEF1A2. Recently, Jin et al. developed a proteolysis-targeting chimera (PROTAC) [65][66][67] strategy by tethering metarrestin with various ligands for the von Hippel–Lindau (VHL) E3 ligase [68]. Thus obtained heterobifunctional molecules were designed to recruit eEF1A2, the binding target of metarrestin, to the ubiquitin/proteasome system (UPS) for selective degradation. As one of these first-in-class eEF1A2 degraders, 27 (Figure 5) was shown to degrade eEF1A2 in three cancer cells in a dose-dependent manner, thus holding promise for the treatment of eEF1A2-mediated carcinogenesis.
6. 2-Phenyl-3-Hydroxy-4(1H)-Quinolinones
2-Phenyl-3-hydroxy-4(1H)-quinolinones (abbreviated as 3-HQs) such as 28 (Figure 6) are aza-analogues of previously reported eEF1A-targeting anticancer flavonoids [69]. Based on a homology model of human eEF1A1, Hlavac et al. carried out docking calculations to identify the binding site for gamendazole, a known eEF1A1 inhibitor for male contraception [70]. Encouragingly, they found that these 3-HQs fit into the same gamendazole-binding site on the surface of eEF1A1. Such direct interaction between eEF1A1 and 3-HQs was verified through pull-down assay using biotinylated 3-HQ derivatives [71]. Having validated the constructed eEF1A1 model, the authors performed virtual screening of in silico designed 3-HQs with varying substituents R1, R2, and R3 (illustrated in the structure of 28). The six highest-scored and synthetically accessible compounds were chosen for wet-lab preparation. Their binding to eEF1A1 was quantitatively characterized with isothermal titration calorimetry (ITC), which provided thermodynamic information consistent with docking calculation results. Biological evaluation discovered 29, one of these rationally designed eEF1A1 inhibitors, with optimal inhibitory activity against several cancer cell lines but low toxicity toward a normal cell line [71].

Figure 6. Structures of eEF1A1-targeting anticancer 3-HQ derivative 29 and its prototype 28.
7. Cordyheptapeptide A
Cordyheptapeptide A (30), a partially N-methylated cyclic heptapeptide with anticancer activity, was originally isolated from the insect pathogenic fungus Cordyceps [72][73]. Although a solution-phase total synthesis of 30 was reported before [74], its SAR and mechanism of action were unclear. Lokey et al. developed a high-throughput solid-phase peptide synthesis (SPPS) to access a library of side-chain- and backbone-modified analogues [75]. They observed the following SAR trends: (1) all side chains are critical to its antiproliferative activity [76]; (2) halogenation at the aromatic side chain of residue 2 or 5 deteriorates activity at varying degrees; (3) whereas removal of the N-methyl moiety at residue 2 or 6 impairs activity, this is not the case for residue 4, for which changing sarcosine to glycine tends to improve activity, and when coupled with ortho-fluorination at residue 5, such N-demethylation brings about equipotent variant 31 with a 39-fold improvement in aqueous solubility (table inset in Figure 7). According to molecular dynamics simulations, the enhancement in activity stems from more conformational flexibility of its glycine-carrying scaffold, which is accordingly more accessible to target-binding conformations than the parent natural product 30.
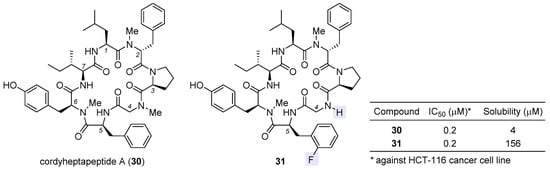
Figure 7. Structures of natural cordyheptapeptide A (30) and synthetic analogue 31, with the inset table comparing their IC50 values against HCT-116 cancer cell line and aqueous solubility.
To find out the mechanism of action of 30, the authors determined its cytotoxicity profile via the NCI60 human tumor cell line assay [75]. Analyzed by the COMPARE algorithm, this profile was best correlated with that of phyllanthoside, a known eukaryotic protein-synthesis inhibitor [77]. Consistently, cytological profiling (CP) [78] indicated that 30 clustered most closely with protein-synthesis inhibitors such as didemnin B and ternatin but deviated significantly from microtubule inhibitors and poly (ADP-ribose) polymerase (PARP) inhibitors. Combining the results from both phenotypic experiments, 30 is quite likely a protein-synthesis inhibitor. This inference was confirmed by bioorthogonal noncanonical amino acid tagging (BONCAT) [79], which proved that the agent primarily blocks protein synthesis and has a secondary influence on DNA synthesis. Suspecting its target to be eEF1A, 30 was evaluated in the HCT-116 cancer cells with a point mutation of eEF1A (A399V). Previously, the same mutation was reported to confer resistance to eEF1A-targeting didemnin B [80], ternatin [24], and nannocystin A [32]. Indeed, the activity dropped remarkably in the mutant cells, thus providing genetic evidence that supports eEF1A as the target of 30.
