Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Luca Gentilucci | -- | 3412 | 2023-02-19 16:48:20 | | | |
| 2 | Sirius Huang | Meta information modification | 3412 | 2023-02-20 02:57:30 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Santino, F.; He, T.; Bedini, A.; Francescato, M.; Gentilucci, L. κ-Opioid Receptor Agonists. Encyclopedia. Available online: https://encyclopedia.pub/entry/41402 (accessed on 25 June 2026).
Santino F, He T, Bedini A, Francescato M, Gentilucci L. κ-Opioid Receptor Agonists. Encyclopedia. Available at: https://encyclopedia.pub/entry/41402. Accessed June 25, 2026.
Santino, Federica, Tingting He, Andrea Bedini, Marco Francescato, Luca Gentilucci. "κ-Opioid Receptor Agonists" Encyclopedia, https://encyclopedia.pub/entry/41402 (accessed June 25, 2026).
Santino, F., He, T., Bedini, A., Francescato, M., & Gentilucci, L. (2023, February 19). κ-Opioid Receptor Agonists. In Encyclopedia. https://encyclopedia.pub/entry/41402
Santino, Federica, et al. "κ-Opioid Receptor Agonists." Encyclopedia. Web. 19 February, 2023.
Copy Citation
The κ-opioid receptor (KOR) belongs to the class of inhibitory G protein-coupled receptors (GPCRs), widely expressed throughout the central nervous system and peripheral tissues. Due to the implications of KOR activation, KOR agonists have attracted recent attention for their ability to produce analgesia without the harmful side effects typically associated with MOR activation. In addition, KOR agonists show the potential for the treatment of pruritis, multiple sclerosis, Alzheimer’s disease, immune mediated diseases such as osteoarthritis, atopic dermatitis, food allergy, gastrointestinal diseases, cancer, hypoxia and ischemia.
κ-opioid receptor
biased agonist
partial agonist
antinociception
dysphoria
1. Introduction
The κ-opioid receptor (KOR) belongs to the class of inhibitory GPCRs, widely expressed throughout the CNS and peripheral tissues [1][2]. Together with the μ-, δ- and the nociceptin-opioid receptors (MOR, DOR, NOR) [3], KOR is involved in modulation of pain, reward, mood state, and cognitive functions [4][5]. The endogenous ligands of KOR are the Dynorphins [6], a family of neuropeptides of varying lengths that are formed from the precursor prodynorphin. KOR activation elicits potent antinociceptive effects in various models of acute (mechanical, thermal, and chemical), inflammatory, neuropathic, and cancer pain.
At present, the large majority of therapeutic painkillers are naturally occurring opiate MOR agonists, such as morphine, codeine, or semi-synthetic opioid derivatives, e.g., oxycodone, or fully synthetic molecules such as fentanyl or tramadol. On the other hand, the use of these drugs is accompanied by severe side effects, i.e., addiction and high abuse potential, slowed breathing, constipation, nausea, confusion, and drowsiness. Agonist activity on DOR does not lead to the same adverse effects associated with MOR agonists, but DOR agonists were found to display proconvulsive activity. Interestingly, the analgesic effects mediated by NOR are more complex than those elicited by the other opioid receptors. Indeed, agonism at NOR was shown to produce either anti- or pro-nociceptive effects depending on the route of administration and dosage [3].
Due to the implications of KOR activation, KOR agonists have attracted recent attention for their ability to produce potent analgesic effects without the harmful side effects typically associated with MOR activation [7][8]. KOR agonists present low abuse potential and may also prevent hyperalgesia produced by chronic use of MOR-targeting drugs [9]. In addition to analgesia, KOR agonists showed potential for the treatment of pruritis, multiple sclerosis, Alzheimer’s disease, immune mediated diseases such as osteoarthritis, atopic dermatitis, food allergy, gastrointestinal (GIT) diseases, cancer, and hypoxia and ischemia [9].
Despite their potential efficacy, at present no KOR agonist is used to treat pain in humans, mostly due to relevant side effects, i.e., severe dysphoria, neuropathy-induced astrocyte proliferation and subsequent hyperalgesia, sedation, coordination impairment, and anhedonia. Albeit not exploited as analgesics, the many selective KOR agonists discovered so far have aroused much attention for their potential to treat abuse conditions, in particular those of cocaine or alcohol [10]. In fact, it appears from several studies that dynorphin/KOR systems are connected with the dopamine pathway [11]. More specifically, KOR activation is believed to inhibit dopamine release and avoid the hypodopaminergic state responsible for the hypersensitivity to emotional distress during drug withdrawal and abstinence. KOR agonists and drugs of abuse seem to have opposite effects on both abuse related neurochemical and behavioral end points [12]. These effects can be attributed to the ability of KOR agonists to form oligomers with the dopamine transporter (DAT) and decrease dopamine levels in the nucleus accumbens.
2. KOR Agonists
Selected examples of molecules with a clear activity at KOR are reviewed herein. Molecules such as for instance Noribogaine, known as a potent serotonin reuptake inhibitor, which also acts as a moderate KOR agonist and weak MOR agonist or weak partial agonist, are not included.
2.1. Small Molecules
2.1.1. Morphinans
Nalfurafine is a selective KOR agonist approved for clinical use as an antipruritic. It is considered a biased agonist because an in vitro experiment resulted in its preferred activation of β-arrestin pathway signaling [13]. Unexpectedly, even if this pathway usually elicits side effects, in vivo it does not appear to be responsible for KOR agonist-induced aversion.
Nalmefene (NMF) is a partial KOR agonist and potent MOR antagonist (Figure 1) utilized for opioid and alcohol addiction [14], whose most commonly occurring adverse events are nausea, insomnia, and dizziness.
