Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ali Shilleh | -- | 2347 | 2023-03-03 10:45:58 | | | |
| 2 | Rita Xu | Meta information modification | 2347 | 2023-03-03 11:00:53 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Shilleh, A.H.; Russ, H.A. Cell Replacement Therapy for Type 1 Diabetes Patients. Encyclopedia. Available online: https://encyclopedia.pub/entry/41849 (accessed on 28 July 2026).
Shilleh AH, Russ HA. Cell Replacement Therapy for Type 1 Diabetes Patients. Encyclopedia. Available at: https://encyclopedia.pub/entry/41849. Accessed July 28, 2026.
Shilleh, Ali H., Holger A. Russ. "Cell Replacement Therapy for Type 1 Diabetes Patients" Encyclopedia, https://encyclopedia.pub/entry/41849 (accessed July 28, 2026).
Shilleh, A.H., & Russ, H.A. (2023, March 03). Cell Replacement Therapy for Type 1 Diabetes Patients. In Encyclopedia. https://encyclopedia.pub/entry/41849
Shilleh, Ali H. and Holger A. Russ. "Cell Replacement Therapy for Type 1 Diabetes Patients." Encyclopedia. Web. 03 March, 2023.
Copy Citation
Cell replacement therapy using stem-cell-derived insulin-producing β-like cells (sBCs) has been proposed as a practical cure for patients with type one diabetes (T1D). sBCs can correct diabetes in preclinical animal models, demonstrating the promise of this stem cell-based approach.
cell replacement therapy
type 1 diabetes
stem-cell-derived β-like cells
autoimmune diabetes
1. Introduction
The pancreas consists of two main compartments, the exocrine and endocrine tissue, both with distinct functions. Exocrine tissue consists predominantly of acinar cells that release digestive enzymes into the duodenum via a ductal system, making up most of the cell mass found in the organ. The pancreas also contains endocrine cells that are organized together into highly vascularized cell clusters called the islets of Langerhans. Endocrine cells within islets secret hormones that exquisitely regulate and maintain blood sugar levels within a tight physiological range. Representing only about ~1–2% of the organ tissue, the main endocrine cells are insulin-producing β-cells, glucagon-producing α-cells, somatostatin-producing δ-cells, polypeptide-producing PP cells, and ghrelin-producing ε-cells [1]. Out of all endocrine cells, only β-cells express and secrete insulin in response to elevations in blood glucose levels. β-cell dysfunction or loss is key in contributing toward the development of diabetes, and much research has focused on this fascinating cell type.
Diabetes presents as two major subtypes. In both, the inadequate release of insulin results in hyperglycemia that can be life-threatening. The most common diabetes form, type 2 diabetes (T2D), affecting 462 million people globally, is characterized by the insulin resistance of peripheral tissues and (subsequent) β-cell dysfunction, exhaustion, and loss [2]. In type 1 diabetes (T1D), the patient’s own insulin-producing β-cells are specifically destroyed through an autoimmune-mediated attack predominantly of T-cells, resulting in insulin deficiency. Type 1 diabetes (T1D) is a chronic condition that affects 1 in 500 Americans by the age of 15 [3]. Current treatment for both T1D and late-stage T2D consists of injecting endogenous insulin. Exogenous insulin replacement therapy falls short of recapitulating the exact physiological function of a β-cell, and patients are susceptible to acute and long-term complications [4][5][6]. Hypoglycemic conditions, induced by injecting too much insulin, can result in a life-threatening coma and are a constant risk for patients living with T1D and a practical cure that would alleviate the risks and concerns of current insulin therapy is desperately needed.
2. Current and Potential Cell Replacement Strategies for T1D Patients
Islet Transplantation to Restore β-Cell Mass
A proof of principal for a potential practical cure has been shown with the establishment of the Edmond protocol in 2000. In this protocol, isolated allogenic cadaveric islets are infused in the portal vein of long-standing T1D patients that receive non-steroid immunosuppression [7][8]. Importantly, islet recipients achieve on average ~35 months of insulin independence [9]. Subsequently, islet transplantation was often performed in conjunction with kidney transplantation [10]. However, there are several challenges associated with this procedure that prevent it from becoming widely accessible for patients. A major drawback with islet transplantation is the limited availability of high-purity isolated human cadaveric donor islet material. This is required to restore euglycemia in patients. Typically, each patient receives 10,000 islet equivalents (IEQs) per kilogram of body weight, an amount that usually needs to be extracted from two donor pancreases. In addition, initial clinical trials showed some patients requiring multiple islet infusions throughout the study, further highlighting the need for an abundant source of functional insulin-producing cells. The chronic immune suppression of patients, especially in children and adolescents, is problematic due to long-term complications, including severe and chronic infections and malignancy. In addition, studies have shown that immune suppressive agents impair β-cell function and survival using animal models [11][12]. Indeed, functional cadaveric islet grafts are frequently lost within 2–5 years due to recurring autoimmunity, side effects of immunosuppressants, and other unknown mechanisms [9][13]. The lack of sufficient donor islets has prompted the search for alternative and abundant sources of functional β(-like) cells for replacement therapy purposes, and much progress has been made using different approaches.
Since porcine insulin has been shown to be physiologically well-matched to humans, the xenotransplantation of porcine islets has been considered an effective strategy to provide adequate amounts of islet material to treat T1D patients. However, immunological responses, such as instant blood-mediated inflammatory reaction (IBMIR) [14][15][16], hyperacute, and cellular rejection, remain major hurdles to overcome and improve porcine islet survival [17][18][19][20][21]. Therefore, several strategies have been explored to overcome immune complications in this setting. The development of genetically modified pigs lacking the expression of certain surface proteins that play key roles in immune rejection upon porcine islet transplantation demonstrates promising results in improving porcine islet survival through combating IBMIR and hyperacute rejection. Preclinical studies also revealed that the blockade of co-stimulatory cell surface molecules suppresses T cell activation, hampers cellular rejection, and improves islet survival in vivo. With no evidence of porcine endogenous retrovirus (PERV) transmission, clinical studies of porcine islet xenotransplantation in T1D patients showed initial successes; however, most recipients failed to maintain long-term normal glycemic levels. Encapsulating pig islets has been suggested as effective in reducing xenogeneic immune rejection and prolonging graft survival and was tested in two clinical studies in T1D patients but showed only minimal reduction in their daily insulin needs [22][23][24][25][26].