8. BE-43547A2
Isolated from Streptomyces sp. in 1998, BE-43547A1 (32, Figure 8), BE-43547A2 (33), and other congeners are a series of macrocyclic depsipeptides differing in the C21 side chain [81]. These compounds belong to the amidopentadienoate-containing cyclolipodepsipeptide (APD-CLD) natural products that feature an electrophilic 4-amido-2,4-pentadienoate (APD, highlighted red) functionality as well as a lipophilic side chain [82]. Poulsen et al. developed the first total synthesis of ent-32 and re-isolated the authentic 32 from the fermentation broth of a BE-43547-producing microorganism, thereby establishing the absolute stereo-configuration of the BE-43547 family [83]. Importantly, 32 and 33 exhibited superior hypoxia-selective cytotoxicity in PANC-1 pancreatic cancer cells than rakicidin A, another APD-CLD natural product they investigated earlier [84][85][86][87]. Chen et al. developed a total synthesis of 33 and reported that this agent selectively targets pancreatic cancer stem cells (PCSCs) [88][89]. Preparation of more analogues [90][91] led to the following SAR results (illustrated in Figure 8): (1) the exocyclic alkene at C8 within the APD unit must be reserved; (2) the macrolide cannot be changed to the corresponding macrolactam, or in other words, the O35 cannot be replaced with a nitrogen atom; (3) the (S)-hydroxyl group at C15 is critical to activity; (4) a lipophilic side chain at C21 is necessary but variable.
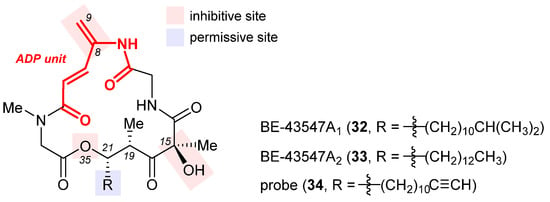
Figure 8. Structures of BE-43547A1 (32), BE-43547A2 (33), and the clickable probe 34. The SAR illustrated in their structures consists of inhibitive (red) and permissive (blue) sites: the former must be reserved for high activity, whereas the latter can tolerate moderate change.
The excellent hypoxia-selective toxicity of APD-CLD natural products such as rakicidin A and the BE-43547A members intrigued Poulsen et al. to elucidate their mechanism of action, which is distinguished from conventional hypoxia-activated compounds. They showed that these APD-CLDs induced rapid and hypoxia-selective impairment of mitochondrial structure and function, thereby driving a peculiar form of non-apoptotic cancer cell death in a hypoxic milieu [92]. To discover the molecular target of 33, Chen et al. synthesized a clickable probe 34 [91]. Biotinylation of 34 via in situ click cycloaddition followed by pull-down assay indicated eEF1A as a binding target, and the isoform was determined to be eEF1A1 via immunoblotting. Having located the cysteine234 residue of eEF1A1 as the most probable binding site of 33 according to LC-MS/MS analysis, they engineered three types of pancreatic cancer cells with (1) eEF1A1 knockdown (KD), (2) eEF1A1 recovered from KD (RE-KD), and (3) C234-mutant eEF1A1 constructed from KD (RE-C234S).
References
- Zhang, J.-N.; Xia, Y.-X.; Zhang, H.-J. Natural cyclopeptides as anticancer agents in the last 20 years. Int. J. Mol. Sci. 2021, 22, 3973.
- Rinehart, K.L., Jr.; Gloer, J.B.; Hughes, R.G., Jr.; Renis, H.E.; McGovren, J.P.; Swynenberg, E.B.; Stringfellow, D.A.; Kuentzel, S.L.; Li, L.H. Didemnins: Antiviral and antitumor depsipeptides from a caribbean tunicate. Science 1981, 212, 933–935.
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudencio, S.P.; Costa-Lotufo, L.V. Enriching cancer pharmacology with drugs of marine origin. Brit. J. Pharmacol. 2020, 177, 3–27.