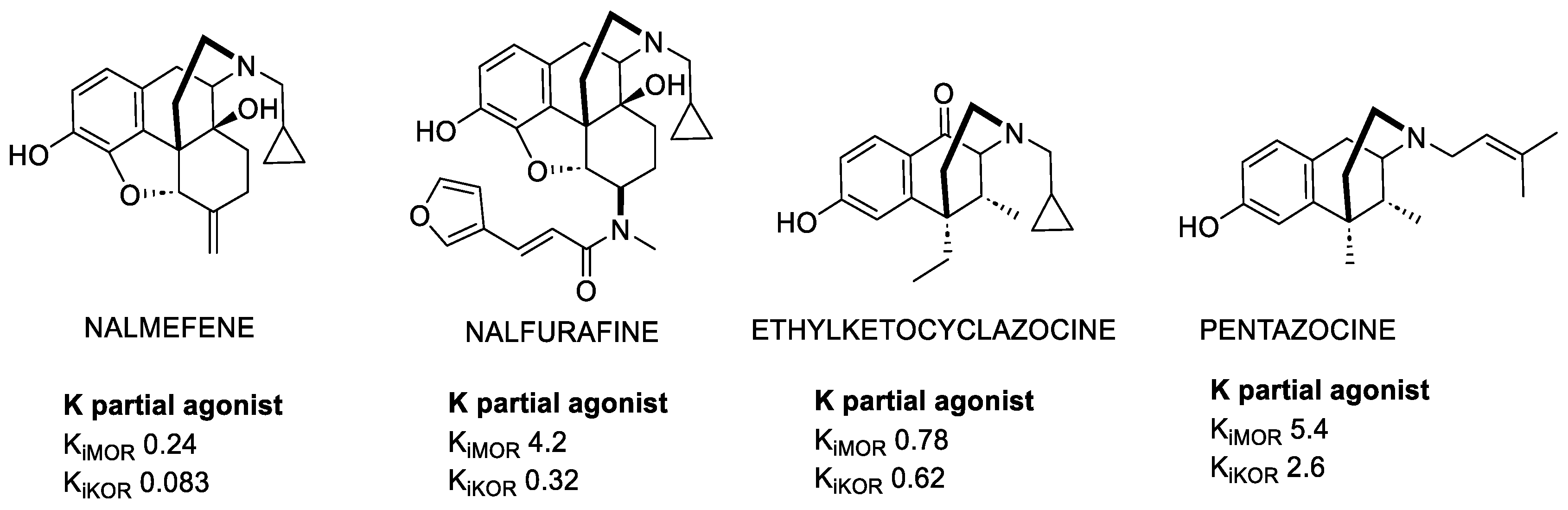
Figure 1. Examples of relevant morphinans and benzomorphans. Ki values are also shown (nM).
2.1.2. Benzomorphans
The κ-type receptor owes its name to ketocyclazocine, the first selective KOR ligand [15], which elicited analgesia and sedation. Ketocyclazocine belongs to the benzomorphans class, obtained by morphine skeleton reduction. Despite ketocyclazocine resulting to be less potent than morphine, Benzomorphans (Figure 1) were studied for many years for their interesting mixed KOR agonist/MOR antagonist profile. Their clinical development was precluded because they also induce respiratory depression and other side effects.
In contrast, Pentazocine is still under investigation, and recent studies showed that it was more potent in activating p38 MAPK mediated by h-KOR than the rat KOR, and it was also shown to be a potent promoter of β-arrestin 2 recruitment [16]. Studies on Pentazocine led to the identification of Bramazocine as a potent KOR agonist. Despite its activity profile, it does not seem to elicit morphine-like adverse actions, and it can be used for its analgesic and diuretic properties. Nonetheless, its psychotomimetic side effects limit its use as a clinical analgesic, but still allow for its use in the treatment of addictions [17].
2.1.3. Arylacetamides
The class of KOR-selective Arylacetamide derivatives (Figure 2) was described for the first time in 1982 [18]. Among them, U50,488 emerged for novel structure, potency, and selectivity, with analgesic, antipruritic, and other effects, Unfortunately, U50,488 and its subsequent derivatives showed all the side effects correlated with the activation of KOR, and also a modest permeability of the blood–brain barrier (BBB).
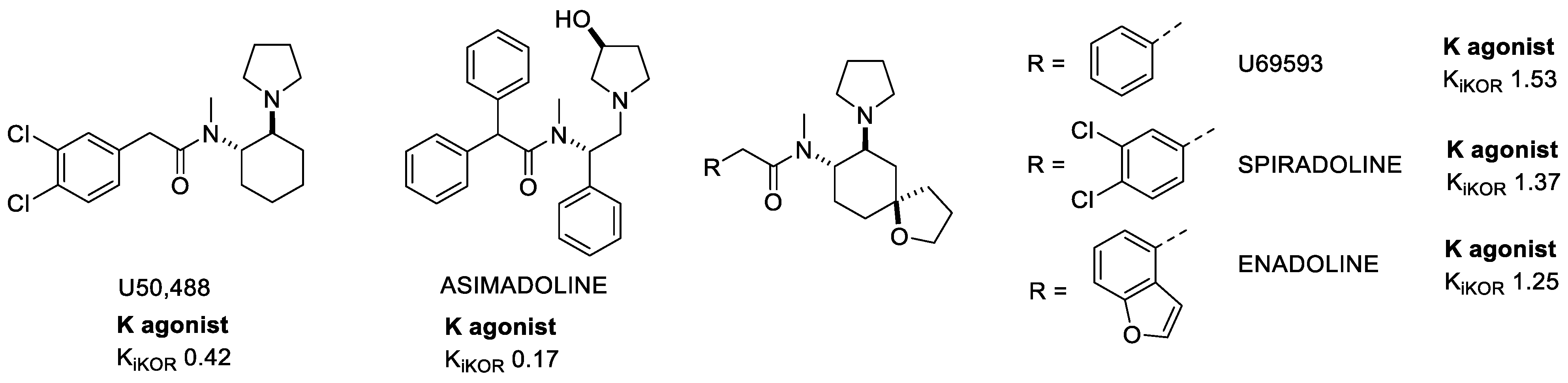
Figure 2. Examples of Arylacetamide KOR agonists. Ki values are also shown (nM).