3. Alternative Approaches to Increase Functional β-Cell Mass
Other strategies have aimed at inducing β-cell replication or neogenesis via the transdifferentiation of pancreatic non-β cells to replenish the β-cell mass. Recently, DYRK1A inhibition in conjugation with other pathway manipulations has been shown to be effective in inducing increased β-cell proliferation [27][28][29][30][31][32]. If safe β-cell-specific delivery modalities can be identified, inducing the proliferation of remaining β-cells in diabetic patients might provide a viable therapeutic strategy [33].
Transdifferentiation refers to the change in functional cell phenotype of a differentiated cell into another rather than the differentiation of a less specialized stem cell into a functional cell type. Pancreatic duct ligation (PDL) has been shown to promote β-cell transdifferentiation from ductal, acinar, and alpha cells in mice [34][35][36][37], although some observations could not be repeated in another study [38]. Similarly, the overexpression of MafA, Pdx1, and Ngn3 can trigger β-cell transdifferentiation predominantly from mouse acinar cells in vivo [39]. Recently, a group induced alpha cell transdifferentiation into β-cells in vivo by infusing adeno-associated viruses carrying Pdx1 and MafA into the pancreatic duct of NOD mice [40]. However, such experimental strategies, while carrying the potential for endogenous β-cell repopulation in T1D patients, are awaiting translation to human systems and/or clinical settings.
4. Stem-Cell-Derived β-Cells as an Abundant Cell Source
One attractive approach that has advanced rapidly and shows tremendous potential as an abundant source of functional insulin-producing cells for clinical use is the direct differentiation of human pluripotent stem cells (hPSCs) into stem cell-derived β-like cells (sBCs) [41][42][43][44]. Mouse development studies have identified critical transcription factors and signaling events during pancreas organogenesis, and subsequent work defined the necessary culture conditions to mimic key development stages to direct the differentiation of sBC from pluripotent stem cells [45][46][47][48]. Specifically, several groups focused their efforts on utilizing recombinant proteins and small molecules to generate subsequent cell types resulting in pancreatic cells: definitive endoderm generation [49][50], posterior gut specification [50][51][52][53], formation of pancreatic bipotent progenitors [54][55][56], and endocrine differentiation [41][42][43][50][57]. Although sBCs generated with early protocols were glucose-responsive, cells still displayed features of immature, fetal-like β-cells, and thus performed poorly in dynamic glucose-stimulated insulin secretion (dGSIS) perifusion assays. Several methods, such as the manipulation of key signaling pathways [58][59][60], media composition and in vitro culture extension [61][62], and the use of surface markers and fluorescence-activated cell sorting (FACS) to enrich reaggregated sBCs resulted in a more mature β-cell phenotype that closely resembles primary adult islets [22][44][62][63][64]. Clear criteria defining a mature, functional β-cell that allows distinction from β-cell surrogates has recently been discussed in detail elsewhere [65]. Interestingly, sBC maturation also seems to be accomplished upon transplantation, which in return restored euglycemia and reversed diabetes in preclinical mouse models [41][42][43][66][67]. However, the early events taking place during the immediate engraftment of sBC have not been studied in detail. A considerable body of work has shown that the majority of functional β-cell mass is lost from human islets upon engraftment, suggesting that such drastic effects may also apply to sBCs due to unknown underlying cellular and molecular mechanisms [68]. Potential mechanisms that might occur are: (i) cell death, (ii) dedifferentiation, and (iii) transdifferentiation (Figure 1). Recent work identified distinct human β-cell subpopulations in sBC and human islets in vitro and provided the possibility of (iv) β-subtype interconversion upon engraftment as an additional mechanism. Hence, there is a critical knowledge gap in the research field concerning the fate of sBC upon engraftment. Expanding the knowledge of the contributing mechanisms would expedite the progress in promoting the current approaches of delivering sBC as an effective cell replacement therapy for T1D patients. In addition, sBC cell therapy might represent an attractive treatment modality for T2D patients due to the absence of reoccurring autoimmunity if allogeneic rejection can be avoided in a localized manner.
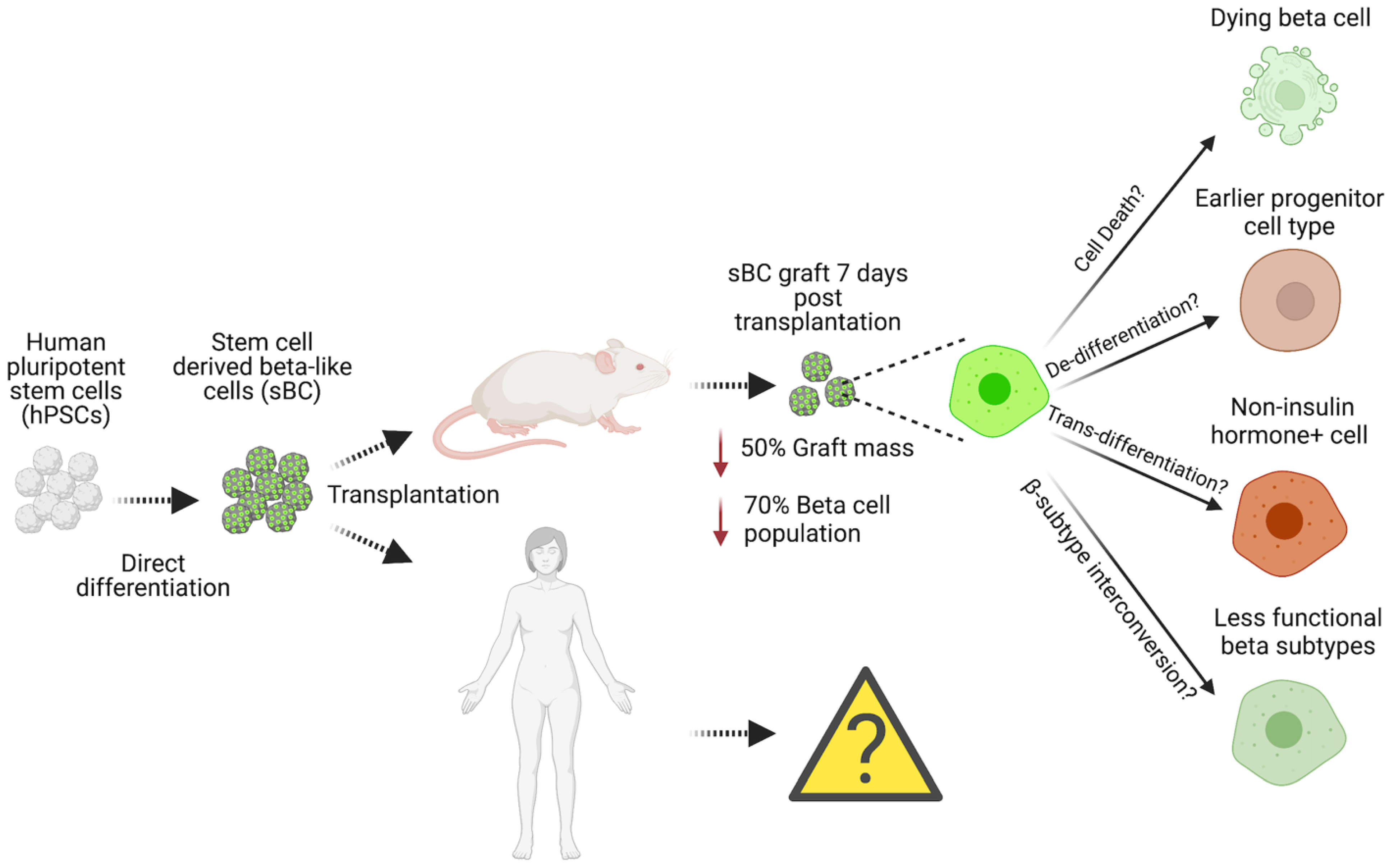
Figure 1. Reported and potential molecular and cellular mechanisms driving human pancreatic β-cell loss upon transplantation.