- Vervoort, H.; Fenical, W.; de Epifanio, R. Tamandarins A and B: New cytotoxic depsipeptides from a Brazilian ascidian of the family Didemnidae. J. Org. Chem. 2000, 65, 782–792.
- Lee, J.; Currano, J.N.; Carroll, P.J.; Joullie, M.M. Didemnins, tamandarins and related natural products. Nat. Prod. Rep. 2012, 29, 404–424.
- Leisch, M.; Egle, A.; Greil, R. Plitidepsin: A potential new treatment for relapsed/refractory multiple myeloma. Future Oncol. 2019, 15, 109–120.
- Chun, H.G.; Davies, B.; Hoth, D.; Suffness, M.; Plowman, J.; Flora, K.; Grieshaber, C.; Leyland-Jones, B. Didemnin B. The first marine compound entering clinical trials as an antineoplastic agent. Investig. New Drugs 1986, 4, 279–284.
- Vera, M.D.; Joullie, M.M. Natural products as probes of cell biology: 20 years of didemnin research. Med. Res. Rev. 2002, 22, 102–145.
- Le Tourneau, C.; Raymond, E.; Faivre, S. Aplidine: A paradigm of how to handle the activity and toxicity of a novel marine anticancer poison. Curr. Pharm. Des. 2007, 13, 3427–3439.
- Muñoz-Alonso, M.J.; González-Santiago, L.; Martínez, T.; Losada, A.; Galmarini, C.M.; Muñoz, A. The mechanism of action of plitidepsin. Curr. Opin. Investig. Drugs 2009, 10, 536–542.
- Danu, A.; Willekens, C.; Ribrag, V. Plitidepsin: An orphan drug. Expert Opin. Orphan Drugs 2013, 1, 569–580.
- Alonso-Alvarez, S.; Pardal, E.; Sanchez-Nieto, D.; Navarro, M.; Caballero, M.D.; Mateos, M.V.; Martin, A. Plitidepsin: Design, development, and potential place in therapy. Drug Des. Dev. Ther. 2017, 11, 253–264.
- Potts, M.B.; Kim, H.S.; Fisher, K.W.; Hu, Y.; Carrasco, Y.P.; Bulut, G.B.; Ou, Y.-H.; Herrera-Herrera, M.L.; Cubillos, F.; Mendiratta, S.; et al. Using functional signature ontology (FUSION) to identify mechanisms of action for natural products. Sci. Signal. 2013, 6, ra90.
- Potts, M.B.; McMillan, E.A.; Rosales, T.I.; Kim, H.S.; Ou, Y.-H.; Toombs, J.E.; Brekken, R.A.; Minden, M.D.; MacMillan, J.B.; White, M.A. Mode of action and pharmacogenomic biomarkers for exceptional responders to didemnin B. Nat. Chem. Biol. 2015, 11, 401–408.
- Losada, A.; Munoz-Alonso, M.J.; Garcia, C.; Sanchez-Murcia, P.A.; Martinez-Leal, J.F.; Dominguez, J.M.; Lillo, M.P.; Gago, F.; Galmarini, C.M. Translation Elongation Factor eEF1A2 is a Novel Anticancer Target for the Marine Natural Product Plitidepsin. Sci. Rep. 2016, 6, 35100.
- Losada, A.; Munoz-Alonso, M.J.; Martinez-Diez, M.; Gago, F.; Dominguez, J.M.; Martinez-Leal, J.F.; Galmarini, C.M. Binding of eEF1A2 to the RNA-dependent protein kinase PKR modulates its activity and promotes tumor cell survival. Br. J. Cancer 2018, 119, 1410–1420.
- Losada, A.; Berlanga, J.J.; Molina-Guijarro, J.M.; Jimenez-Ruiz, A.; Gago, F.; Aviles, P.; de Haro, C.; Martinez-Leal, J.F. Generation of endoplasmic reticulum stress and inhibition of autophagy by plitidepsin induces proteotoxic apoptosis in cancer cells. Biochem. Pharmacol. 2020, 172, 113744.
- Kakeya, H.; Zhang, H.P.; Kobinata, K.; Onose, R.; Onozawa, C.; Kudo, T.; Osada, H. Cytotrienin A, a novel apoptosis inducer in human leukemia HL-60 cells. J. Antibiot. 1997, 50, 370–372.
- Zhang, H.P.; Kakeya, H.; Osada, H. Novel triene-ansamycins, cytotrienins A and B, inducing apoptosis on human leukemia HL-60 cells. Tetrahedron Lett. 1997, 38, 1789–1792.