Asimadoline was found to inhibit nociception via activation of KORs expressed on the peripheral endings of nociceptors in the colon, suggesting a peripherally restricted action which might be useful for a variety of painful conditions in the viscera, such as irritable bowel syndrome (IBS) [19].
With the attempt to further improve the structure of U50,488, a spiroester was added to the cyclohexane ring, leading to a special branch of the Arylacetamide family of which U69593 was the precursor. U69593 produced antinociception without affecting GIT mobility, suggesting a central but not a peripheral activity. Both U50,488 and U69593 activated, with the same efficacy, G-protein and β-arrestin signaling, so they have been classified as full unbiased KOR agonists [20].
Further modifications lead to Spiradoline (U62-066) and Enadoline (CI-977) molecules being capable of crossing the BBB, with reasonable efficacy and with an analgesic potency comparable to morphine, however with reduced respiratory depression. Unfortunately, they still have dose related central effects such as anhedonia, dysphoria, and hallucination-like effects so it was impossible to carry on further clinical investigations [21].
Diverse alkyl- or aryl-substituted ethyl groups were introduced over the years to the base structure in order to achieve higher analgesic efficacy with moderate addictive potential. Among the derivatives it is worth mentioning ICI 199,441, GR89,696, R-84760. Unfortunately, none of these molecules were able to complete the clinical trials due to adverse reactions.
2.1.4. Triazoles
As a result of two different screenings, in 2013 Zhou et al. discovered two new classes of KOR selective agonists: triazoles and isoquinolines [22] (Figure 3). Both of them showed high binding affinity with a preferential activation of G-protein and minimal effects on β-arrestin recruitment and downstream ERK1/2 activation. Further studies revealed that triazole, in particular, induced antinociception, without altering locomotion nor provoking dysphoria or aversion. Triazole 1.1 retained the antinociceptive and antipruritic efficacies of conventional KOR agonists, yet it did not induce sedation or reductions in dopamine release in vivo, nor did it produce dysphoria as determined by intracranial self-stimulation in rats [23].
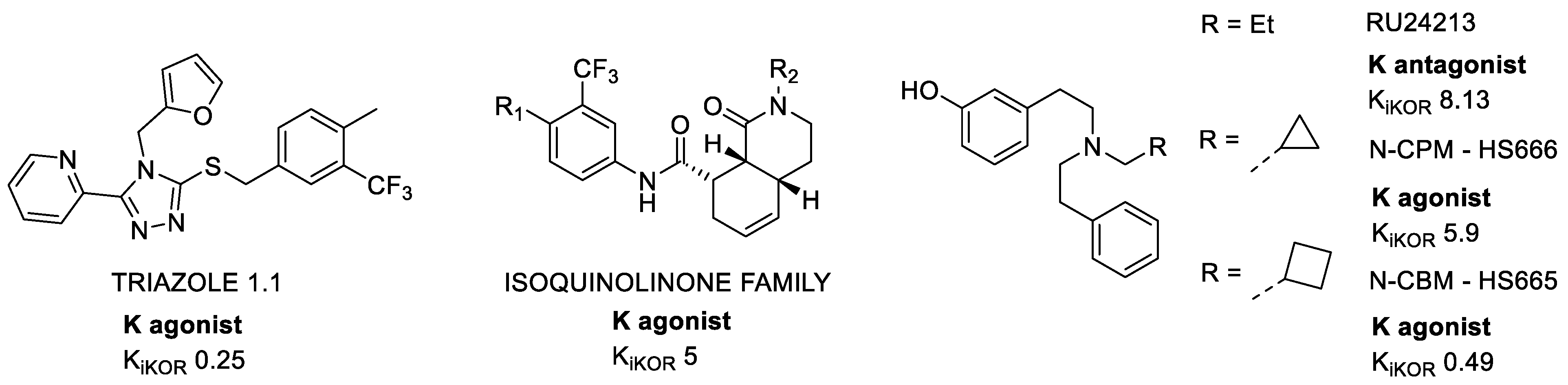
Figure 3. Examples of KOR agonists: triazoles, isoquinolines, and diphenethylamines. Ki values are also shown (nM).
2.1.5. Diphenethylamines
These simple molecules were preliminarily described in 1978 as potential anti-Parkinson’s drugs (Figure 3) [24]. The first synthetized molecule was RU24213, and after it libraries of diverse molecules were designed. The first strategy was the extension of the N-alkyl substituent. In vitro binding studies showed good KOR affinity and potency, but weak selectivity with a decreasing trend as the number of carbons increased [25].
Subsequently, the introduction of bulkier substituents allowed for an increase in KOR affinity, selectivity, agonist potency, and efficacy, giving N-cyclobutylmethyl (N-CBM, HS665) and N-cyclopropylmethyl (N-CPM, HS666) [26]. HS665 and HS666 were shown to produce potent antinociception, mediated by central KORs. In particular, HS666 showed reduced liability for aversive effects, and this was correlated with its lower efficacy in the β-arrestin2 signaling pathway [27].
Considering the inclusion in recent years of fluorine in drug candidates, in 2017 Erli et al. expanded the SAR studies on the original series, e.g., by examining any effects of fluorine at position 2 of diphenethylamines, and by introducing bulkier N-substituents, plus additional hydroxyl groups at positions 3′ and 4′. It was demonstrated that some analogues gained sub-nanomolar affinity and excellent KOR selectivity, acting as full or partial agonists or as antagonists [28].
2.1.6. Salvinorins
Salvinorin A (SalA) (Figure 4) is a terpenoid isolated in 1982 by Ortega et al. from Salvia Divinorum, a herbal plant native to the southwestern region of Mexico [29]. Two years later, Valdes et al. extracted Salvinorin B [30] and in 2001 also Salvinorin C [31]. These compounds were found to be selective KOR agonists with analgesic and hallucinogenic effects. All of these compounds immediately gained interest because of their lack of structural similarity to the other psychotomimetic substances. Indeed, unlike the conventional opioid ligands, salvinorins do not contain any nitrogen atoms.