5. Engrafted β-Cell Loss via Cell Death
Classically, the main mechanism associated with islet cell death in vitro and in vivo is apoptosis. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) has shown that a considerable proportion of isolated human islets harvested from donor pancreases are lost in vitro within 5 days of culture due to apoptosis [69]. The high propensity of β-cells to undergo apoptosis within islet preparations might be due to the increased metabolic rate that is not met under cell culture conditions. Later studies determined the caspase cascade as the major intrinsic mediator of apoptosis in cultured islets after being exposed to toxic levels of glucose [70]. Glucotoxicity downregulated BCL-2 (anti-apoptotic protein) expression in isolated islets, which acts as an intrinsic signal to activate caspase-associated apoptosis [71][72]. Similarly, after 24 h of transplantation, TUNEL staining showed that approximately a quarter of all β-cells in human islets engrafted in the kidney capsule of immunodeficient mice are lost due to apoptosis [73]. Overall, although the exact mechanisms contributing to primary β-cell death upon transplantation are poorly understood, the current literature predominantly attributes the observed loss to ischemia and nutrient deprivation. Mediated by endothelial cells, in situ, pancreatic islets are highly vascularized and are under a continuous supply of oxygen and nutrients, ensuring optimal function. However, this supply is lost during the islet isolation process, which involves the use of digestive enzymes and mechanical force to separate the islets from the native organ [74]. Due to loss of blood flow and imperfect culturing conditions, endothelial cells, which are critical in providing cellular matrix proteins that fine-tune the function of β-cells, eventually die in vitro [75]. Since blood flow is abolished after isolation, islets are under an acute nutrient deficiency and exposed to oxidative stress mediated by hypoxia. Isolated islets depend on passive nutrition diffusion to satisfy the activities of the highly metabolic β-cells. Thus, culture conditions are insufficient in supplying uniform O2 levels to all β-cells, especially cells located at the core of the islet, negatively effecting β-cell survival in vitro, with necrotic cores present, especially in larger islets due to low oxygen accessibility [76][77][78]. Similarly transplanted human and rodent islets have been shown to have reduced graft oxygen tension in the initial stages of engraftment and to suffer a drastic loss of β-cells in vivo [79][80][81][82][83][84]. Therefore, several in vitro pre-transplant priming methods have been adopted to improve islet survival in transplants, such as oxygenation treatment, culture in hyperoxic conditions, and modulation of seeding density; however, these strategies have failed to be exceedingly successful [85][86][87]. Further mechanistic analysis revealed several signaling pathways, such as anaerobic glycolysis and hypoxia-inducible factor (HIF)-related pathways, to be associated with β-cell survival under hypoxic conditions; however, further investigation is required [84][88][89][90][91][92][93]. Finally, other necrotic-regulated mechanisms such as pyroptosis [94][95], ferroptosis [96][97][98][99][100][101], and necroptosis [102][103][104][105][106][107] have been implicated to contribute toward β-cell death during islet isolation, culture, and transplantation; however, these mechanisms have not been comprehensively elucidated as of yet.
Most in vivo sBC studies have focused on the metabolic action and long-term therapeutic capacity of engrafted, surviving cells using preclinical animals starting at 3–4 weeks post-transplantation when grafts are fully vascularized. However, most studies have largely neglected the early phase of sBC transplantation. In a recent study, sBCs constitutively expressing luciferase were transplanted subcutaneously or under the kidney capsule of immunodeficient mice, and total graft mass was quantified using bioluminescence [68]. As expected, on the day of transplantation, robust expression of luciferase was detected; however, 7 days post-transplant, this expression was significantly reduced in both sites, indicating substantial graft loss. In addition, the hPSC cell line employed also contains a GFP reporter driven by the insulin promoter, allowing quantification of sBCs before and after transplant. Flow cytometry analysis revealed that approximately 70% of sBCs were lost, while the total graft was only 50% reduced within the first 7 days of engraftment, indicating a preferential loss of sBCs compared to other cells present. The main drivers of graft loss are considered to be ischemia-induced hypoxia and nutrition deprivation. Amino acid supplementation and adjusting the physiological oxygen levels to 5% in culture improved sBC graft survival significantly. In situ, pancreatic islets are abundantly vascularized with a continuous supply of oxygen and nutrients; therefore, this study further highlights the importance of vascularization to sBC survival and function in vivo. Pepper and colleagues showed the pre-vascularization of the subcutaneous site followed by the transplantation of pancreatic endoderm (PEC) cells improved stem cell-derived β-cell functionality and survival in vivo, providing further evidence for the notion that appropriate vascularization is critical for β-cell survival and function [108]. Several groups focused on engrafting sBCs that incorporate endothelial cells alone or in combination with mesenchymal cells [109][110][111][112][113]. In a recent elegant study, micro-vessels isolated from adipose tissue have been shown to improve and accelerate the vascularization of sBC grafts, as well as their survival and function in vivo [114]. Finally, using oxygen-generating biomaterials shows promising results to improve islet survival in vivo that could be applied to future sBC engraftments [109][113][115][116][117][118][119]. Altogether, the literature has provided data suggesting hypoxia and nutrient deprivation as two key contributors to sBC graft decline that can be mitigated by providing better engraftment solutions. Understanding what distinguishes sBCs that survive the first week of engraftment from sBCs that are lost during this period could provide additional means to preserve total functional graft mass.