- Kakeya, H.; Onose, R.; Osada, H. Caspase-mediated activation of a 36-kDa myelin basic protein kinase during anticancer drug-induced apoptosis. Cancer Res. 1998, 58, 4888–4894.
- Watabe, M.; Kakeya, H.; Onose, R.; Osada, H. Activation of MST/Krs and c-Jun N-terminal kinases by different signaling pathways during cytotrienin A-induced apoptosis. J. Biol. Chem. 2000, 275, 8766–8771.
- Yamada, Y.; Taketani, S.; Osada, H.; Kataoka, T. Cytotrienin A, a translation inhibitor that induces ectodomain shedding of TNF receptor 1 via activation of ERK and p38 MAP kinase. Eur. J. Pharmacol. 2011, 667, 113–119.
- Lindqvist, L.; Robert, F.; Merrick, W.; Kakeya, H.; Fraser, C.; Osada, H.; Pelletier, J. Inhibition of translation by cytotrienin A--a member of the ansamycin family. RNA 2010, 16, 2404–2413.
- Carelli, J.D.; Sethofer, S.G.; Smith, G.A.; Miller, H.R.; Simard, J.L.; Merrick, W.C.; Jain, R.K.; Ross, N.T.; Taunton, J. Ternatin and improved synthetic variants kill cancer cells by targeting the elongation factor-1A ternary complex. eLife 2015, 4, e10222.
- Blunt, J.; Cole, T.; Munro, M.; Sun, L.; Weber, J.-F.R.; Ramasamy, K.; Abu Bakar, H.; Abdul Majeed, A.B.B. Bioactive Compounds Derived from Endophytic Aspergillus Fungus Strain Isolated from Garcinia Scortechinii. WO2010062159A1, 3 June 2010.
- Briggs, L.H.; Locker, R.H. 459. Flavonols from the bark of Melicope ternata. Part I. The isolation of four new flavonols, meliternatin, meliternin, ternatin, and wharangin. J. Chem. Soc. 1949, 2157–2162, The term ternatin also refers to a flavone-type natural product, 5-dihydroxy-3,3ʹ,7,8-tetramethoxyflavone (CAS registry number: 571-71-1) first reported in 1949.
- Shimokawa, K.; Mashima, I.; Asai, A.; Yamada, K.; Kita, M.; Uemura, D. (−)-Ternatin, a highly N-methylated cyclic heptapeptide that inhibits fat accumulation: Structure and synthesis. Tetrahedron Lett. 2006, 47, 4445–4448.
- Wang, H.-Y.; Yang, H.; Holm, M.; Tom, H.; Oltion, K.; Al-Khdhairawi, A.A.Q.; Weber, J.-F.F.; Blanchard, S.C.; Ruggero, D.; Taunton, J. Synthesis and single-molecule imaging reveal stereospecific enhancement of binding kinetics by the antitumour eEF1A antagonist SR-A3. Nat. Chem. 2022, 14, 1443–1450.
- Juette, M.F.; Carelli, J.D.; Rundlet, E.J.; Brown, A.; Shao, S.; Ferguson, A.; Wasserman, M.R.; Holm, M.; Taunton, J.; Blanchard, S.C. Didemnin B and ternatin-4 differentially inhibit conformational changes in eEF1A required for aminoacyl-tRNA accommodation into mammalian ribosomes. eLife 2022, 11, e81608.
- Oltion, K.; Carelli, J.D.; Yang, T.; See, S.K.; Wang, H.-Y.; Kampmann, M.; Taunton, J. An E3 ligase network engages GCN1 to promote the degradation of translation factors on stalled ribosomes. Cell 2023, 186, 346–362.
- Hoffmann, H.; Kogler, H.; Heyse, W.; Matter, H.; Caspers, M.; Schummer, D.; Klemke-Jahn, C.; Bauer, A.; Penarier, G.; Debussche, L.; et al. Discovery, Structure Elucidation, and Biological Characterization of Nannocystin A, a Macrocyclic Myxobacterial Metabolite with Potent Antiproliferative Properties. Angew. Chem. Int. Ed. 2015, 54, 10145–10148.
- Krastel, P.; Roggo, S.; Schirle, M.; Ross, N.T.; Perruccio, F.; Aspesi, P., Jr.; Aust, T.; Buntin, K.; Estoppey, D.; Liechty, B.; et al. Nannocystin A: An Elongation Factor 1 Inhibitor from Myxobacteria with Differential Anti-Cancer Properties. Angew. Chem. Int. Ed. 2015, 54, 10149–10154.