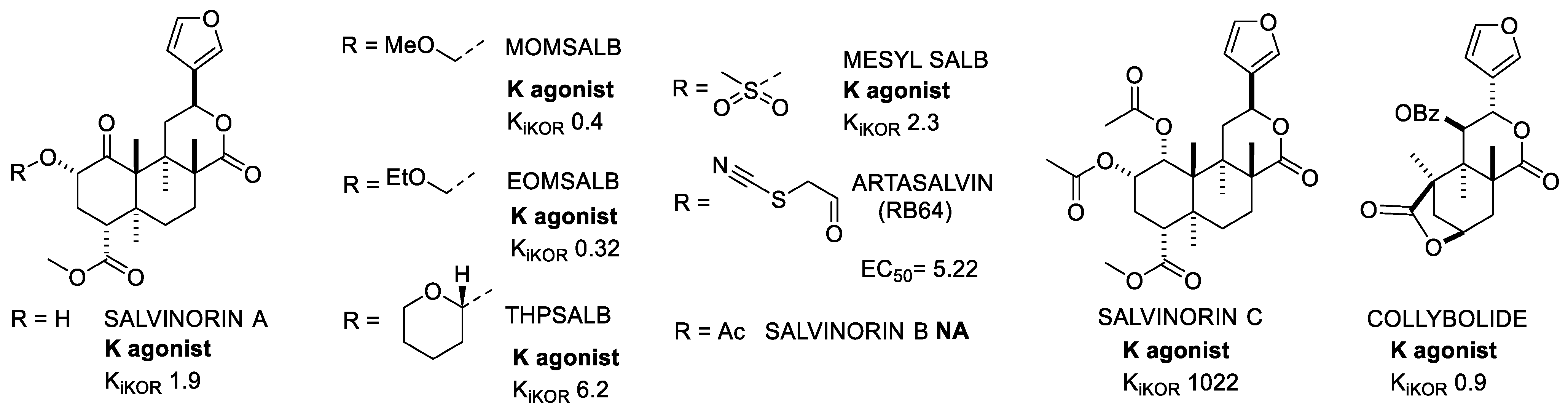
Figure 4. Salvinorin A and selected analogues and derivatives. Ki values are also shown (nM).
Unfortunately, these natural molecules suffered from a short half-life and rapid onset of action; therefore, a large number of analogues were prepared to improve the pharmacokinetic profile, generally by modifying the positions at C2 and C4 of the furan ring [32]. In some cases, e.g., MOM-SalB, EOM SalB, and THP SalB (Figure 4), modifications gave stronger affinities or potencies and improved metabolic stability compared to SalA, but unfortunately this did not translate to increased brain residence time.
Further studies were performed in vivo on anti-addictive, mood, locomotive, and aversion effects. In particular, in 2013, Morani et al. [33] tested MOMSalB, for cocaine-seeking behavior [34] and the study showed a similar effect, compared to SalA, in reducing cocaine primed-induced reinstatement [33].
Mesylation at C2 gave Mesyl SalB, which showed distinct features and differences with respect to the other analogues. When tested for the inhibition of foskolin-stimulated cAMP accumulation, it showed increased potency in vitro. This in vitro result, together with the similar efficacy to U50488 in the recruitment of β-arrestin, suggested a G-protein biased profile. Furthermore, Mesyl SalB had fewer side effects than SalA and U50488 when tested for cocaine addiction, but it maintained pro-depressive effects and showed no antinociception activity [35].
Additionally, RB64 was identified as a biased G-protein KOR agonist with the specific characteristic of inducing less receptor internalization, having longer lasting effects and less unwanted reactions (anhedonia or motor activity alteration), while still inducing aversion [36].
Collybolide was extracted in 1974 from the fungus Collybia Maculata [37], and attracted attention as a non-nitrogenous, potent, and biased KOR agonist. However, more recent studies indicated that collybolide was mischaracterized as a KOR agonist [38].
2.2. Peptides and Peptidomimetics
Dynorphin is one of the endogenous 17-mer neuropeptide ligands of KOR. Other peptides with KOR activity have been identified in fungi and invertebrates, e.g., conotoxins, CJ15,209, and several others have been obtained by chemical synthesis [39]. Native peptides such as dynorphin have the potential to potently activate opioid receptors. However, their potential clinical use is hampered by poor pharmacokinetic properties. As a consequence, starting from the native sequences, a variety of peptidomimetics were designed with the perspective of increasing enzymatic stability and bioavailability [40][41].
2.2.1. Dynorphins
Dynorphin A (DynA)1-17 (YGGFLRRIRPKLKWDNQ), isolated in 1975 from porcine pituitary, is an endogenous neuropeptide capable of modulating pain, addiction, and mood [42]. Several isoforms were soon identified: Dynorphin A(1-8), Dynorphin B(1-13), big Dynorphin, and Leumorphin. Dynorphin A (DynA)(1-17) is regarded as the specific endogenous ligand of KOR [6]. Extensive SAR studies have identified DynA fragments critical to bioactivity. The 17 amino acid full sequence was shortened at its C-terminus to give Dynorphin(1-13), without altering activity. While the N-terminal “message sequence” (YGGF) is essential for binding to all opioid receptors, the C terminal “address sequence” (LRRIRPKLKWDNQ) is required for potency and selectivity. In addition, replacement of the nine C-terminal residues of DynA(1-17) with those of Nociceptin also maintained activity at KOR [43]. From this observation, it was concluded that the essential pharmacophores of DynA(1-17) lie within the N-terminal fragment DynA(1-8) (YGGFLRRI).
2.2.2. Difelikefalin (CR845)
In search of KOR agonists with improved in vivo stability, several research groups designed oligopeptides comprising D-amino acid, and these studies yielded molecules which appeared suitable for assessment of peripherally acting analgesics. Most of these oligopeptides shared a common N-terminal D-Phe-D-Phe [44][45][46][47] including Difelikefalin (CR845) (Figure 5), an all-D-amino acid tetrapeptide developed by Cara Therapeutics with high selectivity for KOR that has been shown to be effective in the treatment of chronic pruritus [48] and post-operative pain after abdominal surgery [49]. Due to its hydrophilic properties, transport across the blood–brain barrier is limited. Although Difelikefalin is devoid of detrimental effects linked to the centrally expressed KOR, its intravenous delivery remains a key obstacle for its widespread use.