References
- Ionescu-Tirgoviste, C.; Gagniuc, P.A.; Gubceac, E.; Mardare, L.; Popescu, I.; Dima, S.; Militaru, M. A 3D map of the islet routes throughout the healthy human pancreas. Sci. Rep. 2015, 5, 14634.
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes—Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111.
- Patterson, C.; Guariguata, L.; Dahlquist, G.; Soltesz, G.; Ogle, G.; Silink, M. Diabetes in the young—A global view and worldwide estimates of numbers of children with type 1 diabetes. Diabetes Res. Clin. Pract. 2014, 103, 161–175.
- Patrick, A.W.; Williams, G. Adverse effects of exogenous insulin. Clinical features, management and prevention. Drug Saf. 1993, 8, 427–444.
- Carlson, M.G.; Campbell, P.J. Intensive insulin therapy and weight gain in IDDM. Diabetes 1993, 42, 1700–1707.
- Sjølie, A.K. Ocular complications in insulin treated diabetes mellitus. An epidemiological study. Acta Ophthalmol. Suppl. 1985, 172, 1–77.
- Ryan, E.A.; Lakey, J.R.; Rajotte, R.V.; Korbutt, G.S.; Kin, T.; Imes, S.; Rabinovitch, A.; Elliott, J.F.; Bigam, D.; Kneteman, N.M.; et al. Clinical outcomes and insulin secretion after islet transplantation with the Edmonton protocol. Diabetes 2001, 50, 710–719.
- Shapiro, A.M.; Lakey, J.R.; Ryan, E.A.; Korbutt, G.S.; Toth, E.; Warnock, G.L.; Kneteman, N.M.; Rajotte, R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000, 343, 230–238.
- Shapiro, A.M.; Ricordi, C.; Hering, B.J.; Auchincloss, H.; Lindblad, R.; Robertson, R.P.; Secchi, A.; Brendel, M.D.; Berney, T.; Brennan, D.C.; et al. International trial of the Edmonton protocol for islet transplantation. N. Engl. J. Med. 2006, 355, 1318–1330.
- Markmann, J.F.; Rickels, M.R.; Eggerman, T.L.; Bridges, N.D.; Lafontant, D.E.; Qidwai, J.; Foster, E.; Clarke, W.R.; Kamoun, M.; Alejandro, R.; et al. Phase 3 trial of human islet-after-kidney transplantation in type 1 diabetes. Am. J. Transplant. 2021, 21, 1477–1492.
- Niclauss, N.; Bosco, D.; Morel, P.; Demuylder-Mischler, S.; Brault, C.; Milliat-Guittard, L.; Colin, C.; Parnaud, G.; Muller, Y.D.; Giovannoni, L.; et al. Influence of donor age on islet isolation and transplantation outcome. Transplantation 2011, 91, 360–366.
- Rangel, E.B. Tacrolimus in pancreas transplant: A focus on toxicity, diabetogenic effect and drug-drug interactions. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1585–1605.
- Barton, F.B.; Rickels, M.R.; Alejandro, R.; Hering, B.J.; Wease, S.; Naziruddin, B.; Oberholzer, J.; Odorico, J.S.; Garfinkel, M.R.; Levy, M.; et al. Improvement in outcomes of clinical islet transplantation: 1999–2010. Diabetes Care 2012, 35, 1436–1445.
- Goto, M.; Tjernberg, J.; Dufrane, D.; Elgue, G.; Brandhorst, D.; Ekdahl, K.N.; Brandhorst, H.; Wennberg, L.; Kurokawa, Y.; Satomi, S.; et al. Dissecting the instant blood-mediated inflammatory reaction in islet xenotransplantation. Xenotransplantation 2008, 15, 225–234.
- Johansson, H.; Lukinius, A.; Moberg, L.; Lundgren, T.; Berne, C.; Foss, A.; Felldin, M.; Källen, R.; Salmela, K.; Tibell, A.; et al. Tissue factor produced by the endocrine cells of the islets of Langerhans is associated with a negative outcome of clinical islet transplantation. Diabetes 2005, 54, 1755–1762.
- Nilsson, B. The instant blood-mediated inflammatory reaction in xenogeneic islet transplantation. Xenotransplantation 2008, 15, 96–98.
- Gill, R.G.; Wolf, L.; Daniel, D.; Coulombe, M. CD4+ T cells are both necessary and sufficient for islet xenograft rejection. Transplant. Proc. 1994, 26, 1203.
- Olack, B.J.; Jaramillo, A.; Benshoff, N.D.; Kaleem, Z.; Swanson, C.J.; Lowell, J.A.; Mohanakumar, T. Rejection of porcine islet xenografts mediated by CD4+ T cells activated through the indirect antigen recognition pathway. Xenotransplantation 2002, 9, 393–401.
- Koulmanda, M.; Laufer, T.M.; Auchincloss, H.; Smith, R.N. Prolonged survival of fetal pig islet xenografts in mice lacking the capacity for an indirect response. Xenotransplantation 2004, 11, 525–530.
- Kirchhof, N.; Shibata, S.; Wijkstrom, M.; Kulick, D.M.; Salerno, C.T.; Clemmings, S.M.; Heremans, Y.; Galili, U.; Sutherland, D.E.; Dalmasso, A.P.; et al. Reversal of diabetes in non-immunosuppressed rhesus macaques by intraportal porcine islet xenografts precedes acute cellular rejection. Xenotransplantation 2004, 11, 396–407.
- Lindeborg, E.; Kumagai-Braesch, M.; Möller, E. Phenotypic and functional characterization of human T cell clones indirectly activated against adult pig islet cells. Xenotransplantation 2006, 13, 41–52.
- Aghazadeh, Y.; Nostro, M.C. Cell Therapy for Type 1 Diabetes: Current and Future Strategies. Curr. Diab. Rep. 2017, 17, 37.
- Sakata, N.; Sumi, S.; Yoshimatsu, G.; Goto, M.; Egawa, S.; Unno, M. Encapsulated islets transplantation: Past, present and future. World J. Gastrointest. Pathophysiol. 2012, 3, 19–26.
- Matsumoto, S.; Tan, P.; Baker, J.; Durbin, K.; Tomiya, M.; Azuma, K.; Doi, M.; Elliott, R.B. Clinical porcine islet xenotransplantation under comprehensive regulation. Transplant. Proc. 2014, 46, 1992–1995.
- Matsumoto, S.; Abalovich, A.; Wechsler, C.; Wynyard, S.; Elliott, R.B. Clinical Benefit of Islet Xenotransplantation for the Treatment of Type 1 Diabetes. EBioMedicine 2016, 12, 255–262.