- Liao, L.; Zhou, J.; Xu, Z.; Ye, T. Concise Total Synthesis of Nannocystin A. Angew. Chem. Int. Ed. 2016, 55, 13263–13266.
- Huang, J.; Wang, Z. Total Syntheses of Nannocystins A and A0, Two Elongation Factor 1 Inhibitors. Org. Lett. 2016, 18, 4702–4705.
- Yang, Z.; Xu, X.; Yang, C.H.; Tian, Y.; Chen, X.; Lian, L.; Pan, W.; Su, X.; Zhang, W.; Chen, Y. Total Synthesis of Nannocystin A. Org. Lett. 2016, 18, 5768–5770.
- Zhang, Y.H.; Liu, R.; Liu, B. Total synthesis of nannocystin Ax. Chem. Commun. 2017, 53, 5549–5552.
- Poock, C.; Kalesse, M. Total Synthesis of Nannocystin Ax. Org. Lett. 2017, 19, 4536–4539.
- Liu, Q.; Hu, P.; He, Y. Asymmetric Total Synthesis of Nannocystin A. J. Org. Chem. 2017, 82, 9217–9222.
- Meng, Z.; Souillart, L.; Monks, B.; Huwyler, N.; Herrmann, J.; Mueller, R.; Furstner, A. A “Motif-Oriented” Total Synthesis of Nannocystin Ax. Preparation and Biological Assessment of Analogues. J. Org. Chem. 2018, 83, 6977–6994.
- Wang, Z. The Chemical Syntheses of Nannocystins. Synthesis 2019, 51, 2252–2260.
- Zhang, W. From Target-Oriented to Motif-Oriented: A Case Study on Nannocystin Total Synthesis. Molecules 2020, 25, 5327.
- Fürstner, A. Lessons from Natural Product Total Synthesis: Macrocyclization and Postcyclization Strategies. Acc. Chem. Res. 2021, 54, 861–874.
- Paul, D.; Das, S.; Saha, S.; Sharma, H.; Goswami, R.K. Intramolecular Heck Reaction in Total Synthesis of Natural Products: An Update. Eur. J. Org. Chem. 2021, 2021, 2057–2076.
- Zhang, W. Heck macrocyclization in natural product total synthesis. Nat. Prod. Rep. 2021, 38, 1109–1135.
- Tian, Y.; Xu, X.; Ding, Y.; Hao, X.; Bai, Y.; Tang, Y.; Zhang, X.; Li, Q.; Yang, Z.; Zhang, W.; et al. Synthesis and biological evaluation of nannocystin analogues toward understanding the binding role of the (2R,3S)-Epoxide in nannocystin A. Eur. J. Med. Chem. 2018, 150, 626–632.
- Tian, Y.; Ding, Y.; Xu, X.; Bai, Y.; Tang, Y.; Hao, X.; Zhang, W.; Chen, Y. Total synthesis and biological evaluation of nannocystin analogues modified at the polyketide phenyl moiety. Tetrahedron Lett. 2018, 59, 3206–3209.
- Tian, Y.; Wang, J.; Liu, W.; Yuan, X.; Tang, Y.; Li, J.; Chen, Y.; Zhang, W. Stereodivergent total synthesis of Br-nannocystins underpinning the polyketide (10R,11S) configuration as a key determinant of potency. J. Mol. Struct. 2019, 1181, 568–578.
- Liu, Q.; Yang, X.; Ji, J.; Zhang, S.-L.; He, Y. Novel nannocystin A analogues as anticancer therapeutics: Synthesis, biological evaluations and structure-activity relationship studies. Eur. J. Med. Chem. 2019, 170, 99–111.
- Sun, C.; Liu, R.; Xia, M.; Hou, Y.; Wang, X.; Lu, J.-J.; Liu, B.; Chen, X. Nannocystin Ax, a natural elongation factor 1α inhibitor from Nannocystis sp., suppresses epithelial-mesenchymal transition, adhesion and migration in lung cancer cells. Toxicol. Appl. Pharmacol. 2021, 420, 115535.
- Hou, Y.; Liu, R.; Xia, M.; Sun, C.; Zhong, B.; Yu, J.; Ai, N.; Lu, J.-J.; Ge, W.; Liu, B.; et al. Nannocystin ax, an eEF1A inhibitor, induces G1 cell cycle arrest and caspase-independent apoptosis through cyclin D1 downregulation in colon cancer in vivo. Pharmacol. Res. 2021, 173, 105870.