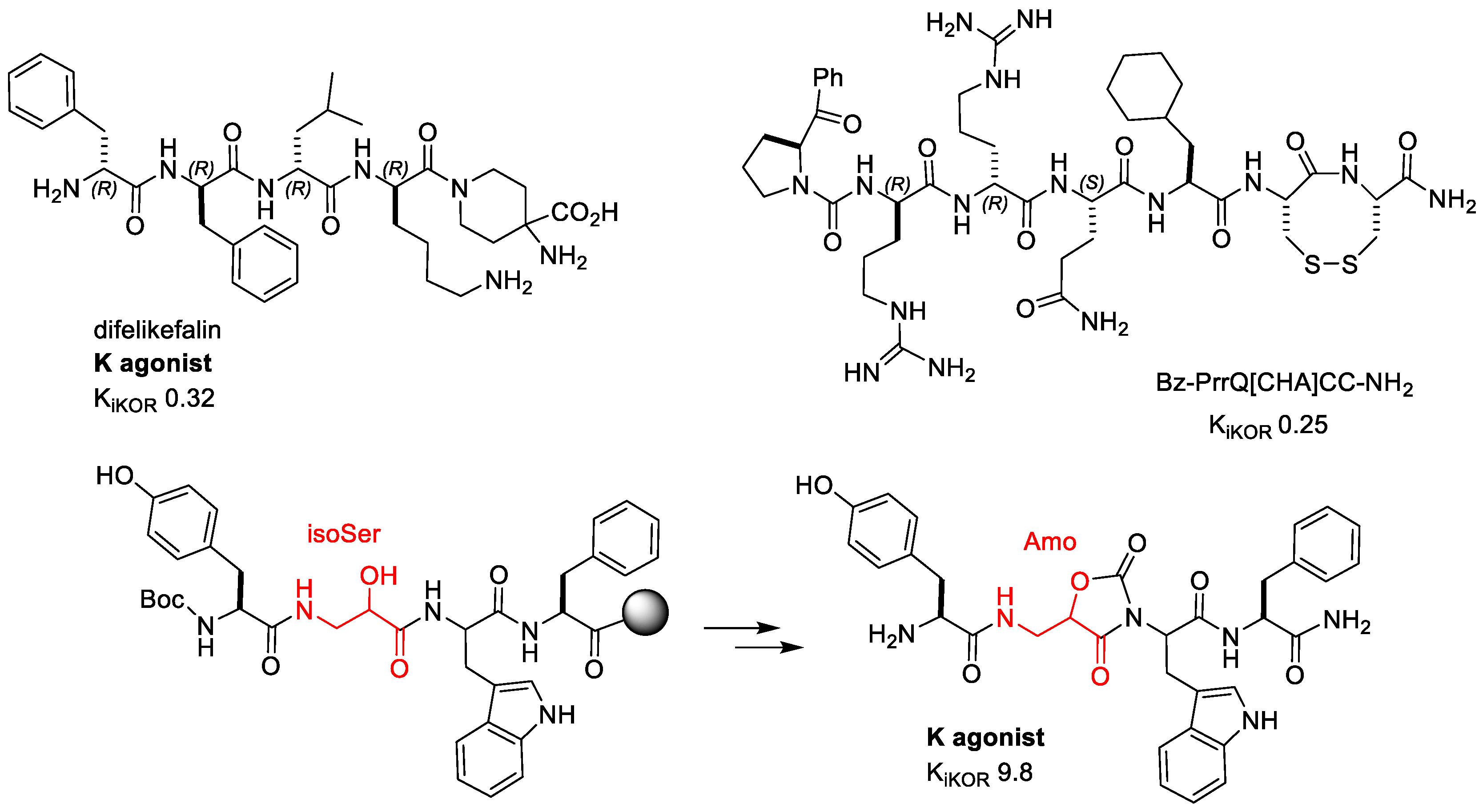
Figure 5. Peptidic and peptidomimetic KOR agonists.
2.2.3. Conorphins
Marine cone snails are equipped with a venom apparatus that typically contains 1000 unique venom peptides called Conotoxins or Conopeptides, usually characterized by a S–S bond-cyclic framework. Among these peptides, ω-Conotoxin MVIIA (Ziconotide), an inhibitor of N-type VGCCs, is on the market for the treatment of chronic pain [50]. A new KOR agonist named Conorphin-T (NCCRRQICC) was identified from the screening of a Conus venom peptide library across the opioid receptors. Estensive SAR studies generated a new class of enzymatically stable molecules with exceptional plasma stability, excellent affinity, and high KOR selectivity [51]. The lead NCCRRQICC, which had a short plasma half-life (t1/2 15 min), was systematically altered to produce the octapeptide Bz-PrrQ[CHA]CC-NH2 (Figure 5), which showed sub-nM affinity, good efficacy (Ki 0.25 nM, EC50 0.36 nM), and improved plasma stability (t1/2 240 min). The Conorphin agonist inhibited nociceptors in the colon, in a mouse tissue model of chronic visceral hypersensitivity, suggesting the potential of KOR agonists for the treatment of chronic abdominal pain [51].
2.2.4. Helianorphins
Gruber et al. utilized cyclic peptides from plants as templates for the design of a stable KOR peptide ligand, acting as a potent analgesic for enteric administration [52]. Specifically, different Dyn(1−13) epitopes were grafted with parts of the peptide SFTI-1, one of the main peptides in the extract of sunflower seeds. SFTI-1 is a bicyclic, “8”-shaped peptide, due to the presence of one disulfide bond, which can be divided in binding and cyclization loops. Dyn(1−13) was incorporated into either the cyclization loop or the binding loop, or it was split into two fragments and inserted into both loops. This strategy allowed for identification of Helianorphin-19, c[CYGGFLRRCIRPKLK], a selective KOR ligand with good affinity (for KOR, Ki 21 nM, EC50 45 nM). Helianorphin-19 was found to preferentially activate G-protein over β-arrestin 2, and to be active in a CVH mouse model of abdominal pain; this is possibly correlated to its stability in the GIT tract as estimated in simulated gastric fluid, while it did not affect motor coordination nor induce central analgesia.