- Morozov, V.A.; Wynyard, S.; Matsumoto, S.; Abalovich, A.; Denner, J.; Elliott, R. No PERV transmission during a clinical trial of pig islet cell transplantation. Virus Res. 2017, 227, 34–40.
- Rachdi, L.; Kariyawasam, D.; Aïello, V.; Herault, Y.; Janel, N.; Delabar, J.M.; Polak, M.; Scharfmann, R. Dyrk1A induces pancreatic β cell mass expansion and improves glucose tolerance. Cell Cycle 2014, 13, 2221–2229.
- Wang, P.; Alvarez-Perez, J.C.; Felsenfeld, D.P.; Liu, H.; Sivendran, S.; Bender, A.; Kumar, A.; Sanchez, R.; Scott, D.K.; Garcia-Ocaña, A.; et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat. Med. 2015, 21, 383–388.
- Dirice, E.; Walpita, D.; Vetere, A.; Meier, B.C.; Kahraman, S.; Hu, J.; Dančík, V.; Burns, S.M.; Gilbert, T.J.; Olson, D.E.; et al. Inhibition of DYRK1A Stimulates Human β-Cell Proliferation. Diabetes 2016, 65, 1660–1671.
- Shen, W.; Taylor, B.; Jin, Q.; Nguyen-Tran, V.; Meeusen, S.; Zhang, Y.Q.; Kamireddy, A.; Swafford, A.; Powers, A.F.; Walker, J.; et al. Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Nat. Commun. 2015, 6, 8372.
- Wang, P.; Karakose, E.; Liu, H.; Swartz, E.; Ackeifi, C.; Zlatanic, V.; Wilson, J.; González, B.J.; Bender, A.; Takane, K.K.; et al. Combined Inhibition of DYRK1A, SMAD, and Trithorax Pathways Synergizes to Induce Robust Replication in Adult Human Beta Cells. Cell Metab. 2019, 29, 638–652.e635.
- Ackeifi, C.; Wang, P.; Karakose, E.; Manning Fox, J.E.; González, B.J.; Liu, H.; Wilson, J.; Swartz, E.; Berrouet, C.; Li, Y.; et al. GLP-1 receptor agonists synergize with DYRK1A inhibitors to potentiate functional human β cell regeneration. Sci. Transl. Med. 2020, 12, eaaw9996.
- Wang, P.; Karakose, E.; Choleva, L.; Kumar, K.; DeVita, R.J.; Garcia-Ocaña, A.; Stewart, A.F. Human Beta Cell Regenerative Drug Therapy for Diabetes: Past Achievements and Future Challenges. Front. Endocrinol. 2021, 12, 671946.
- Chung, C.H.; Hao, E.; Piran, R.; Keinan, E.; Levine, F. Pancreatic β-cell neogenesis by direct conversion from mature α-cells. Stem Cells 2010, 28, 1630–1638.
- Wang, R.N.; Klöppel, G.; Bouwens, L. Duct- to islet-cell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia 1995, 38, 1405–1411.
- Pan, F.C.; Bankaitis, E.D.; Boyer, D.; Xu, X.; Van de Casteele, M.; Magnuson, M.A.; Heimberg, H.; Wright, C.V. Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 2013, 140, 751–764.
- Xu, X.; D’Hoker, J.; Stangé, G.; Bonné, S.; De Leu, N.; Xiao, X.; Van de Casteele, M.; Mellitzer, G.; Ling, Z.; Pipeleers, D.; et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 2008, 132, 197–207.
- Cavelti-Weder, C.; Shtessel, M.; Reuss, J.E.; Jermendy, A.; Yamada, T.; Caballero, F.; Bonner-Weir, S.; Weir, G.C. Pancreatic duct ligation after almost complete β-cell loss: Exocrine regeneration but no evidence of β-cell regeneration. Endocrinology 2013, 154, 4493–4502.
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008, 455, 627–632.
- Xiao, X.; Guo, P.; Shiota, C.; Zhang, T.; Coudriet, G.M.; Fischbach, S.; Prasadan, K.; Fusco, J.; Ramachandran, S.; Witkowski, P.; et al. Endogenous Reprogramming of Alpha Cells into Beta Cells, Induced by Viral Gene Therapy, Reverses Autoimmune Diabetes. Cell Stem Cell 2018, 22, 78–90.e4.
- Pagliuca, F.W.; Millman, J.R.; Gürtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic β cells in vitro. Cell 2014, 159, 428–439.
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133.
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772.
- Veres, A.; Faust, A.L.; Bushnell, H.L.; Engquist, E.N.; Kenty, J.H.; Harb, G.; Poh, Y.C.; Sintov, E.; Gürtler, M.; Pagliuca, F.W.; et al. Charting cellular identity during human in vitro β-cell differentiation. Nature 2019, 569, 368–373.
- Hebrok, M. Hedgehog signaling in pancreas development. Mech. Dev. 2003, 120, 45–57.
- Murtaugh, L.C.; Melton, D.A. Genes, signals, and lineages in pancreas development. Annu. Rev. Cell Dev. Biol. 2003, 19, 71–89.
- Pan, F.C.; Wright, C. Pancreas organogenesis: From bud to plexus to gland. Dev. Dyn. 2011, 240, 530–565.
- Seymour, P.A.; Sander, M. Historical perspective: Beginnings of the beta-cell: Current perspectives in beta-cell development. Diabetes 2011, 60, 364–376.
- D’Amour, K.A.; Agulnick, A.D.; Eliazer, S.; Kelly, O.G.; Kroon, E.; Baetge, E.E. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 2005, 23, 1534–1541.
- D’Amour, K.A.; Bang, A.G.; Eliazer, S.; Kelly, O.G.; Agulnick, A.D.; Smart, N.G.; Moorman, M.A.; Kroon, E.; Carpenter, M.K.; Baetge, E.E. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006, 24, 1392–1401.
- Kroon, E.; Martinson, L.A.; Kadoya, K.; Bang, A.G.; Kelly, O.G.; Eliazer, S.; Young, H.; Richardson, M.; Smart, N.G.; Cunningham, J.; et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008, 26, 443–452.
- Gouon-Evans, V.; Boussemart, L.; Gadue, P.; Nierhoff, D.; Koehler, C.I.; Kubo, A.; Shafritz, D.A.; Keller, G. BMP-4 is required for hepatic specification of mouse embryonic stem cell-derived definitive endoderm. Nat. Biotechnol. 2006, 24, 1402–1411.