- Zhang, H.; Tian, Y.; Yuan, X.; Xie, F.; Yu, S.; Cai, J.; Sun, B.; Shan, C.; Zhang, W. Site-directed Late-Stage Diversification of Macrocyclic Nannocystins Facilitating Anticancer SAR and Mode of Action Studies. RSC Med. Chem. 2023, 14, 299–312.
- Jia, L.; Ge, X.; Du, C.; Chen, L.; Zhou, Y.; Xiong, W.; Xiang, J.; Li, G.; Xiao, G.; Fang, L.; et al. EEF1A2 interacts with HSP90AB1 to promote lung adenocarcinoma metastasis via enhancing TGF-β/SMAD signalling. Brit. J. Cancer 2021, 124, 1301–1311.
- Pollock, C.; Huang, S. The perinucleolar compartment. Cold Spring Harbor Perspect. Biol. 2010, 2, a000679.
- Kamath, R.V.; Thor, A.D.; Wang, C.; Edgerton, S.M.; Slusarczyk, A.; Leary, D.J.; Wang, J.; Wiley, E.L.; Jovanovic, B.; Wu, Q.; et al. Perinucleolar compartment prevalence has an independent prognostic value for breast cancer. Cancer Res. 2005, 65, 246–253.
- Norton, J.T.; Pollock, C.B.; Wang, C.; Schink, J.C.; Kim, J.J.; Huang, S. Perinucleolar compartment prevalence is a phenotypic pancancer marker of malignancy. Cancer 2008, 113, 861–869.
- Norton, J.T.; Wang, C.; Gjidoda, A.; Henry, R.W.; Huang, S. The Perinucleolar Compartment Is Directly Associated with DNA. J. Biol. Chem. 2009, 284, 4090–4101.
- Huang, S.; Deerinck, T.J.; Ellisman, M.H.; Spector, D.L. The Dynamic Organization of the Perinucleolar Compartment in the Cell Nucleus. J. Cell Biol. 1997, 137, 965–974.
- Norton, J.T.; Titus, S.A.; Dexter, D.; Austin, C.P.; Zheng, W.; Huang, S. Automated High-Content Screening for Compounds That Disassemble the Perinucleolar Compartment. J. Biomol. Screen. 2009, 14, 1045–1053.
- Frankowski, K.J.; Wang, C.; Patnaik, S.; Schoenen, F.J.; Southall, N.; Li, D.; Teper, Y.; Sun, W.; Kandela, I.; Hu, D.; et al. Metarrestin, a perinucleolar compartment inhibitor, effectively suppresses metastasis. Sci. Transl. Med. 2018, 10, eaap8307.
- Frankowski, K.J.; Patnaik, S.; Wang, C.; Southall, N.; Dutta, D.; De, S.; Li, D.; Dextras, C.; Lin, Y.-H.; Bryant-Connah, M.; et al. Discovery and Optimization of Pyrrolopyrimidine Derivatives as Selective Disruptors of the Perinucleolar Compartment, a Marker of Tumor Progression toward Metastasis. J. Med. Chem. 2022, 65, 8303–8331.
- Vilimas, T.; Wang, A.Q.; Patnaik, S.; Hughes, E.A.; Singleton, M.D.; Knotts, Z.; Li, D.; Frankowski, K.; Schlomer, J.J.; Guerin, T.M.; et al. Pharmacokinetic evaluation of the PNC disassembler metarrestin in wild-type and Pdx1-Cre; LSL-KrasG12D/+; Tp53R172H/+ (KPC) mice, a genetically engineered model of pancreatic cancer. Cancer Chemoth. Pharm. 2018, 82, 1067–1080.
- Padilha, E.C.; Shah, P.; Wang, A.Q.; Singleton, M.D.; Hughes, E.A.; Li, D.; Rice, K.A.; Konrath, K.M.; Patnaik, S.; Marugan, J. Metabolism and pharmacokinetics characterization of metarrestin in multiple species. Cancer Chemother. Pharm. 2020, 85, 805–816.
- Bourdi, M.; Rudloff, U.; Patnaik, S.; Marugan, J.; Terse, P.S. Safety assessment of metarrestin in dogs: A clinical candidate targeting a subnuclear structure unique to metastatic cancer cells. Regul. Toxicol. Pharm. 2020, 116, 104716.