2.2.5. H-Tyr-Amo-Trp-PheNH2
Bedini et al. synthesized a mini-library of diastereomeric and constrained analogues of the endogenous, highly selective MOR agonist Endomorphin-1 (EM1), H-Tyr-Pro-Trp-PheNH2 [53][54][55] by introducing β2-homo-Freidinger lactam-like scaffolds at position 2 [56] (Figure 5). The 5-(aminomethyl) oxazolidine-2,4-dione (Amo) scaffolds were obtained by in-peptide cyclization of isoserine-Trp [57]. Intriguingly, the all-(S) configured H-Tyr-Amo-Trp-PheNH2 displayed high KOR affinity (Ki 9.8 nM) and high selectivity. The peptidomimetic inhibited forskolin-induced cAMP accumulation as a partial agonist, not activating β-arrestin signaling. In the tail-immersion test, the peptidomimetic was determined to have a relevant analgesic effect (20 mg/kg, ip: 60% MPE at 15 min, and still 42% MPE at 30 min), and the effects were counteracted by the preemptive administration of the KOR selective antagonist nor-BNI, thus confirming, also in vivo, a KOR-mediated activity.
2.2.6. CJ-15,209 and Derivatives
In 2004, Saito et al. isolated from the fermentation broth of the fungus Ctenomyces serratus ATCC15502 the cyclotetrapeptide c[Phe-D-Pro-Phe-Trp] (CJ-15,208) (Figure 6). CJ-15,208 was a modestly selective KOR/MOR ligand (IC50 nM: KOR, 47; MOR, 260 DOR, 2600). Initially, the researchers observed antagonist activity against the KOR agonist asimadoline in the rabbit vas deferens smooth muscle assay (EC50 1300 nM) [58]. Later, the antagonism was confirmed by the [32S]GTPγS functional test [59]. An Ala-scan highlighted the importance of the residues Phe3 and Trp4; indeed, c[Ala-D-Pro-Phe-Trp] (Figure 6) showed low nanomolar affinity for KOR and MOR, while the other Ala derivatives suffered a substantial loss in binding affinity [60].
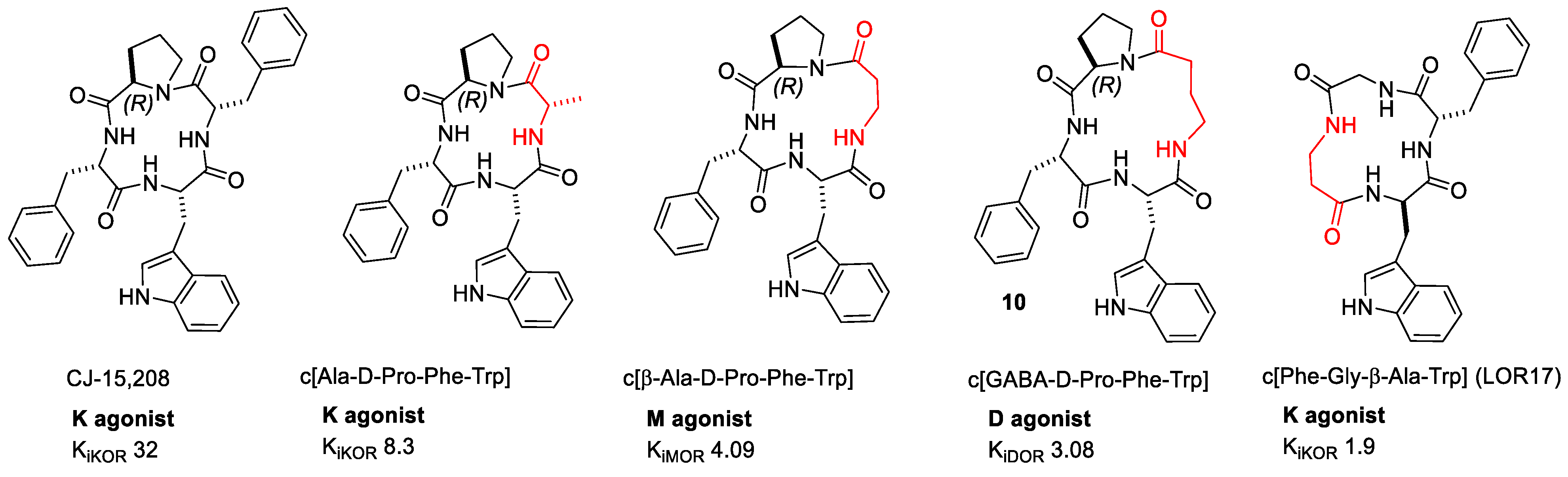
Figure 6. CJ-15,208 and some relevant derivatives. Ki values are also shown (nM).
The reversal of the configuration at Trp gave c[Phe-D-Pro-Phe-D-Trp], a dual KOR/MOR antagonist in the [32S]GTPγS test (IC50 140 nM) [59]. Additionally, in this case, Ala-scan was not tolerated at positions 3 and 4, since c[Ala-D-Pro-Phe-D-Trp] was the only compound equipotent to the parent (Figure 6) [59]. All of the derivatives of CJ-15,208 maintained the same mixed KOR > MOR affinity profile, albeit with different Ki values, and did not exhibit any agonist activity in vitro.