- Hebrok, M.; Kim, S.K.; Melton, D.A. Notochord repression of endodermal Sonic hedgehog permits pancreas development. Genes Dev. 1998, 12, 1705–1713.
- Nostro, M.C.; Sarangi, F.; Yang, C.; Holland, A.; Elefanty, A.G.; Stanley, E.G.; Greiner, D.L.; Keller, G. Efficient generation of NKX6-1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Rep. 2015, 4, 591–604.
- Rezania, A.; Bruin, J.E.; Riedel, M.J.; Mojibian, M.; Asadi, A.; Xu, J.; Gauvin, R.; Narayan, K.; Karanu, F.; O’Neil, J.J.; et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes 2012, 61, 2016–2029.
- Chen, S.; Borowiak, M.; Fox, J.L.; Maehr, R.; Osafune, K.; Davidow, L.; Lam, K.; Peng, L.F.; Schreiber, S.L.; Rubin, L.L.; et al. A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat. Chem. Biol. 2009, 5, 258–265.
- Nostro, M.C.; Sarangi, F.; Ogawa, S.; Holtzinger, A.; Corneo, B.; Li, X.; Micallef, S.J.; Park, I.H.; Basford, C.; Wheeler, M.B.; et al. Stage-specific signaling through TGFβ family members and WNT regulates patterning and pancreatic specification of human pluripotent stem cells. Development 2011, 138, 861–871.
- Velazco-Cruz, L.; Goedegebuure, M.M.; Maxwell, K.G.; Augsornworawat, P.; Hogrebe, N.J.; Millman, J.R. SIX2 Regulates Human β Cell Differentiation from Stem Cells and Functional Maturation In Vitro. Cell Rep. 2020, 31, 107687.
- Zhu, S.; Russ, H.A.; Wang, X.; Zhang, M.; Ma, T.; Xu, T.; Tang, S.; Hebrok, M.; Ding, S. Human pancreatic beta-like cells converted from fibroblasts. Nat. Commun. 2016, 7, 10080.
- Zhou, X.; Nair, G.G.; Russ, H.A.; Belair, C.D.; Li, M.L.; Shveygert, M.; Hebrok, M.; Blelloch, R. LIN28B Impairs the Transition of hESC-Derived β Cells from the Juvenile to Adult State. Stem Cell Rep. 2020, 14, 9–20.
- Velazco-Cruz, L.; Song, J.; Maxwell, K.G.; Goedegebuure, M.M.; Augsornworawat, P.; Hogrebe, N.J.; Millman, J.R. Acquisition of Dynamic Function in Human Stem Cell-Derived β Cells. Stem Cell Rep. 2019, 12, 351–365.
- Docherty, F.M.; Riemondy, K.A.; Castro-Gutierrez, R.; Dwulet, J.M.; Shilleh, A.H.; Hansen, M.S.; Williams, S.P.M.; Armitage, L.H.; Santostefano, K.E.; Wallet, M.A.; et al. ENTPD3 Marks Mature Stem Cell-Derived β-Cells Formed by Self-Aggregation In Vitro. Diabetes 2021, 70, 2554–2567.
- Nair, G.G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.L.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived beta cells. Nat. Cell Biol. 2019, 21, 263–274.
- Molakandov, K.; Berti, D.A.; Beck, A.; Elhanani, O.; Walker, M.D.; Soen, Y.; Yavriyants, K.; Zimerman, M.; Volman, E.; Toledo, I.; et al. Selection for CD26. Front. Endocrinol. 2021, 12, 635405.
- Kaestner, K.H.; Campbell-Thompson, M.; Dor, Y.; Gill, R.G.; Glaser, B.; Kim, S.K.; Sander, M.; Stabler, C.; Stewart, A.F.; Powers, A.C. What is a β cell?—Chapter I in the Human Islet Research Network (HIRN) review series. Mol. Metab. 2021, 53, 101323.
- Augsornworawat, P.; Maxwell, K.G.; Velazco-Cruz, L.; Millman, J.R. Single-Cell Transcriptome Profiling Reveals β Cell Maturation in Stem Cell-Derived Islets after Transplantation. Cell Rep. 2020, 32, 108067.
- Balboa, D.; Barsby, T.; Lithovius, V.; Saarimäki-Vire, J.; Omar-Hmeadi, M.; Dyachok, O.; Montaser, H.; Lund, P.E.; Yang, M.; Ibrahim, H.; et al. Functional, metabolic and transcriptional maturation of human pancreatic islets derived from stem cells. Nat. Biotechnol. 2022, 40, 1042–1055.
- Faleo, G.; Russ, H.A.; Wisel, S.; Parent, A.V.; Nguyen, V.; Nair, G.G.; Freise, J.E.; Villanueva, K.E.; Szot, G.L.; Hebrok, M.; et al. Mitigating Ischemic Injury of Stem Cell-Derived Insulin-Producing Cells after Transplant. Stem Cell Rep. 2017, 9, 807–819.
- Paraskevas, S.; Maysinger, D.; Wang, R.; Duguid, T.P.; Rosenberg, L. Cell loss in isolated human islets occurs by apoptosis. Pancreas 2000, 20, 270–276.
- McKenzie, M.D.; Jamieson, E.; Jansen, E.S.; Scott, C.L.; Huang, D.C.; Bouillet, P.; Allison, J.; Kay, T.W.; Strasser, A.; Thomas, H.E. Glucose induces pancreatic islet cell apoptosis that requires the BH3-only proteins Bim and Puma and multi-BH domain protein Bax. Diabetes 2010, 59, 644–652.
- Federici, M.; Hribal, M.; Perego, L.; Ranalli, M.; Caradonna, Z.; Perego, C.; Usellini, L.; Nano, R.; Bonini, P.; Bertuzzi, F.; et al. High glucose causes apoptosis in cultured human pancreatic islets of Langerhans: A potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes 2001, 50, 1290–1301.
- Costes, S.; Vandewalle, B.; Tourrel-Cuzin, C.; Broca, C.; Linck, N.; Bertrand, G.; Kerr-Conte, J.; Portha, B.; Pattou, F.; Bockaert, J.; et al. Degradation of cAMP-responsive element-binding protein by the ubiquitin-proteasome pathway contributes to glucotoxicity in beta-cells and human pancreatic islets. Diabetes 2009, 58, 1105–1115.
- McCall, M.; Pawlick, R.; Kin, T.; Shapiro, A.M. Anakinra potentiates the protective effects of etanercept in transplantation of marginal mass human islets in immunodeficient mice. Am. J. Transplant. 2012, 12, 322–329.