- Metarrestin (ML-246) in Subjects with Metastatic Solid Tumors. 2020. Available online: https://ClinicalTrials.gov/show/NCT04222413 (accessed on 2 March 2023).
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200.
- Pedrucci, F.; Pappalardo, C.; Marzaro, G.; Ferri, N.; Ferlin, A.; De Toni, L. Proteolysis Targeting Chimeric Molecules: Tuning Molecular Strategies for a Clinically Sound Listening. Int. J. Mol. Sci. 2022, 23, 6630.
- Yao, T.; Xiao, H.; Wang, H.; Xu, X. Recent Advances in PROTACs for Drug Targeted Protein Research. Int. J. Mol. Sci. 2022, 23, 10328.
- Jin, J.; Kabir, M.; Sun, N.; Kaniskan, H.U. Preparation of Heterobifunctional Compounds as Degraders of eEF1A2. WO2022159650A1, 28 July 2022.
- Yao, N.; Chen, C.Y.; Wu, C.Y.; Motonishi, K.; Kung, H.J.; Lam, K.S. Novel flavonoids with antiproliferative activities against breast cancer cells. J. Med. Chem. 2011, 54, 4339–4349.
- Tash, J.S.; Chakrasali, R.; Jakkaraj, S.R.; Hughes, J.; Smith, S.K.; Hornbaker, K.; Heckert, L.L.; Ozturk, S.B.; Hadden, M.K.; Kinzy, T.G.; et al. Gamendazole, an orally active indazole carboxylic acid male contraceptive agent, targets HSP90AB1 (HSP90BETA) and EEF1A1 (eEF1A), and stimulates Il1a transcription in rat sertoli cells. Biol. Reprod. 2008, 78, 1139–1152.
- Burglová, K.; Rylová, G.; Markos, A.; Prichystalova, H.; Soural, M.; Petracek, M.; Medvedikova, M.; Tejral, G.; Sopko, B.; Hradil, P.; et al. Identification of Eukaryotic Translation Elongation Factor 1-α 1 Gamendazole-Binding Site for Binding of 3-Hydroxy-4(1H)-quinolinones as Novel Ligands with Anticancer Activity. J. Med. Chem. 2018, 61, 3027–3036.
- Isaka, M.; Srisanoh, U.; Lartpornmatulee, N.; Boonruangprapa, T. ES-242 derivatives and cycloheptapeptides from Cordyceps sp. strains BCC 16173 and BCC 16176. J. Nat. Prod. 2007, 70, 1601–1604.
- Rukachaisirikul, V.; Chantaruk, S.; Tansakul, C.; Saithong, S.; Chaicharernwimonkoon, L.; Pakawatchai, C.; Isaka, M.; Intereya, K. A cyclopeptide from the insect pathogenic fungus Cordyceps sp. BCC 1788. J. Nat. Prod. 2006, 69, 305–307.
- Kumar, S.; Dahiya, R.; Khokra, S.L.; Mourya, R.; Chennupati, S.V.; Maharaj, S. Total synthesis and pharmacological investigation of cordyheptapeptide A. Molecules 2017, 22, 682.
- Klein, V.G.; Bray, W.M.; Wang, H.-Y.; Edmondson, Q.; Schwochert, J.; Ono, S.; Naylor, M.R.; Turmon, A.C.; Faris, J.H.; Okada, O.; et al. Identifying the Cellular Target of Cordyheptapeptide A and Synthetic Derivatives. ACS Chem. Biol. 2021, 16, 1354–1364.
- Naylor, M.R.; Ly, A.M.; Handford, M.J.; Ramos, D.P.; Pye, C.R.; Furukawa, A.; Klein, V.G.; Noland, R.P.; Edmondson, Q.; Turmon, A.C.; et al. Lipophilic Permeability Efficiency Reconciles the Opposing Roles of Lipophilicity in Membrane Permeability and Aqueous Solubility. J. Med. Chem. 2018, 61, 11169–11182.
- Chan, J.; Khan, S.N.; Harvey, I.; Merrick, W.; Pelletier, J. Eukaryotic protein synthesis inhibitors identified by comparison of cytotoxicity profiles. RNA 2004, 10, 528–543.
- Schulze, C.J.; Bray, W.M.; Woerhmann, M.H.; Stuart, J.; Lokey, R.S.; Linington, R.G. “Function-first” lead discovery: Mode of action profiling of natural product libraries using image-based screening. Chem. Biol. 2013, 20, 285–295.