Unexpectedly, in vivo tests showed contrasting activities when compared to the parent peptides. As expected, c[Phe-D-Pro-Phe-D-Trp] behaved as a KOR antagonist also in vivo and prevented the stress-induced reinstatement of extinguished cocaine-seeking behavior [61]. In contrast, the natural isomer CJ-15,208 exhibited robust antinociceptive activity in the warm-water tail withdrawal test, following icv administration [61]. Intriguingly, the Ala analog c[Ala-D-Pro-Phe-D-Trp] also produced potent OR-mediated antinociception in vivo [62]. Finally, CJ-15,208 and c[Phe-D-Pro-Phe-D-Trp] [63][64] and other phenylalanine stereoisomers [65] were found to be orally active and appeared to penetrate the CNS.
The structures of CJ-15,208 and all its derivatives appear clearly correlated to that of c[Phe-Gly-Tyr-D-Pro-D-Trp] [66], a cyclopentapeptide (CPP) discovered independently from CJ-15,208, designed as a cyclic analogue of the EM1. The CPP was found to be a MOR ligand (Ki 10−8 M), partial agonist in the cAMP functional assay. After systemic administration, it produced antinociception in a mouse model of visceral pain, while the parent EM1 was completely ineffective [67]. Subsequent modifications gave c[Phe-Gly-Tyr-Gly-D-Trp] and c[Phe-D-isoAsp-β-Ala-D-Trp], which showed 10-fold improved MOR affinity, while maintaining the agonist profile [68]. Reduction in molecular complexity yielded the MOR-selective agonist tripeptide Ac-D-Trp-Phe-GlyNH2 [69]. The introduction of different substituents at the indole of D-Trp influenced BBB permeability, allowing a measurable MOR-mediated central antinociception in the mouse warm-water tail withdrawal test after ip administration [70].
These Trp-containing macrocycles are clearly an alternative, with respect to the classic opioid peptides, as they lack the protonable amino group of Tyr1, commonly regarded as the fundamental “message” pharmacophore [71][72]. Despite the close structural similarities, the two families of opioid peptides showed distinct receptor selectivity and in vivo activity. This led us to presume a correlation between bioactivity and 3D displays of the pharmacophores, which depend in turn on ring size, stereochemistry, and secondary structures.
This hypothesis led to expansion of the ring of CJ-15,208 to the 13-membered c[β-Ala-D-Pro-Phe-Trp], which turned out to be a highly selective MOR ligand (Ki 4.1 nM). Surprisingly, a further enlargement of ring size by introducing GABA at position 1 produced a different receptor selectivity. The 14-membered c[GABA-D-Pro-Phe-Trp] (GABA, γ-aminobutyric acid acid), showed very scarce MOR affinity and gained high DOR affinity (Ki 3.08 nM) [73]. c[β-Ala-D-Pro-Phe-Trp] inhibited forskolin-induced cAMP production (IC50 6.1 nM and Emax 90%), suggestive of full agonism, while the DOR≫MOR ligand c[GABA-D-Pro-Phe-Trp] inhibited forskolin-induced cAMP accumulation in HEK/MOR (IC50 183 nM). Interestingly, the latter did not alter forskolin-induced cAMP accumulation in HEK/DOR, but significantly antagonized, in a concentration-related manner, the inhibition of forskolin induced cAMP accumulation by 10 μM DPDPE (IC50 7.4 nM). In a mouse model of visceral pain, c[β-Ala-D-Pro-Phe-Trp] elicited peripheral preemptive antinociception (0.5–10 mg/kg, ip: ED50 0.64 mg/kg), and the effect was prevented by pretreating the animals with the antagonist NLX—but not by the DOR selective antagonist NTD, or by the KOR selective antagonist—thus confirming that the observed effect was MOR-dependent.
More recently, the same authors designed the minimalist CJ-15,208 analogue c[Phe-Gly-β-Ala-D-Trp] (LOR17). The peptide was a KOR selective agonist in different cell models with nM affinity (Ki 1.19 nM) [74]. In contrast to U50,488, LOR17 displayed functional selectivity toward G-protein signaling and provoked antinociceptive/antihypersensitivity effects in different in vivo models, including neuropathy by oxaliplatin.
The peptides described in this paragraph constitute a distinct family of tryptophan-containing non cationizable opioid peptides. In contrast to most opioids, their atypical bioactivity appears to reside in the minimal pharmacophoric motif Trp-Phe, with indole being fundamental to the ligand-receptor interaction. The diverse preferences for MOR, DOR, and KOR, seem to depend on specific secondary structures [75].
References
- Lemos, C.J.; Chavkin, C. Kappa opioid receptor function. In The Opiate Receptors, 2nd ed.; Pasternak, C.W., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 265–305.
- Liu-Chen, L.-Y.; Inan, S. The Kappa Opioid Receptor, Handbook of Experimental Pharmacology, 1st ed.; Springer: Cham, Switzerland, 2022; Volume 271, p. 577.
- Janecka, A.; Fichna, J.; Janecki, T. Opioid receptors and their ligands. Curr. Top. Med. Chem. 2004, 4, 1–17.
- Carlezon, W.A., Jr.; Béguin, C.; Knoll, A.T.; Cohen, B.M. Kappa-opioid ligands in the study and treatment of mood disorders. Pharm. Ther. 2009, 123, 334–343.
- Clark, S.D. The Role of Dynorphin and the Kappa Opioid Receptor in Schizophrenia and Major Depressive Disorder: A Translational Approach. In The Kappa Opioid Receptor. Handbook of Experimental Pharmacology; Liu-Chen, L.Y., Inan, S., Eds.; Springer: Cham, Switzerland, 2020; Volume 271, pp. 525–546.
- Chavkin, C.; James, I.; Goldstein, A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science 1982, 215, 413–415.
- Lazenka, M.F. Antinociceptive Effects of Kappa-Opioid Receptor Agonists. Handb. Exp. Pharmacol. 2022, 271, 293–313.
- Beck, T.C.; Hapstack, M.A.; Beck, K.R.; Dix, T.A. Therapeutic Potential of Kappa Opioid Agonists. Pharmaceuticals 2019, 12, 95.