- Ricordi, C.; Lacy, P.E.; Finke, E.H.; Olack, B.J.; Scharp, D.W. Automated method for isolation of human pancreatic islets. Diabetes 1988, 37, 413–420.
- Nikolova, G.; Jabs, N.; Konstantinova, I.; Domogatskaya, A.; Tryggvason, K.; Sorokin, L.; Fässler, R.; Gu, G.; Gerber, H.P.; Ferrara, N.; et al. The vascular basement membrane: A niche for insulin gene expression and Beta cell proliferation. Dev. Cell 2006, 10, 397–405.
- Linn, T.; Schmitz, J.; Hauck-Schmalenberger, I.; Lai, Y.; Bretzel, R.G.; Brandhorst, H.; Brandhorst, D. Ischaemia is linked to inflammation and induction of angiogenesis in pancreatic islets. Clin. Exp. Immunol. 2006, 144, 179–187.
- Olsson, R.; Carlsson, P.O. Oxygenation of cultured pancreatic islets. Adv. Exp. Med. Biol. 2006, 578, 263–268.
- Maechler, P.; de Andrade, P.B. Mitochondrial damages and the regulation of insulin secretion. Biochem. Soc. Trans. 2006, 34, 824–827.
- Carlsson, P.O.; Liss, P.; Andersson, A.; Jansson, L. Measurements of oxygen tension in native and transplanted rat pancreatic islets. Diabetes 1998, 47, 1027–1032.
- Carlsson, P.O.; Palm, F.; Andersson, A.; Liss, P. Chronically decreased oxygen tension in rat pancreatic islets transplanted under the kidney capsule. Transplantation 2000, 69, 761–766.
- Carlsson, P.O.; Palm, F.; Andersson, A.; Liss, P. Markedly decreased oxygen tension in transplanted rat pancreatic islets irrespective of the implantation site. Diabetes 2001, 50, 489–495.
- Davalli, A.M.; Ogawa, Y.; Ricordi, C.; Scharp, D.W.; Bonner-Weir, S.; Weir, G.C. A selective decrease in the beta cell mass of human islets transplanted into diabetic nude mice. Transplantation 1995, 59, 817–820.
- Carlsson, P.O.; Palm, F.; Mattsson, G. Low revascularization of experimentally transplanted human pancreatic islets. J. Clin. Endocrinol. Metab. 2002, 87, 5418–5423.
- Stokes, R.A.; Cheng, K.; Deters, N.; Lau, S.M.; Hawthorne, W.J.; O’Connell, P.J.; Stolp, J.; Grey, S.; Loudovaris, T.; Kay, T.W.; et al. Hypoxia-inducible factor-1α (HIF-1α) potentiates β-cell survival after islet transplantation of human and mouse islets. Cell Transplant. 2013, 22, 253–266.
- Rodriguez-Brotons, A.; Bietiger, W.; Peronet, C.; Magisson, J.; Sookhareea, C.; Langlois, A.; Mura, C.; Jeandidier, N.; Pinget, M.; Sigrist, S.; et al. Impact of Pancreatic Rat Islet Density on Cell Survival during Hypoxia. J. Diabetes Res. 2016, 2016, 3615286.
- Komatsu, H.; Kang, D.; Medrano, L.; Barriga, A.; Mendez, D.; Rawson, J.; Omori, K.; Ferreri, K.; Tai, Y.C.; Kandeel, F.; et al. Isolated human islets require hyperoxia to maintain islet mass, metabolism, and function. Biochem. Biophys. Res. Commun. 2016, 470, 534–538.
- Komatsu, H.; Cook, C.; Wang, C.H.; Medrano, L.; Lin, H.; Kandeel, F.; Tai, Y.C.; Mullen, Y. Oxygen environment and islet size are the primary limiting factors of isolated pancreatic islet survival. PLoS ONE 2017, 12, e0183780.
- Garcia-Contreras, M.; Tamayo-Garcia, A.; Pappan, K.L.; Michelotti, G.A.; Stabler, C.L.; Ricordi, C.; Buchwald, P. Metabolomics Study of the Effects of Inflammation, Hypoxia, and High Glucose on Isolated Human Pancreatic Islets. J. Proteome Res. 2017, 16, 2294–2306.
- Maillard, E.; Juszczak, M.T.; Langlois, A.; Kleiss, C.; Sencier, M.C.; Bietiger, W.; Sanchez-Dominguez, M.; Krafft, M.P.; Johnson, P.R.; Pinget, M.; et al. Perfluorocarbon emulsions prevent hypoxia of pancreatic β-cells. Cell Transplant. 2012, 21, 657–669.
- Cantley, J.; Grey, S.T.; Maxwell, P.H.; Withers, D.J. The hypoxia response pathway and β-cell function. Diabetes Obes. Metab. 2010, 12 (Suppl. S2), 159–167.
- Cheng, K.; Ho, K.; Stokes, R.; Scott, C.; Lau, S.M.; Hawthorne, W.J.; O’Connell, P.J.; Loudovaris, T.; Kay, T.W.; Kulkarni, R.N.; et al. Hypoxia-inducible factor-1alpha regulates beta cell function in mouse and human islets. J. Clin. Investig. 2010, 120, 2171–2183.
- Cantley, J.; Walters, S.N.; Jung, M.H.; Weinberg, A.; Cowley, M.J.; Whitworth, T.P.; Kaplan, W.; Hawthorne, W.J.; O’Connell, P.J.; Weir, G.; et al. A preexistent hypoxic gene signature predicts impaired islet graft function and glucose homeostasis. Cell Transplant. 2013, 22, 2147–2159.
- Moritz, W.; Meier, F.; Stroka, D.M.; Giuliani, M.; Kugelmeier, P.; Nett, P.C.; Lehmann, R.; Candinas, D.; Gassmann, M.; Weber, M. Apoptosis in hypoxic human pancreatic islets correlates with HIF-1alpha expression. FASEB J. 2002, 16, 745–747.
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140.
- Wali, J.A.; Gurzov, E.N.; Fynch, S.; Elkerbout, L.; Kay, T.W.; Masters, S.L.; Thomas, H.E. Activation of the NLRP3 inflammasome complex is not required for stress-induced death of pancreatic islets. PLoS ONE 2014, 9, e113128.
- do Amaral, A.S.; Pawlick, R.L.; Rodrigues, E.; Costal, F.; Pepper, A.; Galvão, F.H.; Correa-Giannella, M.L.; Shapiro, A.M. Glutathione ethyl ester supplementation during pancreatic islet isolation improves viability and transplant outcomes in a murine marginal islet mass model. PLoS ONE 2013, 8, e55288.