- Dieterich, D.C.; Link, A.J.; Graumann, J.; Tirrell, D.A.; Schuman, E.M. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl. Acad. Sci. USA 2006, 103, 9482–9487.
- Sánchez-Murcia, P.A.; Cortés-Cabrera, Á.; Gago, F. Structural rationale for the cross-resistance of tumor cells bearing the A399V variant of elongation factor eEF1A1 to the structurally unrelated didemnin B, ternatin, nannocystin A and ansatrienin B. J. Comput. Aided Mol. Des. 2017, 31, 915–928.
- Nishioka, H.; Nakajima, S.; Nagashima, M.; Kojiri, K.; Suda, H. BE-43547 Series Substances, Their Manufacture with Streptomyces Species, and Their Use as Antitumor Agents. JP10147594A, 2 June 1998.
- Poulsen, T.B. Total Synthesis of Natural Products Containing Enamine or Enol Ether Derivatives. Acc. Chem. Res. 2021, 54, 1830–1842.
- Villadsen, N.L.; Jacobsen, K.M.; Keiding, U.B.; Weibel, E.T.; Christiansen, B.; Vosegaard, T.; Bjerring, M.; Jensen, F.; Johannsen, M.; Tørring, T.; et al. Synthesis of ent-BE-43547A1 reveals a potent hypoxia-selective anticancer agent and uncovers the biosynthetic origin of the APD-CLD natural products. Nat. Chem. 2017, 9, 264–272.
- Poulsen, T.B. A concise route to the macrocyclic core of the rakicidins. Chem. Commun. 2011, 47, 12837–12839.
- Clement, L.L.; Tsakos, M.; Schaffert, E.S.; Scavenius, C.; Enghild, J.J.; Poulsen, T.B. The amido-pentadienoate-functionality of the rakicidins is a thiol reactive electrophile–development of a general synthetic strategy. Chem. Commun. 2015, 51, 12427–12430.
- Tsakos, M.; Clement, L.L.; Schaffert, E.S.; Olsen, F.N.; Rupiani, S.; Djurhuus, R.; Yu, W.; Jacobsen, K.M.; Villadsen, N.L.; Poulsen, T.B. Total synthesis and biological evaluation of rakicidin A and discovery of a simplified bioactive analogue. Angew. Chem. Int. Ed. 2016, 55, 1030–1035.
- Tsakos, M.; Jacobsen, K.M.; Yu, W.; Villadsen, N.L.; Poulsen, T.B. The rakicidin family of anticancer natural products–synthetic strategies towards a new class of hypoxia-selective cytotoxins. Synlett 2016, 27, 1898–1906.
- Sun, Y.; Ding, Y.; Li, D.; Zhou, R.; Su, X.; Yang, J.; Guo, X.; Chong, C.; Wang, J.; Zhang, W.; et al. Cyclic Depsipeptide BE-43547A2: Synthesis and Activity against Pancreatic Cancer Stem Cells. Angew. Chem. Int. Ed. 2017, 56, 14627–14631.
- Sun, Y.; Su, X.; Zhou, R.; Wang, D.; Zhao, Y.; Jiang, Y.; Wang, L.; Chen, Y. Total synthesis of BE-43547A2. Tetrahedron 2018, 74, 5955–5964.
- Sun, Y.; Zhou, R.; Xu, H.; Wang, D.; Su, X.; Wang, C.; Ding, Y.; Wang, L.; Chen, Y. Syntheses and biological evaluation of BE-43547A2 analogs modified at O35 ester and C15-OH sites. Tetrahedron 2019, 75, 1808–1818.
- Liu, C.; Wang, L.; Sun, Y.; Zhao, X.; Chen, T.; Su, X.; Guo, H.; Wang, Q.; Xi, X.; Ding, Y.; et al. Probe Synthesis Reveals Eukaryotic Translation Elongation Factor 1 Alpha 1 as the Anti-Pancreatic Cancer Target of BE-43547A2. Angew. Chem. Int. Ed. 2022, 61, e202206953.
- Jacobsen, K.M.; Villadsen, N.L.; Toerring, T.; Nielsen, C.B.; Salomon, T.; Nielsen, M.M.; Tsakos, M.; Sibbersen, C.; Scavenius, C.; Nielsen, R.; et al. APD-Containing Cyclolipodepsipeptides Target Mitochondrial Function in Hypoxic Cancer Cells. Cell Chem. Biol. 2018, 25, 1337–1349.
More
Information
Subjects:
Chemistry, Medicinal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
15 Mar 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No