- Miwa, I.; Ichimura, N.; Sugiura, M.; Hamada, Y.; Taniguchi, S. Inhibition of glucose-induced insulin secretion by 4-hydroxy-2-nonenal and other lipid peroxidation products. Endocrinology 2000, 141, 2767–2772.
- Carobbio, S.; Ishihara, H.; Fernandez-Pascual, S.; Bartley, C.; Martin-Del-Rio, R.; Maechler, P. Insulin secretion profiles are modified by overexpression of glutamate dehydrogenase in pancreatic islets. Diabetologia 2004, 47, 266–276.
- Li, C.; Buettger, C.; Kwagh, J.; Matter, A.; Daikhin, Y.; Nissim, I.B.; Collins, H.W.; Yudkoff, M.; Stanley, C.A.; Matschinsky, F.M. A signaling role of glutamine in insulin secretion. J. Biol. Chem. 2004, 279, 13393–13401.
- Koulajian, K.; Ivovic, A.; Ye, K.; Desai, T.; Shah, A.; Fantus, I.G.; Ran, Q.; Giacca, A. Overexpression of glutathione peroxidase 4 prevents β-cell dysfunction induced by prolonged elevation of lipids in vivo. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E254–E262.
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556.
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223.
- Daly, K.A.; Liu, S.; Agrawal, V.; Brown, B.N.; Johnson, S.A.; Medberry, C.J.; Badylak, S.F. Damage associated molecular patterns within xenogeneic biologic scaffolds and their effects on host remodeling. Biomaterials 2012, 33, 91–101.
- Paredes-Juarez, G.A.; Sahasrabudhe, N.M.; Tjoelker, R.S.; de Haan, B.J.; Engelse, M.A.; de Koning, E.J.P.; Faas, M.M.; de Vos, P. DAMP production by human islets under low oxygen and nutrients in the presence or absence of an immunoisolating-capsule and necrostatin-1. Sci. Rep. 2015, 5, 14623.
- Land, W.G.; Agostinis, P.; Gasser, S.; Garg, A.D.; Linkermann, A. DAMP-Induced Allograft and Tumor Rejection: The Circle Is Closing. Am. J. Transplant. 2016, 16, 3322–3337.
- Land, W.G.; Agostinis, P.; Gasser, S.; Garg, A.D.; Linkermann, A. Transplantation and Damage-Associated Molecular Patterns (DAMPs). Am. J. Transplant. 2016, 16, 3338–3361.
- Itoh, T.; Takita, M.; SoRelle, J.A.; Shimoda, M.; Sugimoto, K.; Chujo, D.; Qin, H.; Naziruddin, B.; Levy, M.F.; Matsumoto, S. Correlation of released HMGB1 levels with the degree of islet damage in mice and humans and with the outcomes of islet transplantation in mice. Cell Transplant. 2012, 21, 1371–1381.
- Pepper, A.R.; Gala-Lopez, B.; Pawlick, R.; Merani, S.; Kin, T.; Shapiro, A.M. A prevascularized subcutaneous device-less site for islet and cellular transplantation. Nat. Biotechnol. 2015, 33, 518–523.
- Vlahos, A.E.; Cober, N.; Sefton, M.V. Modular tissue engineering for the vascularization of subcutaneously transplanted pancreatic islets. Proc. Natl. Acad. Sci. USA 2017, 114, 9337–9342.
- Song, W.; Chiu, A.; Wang, L.H.; Schwartz, R.E.; Li, B.; Bouklas, N.; Bowers, D.T.; An, D.; Cheong, S.H.; Flanders, J.A.; et al. Engineering transferrable microvascular meshes for subcutaneous islet transplantation. Nat. Commun. 2019, 10, 4602.
- Yu, M.; Agarwal, D.; Korutla, L.; May, C.L.; Wang, W.; Griffith, N.N.; Hering, B.J.; Kaestner, K.H.; Velazquez, O.C.; Markmann, J.F.; et al. Islet transplantation in the subcutaneous space achieves long-term euglycaemia in preclinical models of type 1 diabetes. Nat. Metab. 2020, 2, 1013–1020.
- Butler, M.J.; Sefton, M.V. Cotransplantation of adipose-derived mesenchymal stromal cells and endothelial cells in a modular construct drives vascularization in SCID/bg mice. Tissue Eng. Part A 2012, 18, 1628–1641.
- Weaver, J.D.; Headen, D.M.; Aquart, J.; Johnson, C.T.; Shea, L.D.; Shirwan, H.; García, A.J. Vasculogenic hydrogel enhances islet survival, engraftment, and function in leading extrahepatic sites. Sci. Adv. 2017, 3, e1700184.
- Aghazadeh, Y.; Poon, F.; Sarangi, F.; Wong, F.T.M.; Khan, S.T.; Sun, X.; Hatkar, R.; Cox, B.J.; Nunes, S.S.; Nostro, M.C. Microvessels support engraftment and functionality of human islets and hESC-derived pancreatic progenitors in diabetes models. Cell Stem Cell 2021, 28, 1936–1949.e8.
- Coronel, M.M.; Geusz, R.; Stabler, C.L. Mitigating hypoxic stress on pancreatic islets via in situ oxygen generating biomaterial. Biomaterials 2017, 129, 139–151.
- Coronel, M.M.; Liang, J.P.; Li, Y.; Stabler, C.L. Oxygen generating biomaterial improves the function and efficacy of beta cells within a macroencapsulation device. Biomaterials 2019, 210, 1–11.
- Liang, J.P.; Accolla, R.P.; Soundirarajan, M.; Emerson, A.; Coronel, M.M.; Stabler, C.L. Engineering a macroporous oxygen-generating scaffold for enhancing islet cell transplantation within an extrahepatic site. Acta Biomater. 2021, 130, 268–280.
- Santini-González, J.; Simonovich, J.A.; Castro-Gutiérrez, R.; González-Vargas, Y.; Abuid, N.J.; Stabler, C.L.; Russ, H.A.; Phelps, E.A. In vitro generation of peri-islet basement membrane-like structures. Biomaterials 2021, 273, 120808.
- Takahashi, Y.; Sekine, K.; Kin, T.; Takebe, T.; Taniguchi, H. Self-Condensation Culture Enables Vascularization of Tissue Fragments for Efficient Therapeutic Transplantation. Cell Rep. 2018, 23, 1620–1629.
More
Information
Subjects:
Transplantation
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
647
Revisions:
2 times
(View History)
Update Date:
03 Mar 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No