- Dalefield, M.L.; Scouller, B.; Bibi, R.; Kivell, B.M. The Kappa Opioid Receptor: A Promising Therapeutic Target for Multiple Pathologies. Front. Pharmacol. 2022, 13, 837671.
- Karkhanis, A.; Holleran, K.M.; Jones, S.R. Dynorphin/Kappa Opioid Receptor Signaling in Preclinical Models of Alcohol, Drug, and Food Addiction. Int. Rev. Neurobiol. 2017, 136, 53–88.
- Estave, P.M.; Spodnick, M.B.; Karkhanis, A.N. KOR Control over addiction processing: An exploration of the mesolimbic dopamine pathway. In The Kappa Opioid Receptor. Handbook of Experimental Pharmacology; Liu-Chen, L.Y., Inan, S., Eds.; Springer: Cham, Switzerland, 2020; Volume 271.
- Del Pilar Escobar, A.; Casanova, J.P.; Andrés, M.E.; Fuentealba, J.A. Crosstalk Between Kappa Opioid and Dopamine Systems in Compulsive Behaviors. Front. Pharmacol. 2020, 11, 57.
- Inui, S. Nalfurafine hydrochloride to treat pruritus: A review. Clin. Cosmet. Investig. Dermatol. 2015, 8, 249–255.
- Keating, G.M. Nalmefene: A review of its use in the treatment of alcohol dependence. CNS Drugs 2013, 27, 761–772.
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The effects of morphine- and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532.
- Schattauer, S.S.; Kuhar, J.R.; Song, A.; Chavkin, C. Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor. Cell. Signal. 2017, 32, 59–65.
- Dortch-Carnes, J.; Potter, D.E. Bremazocine: A kappa-opioid agonist with potent analgesic and other pharmacologic properties. CNS Drug Rev. 2005, 11, 195–212.
- Szmuszkovicz, J.; VonVoigtlander, P.F. Benzeneacetamide amines: Structurally novel non-m.mu. opioids. J. Med. Chem. 1982, 25, 1125–1126.
- Mangel, A.W.; Williams, V.S.L. Asimadoline in the treatment of irritable bowel syndrome. Expert Opin. Investig. Drugs 2010, 19, 1257–1264.
- Spetea, M.; Schmidhammer, H. Kappa Opioid Receptor Ligands and Pharmacology: Diphenethylamines, a Class of Structurally Distinct, Selective Kappa Opioid Ligands. Handb. Exp. Pharmacol. 2022, 271, 163–195.
- Faouzi, A.; Varga, B.R.; Majumdar, S. Biased Opioid Ligands. Molecules 2020, 25, 4257.
- Zhou, L.; Lovell, K.M.; Frankowski, K.J.; Slauson, S.R.; Phillips, A.M.; Streicher, J.M.; Stahl, E.; Schmid, C.L.; Hodder, P.; Madoux, F.; et al. Development of functionally selective, small molecule agonists at kappa opioid receptors. J. Biol. Chem. 2013, 288, 36703–36716.
- Brust, T.F.; Morgenweck, J.; Kim, S.A.; Rose, J.H.; Locke, J.L.; Schmid, C.L.; Zhou, L.; Stahl, E.L.; Cameron, M.D.; Scarry, S.M.; et al. Biased Agonists of the Kappa Opioid Receptor Suppress Pain and Itch without Causing Sedation or Dysphoria. Sci. Signal. 2016, 9, ra117.
- Nedelec, L.; Dumont, C.; Oberlander, C.; Frechet, D.; Laurent, J.; Boissier, J.R. Synthèse et étude de l’activité dopaminergique de dérivés de la di(phénéthyl)amine. Eur. J. Med. Chem. Chim. Ther. 1978, 13, 553–563.
- Schmidhammer, H.; Erli, F.; Guerrieri, E.; Spetea, M. Development of Diphenethylamines as Selective Kappa Opioid Receptor Ligands and Their Pharmacological Activities. Molecules 2020, 25, 5092.
- Spetea, M.; Berzetei-Gurske, I.P.; Guerrieri, E.; Schmidhammer, H. Discovery and pharmacological evaluation of a diphenethylamine derivative (HS665), a highly potent and selective κ opioid receptor agonist. J. Med. Chem. 2012, 55, 10302–10306.
- Spetea, M.; Eans, S.O.; Ganno, M.L.; Lantero, A.; Mairegger, M.; Toll, L.; Schmidhammer, H.; McLaughlin, J.P. Selective k receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice. Br. J. Pharmacol. 2017, 174, 2444–2456.
- Erli, F.; Guerrieri, E.; Haddou, B.T.; Lantero, A.; Mairegger, M.; Schmidhammer, H.; Spetea, M. Highly Potent and Selective New Diphenethylamines Interacting with the κ-Opioid Receptor: Synthesis, Pharmacology, and Structure-Activity Relationships. J. Med. Chem. 2017, 60, 7579–7590.
- Ortega, A.; Blount, J.F.; Manchand, P.S. Salvinorin, a new trans-neoclerodane diterpene from Salvia divinorum (Labiatae). J. Chem. Soc. Perkin Trans. 1 1982, 2505–2508.
- Valdes, L.J.; Butler, W.B.; Hatfield, G.M.; Paul, A.G.; Koreeda, M. Divinorin A, a Psychotropic Terpenoid, and Divinorin B from the Hallucinogenic Mexican Mint Salvia divinorum. J. Org. Chem. 1984, 49, 4716–4720.
- Valdes, L.J.; Chang, H.M.; Visger, D.C.; Koreeda, M. Salvinorin C, a New Neoclerodane Diterpene from a Bioactive Fraction of the Hallucinogenic Mexican Mint Salvia divinorum. Org. Lett. 2001, 3, 3935–3937.
- Roach, J.J.; Shenvi, R.A. A Review of Salvinorin Analogs and their Kappa-Opioid Receptor Activity. Bioorg. Med. Chem. Lett. 2018, 28, 1436–1445.
- Morani, A.S.; Ewald, A.; Prevatt-Smith, K.M.; Prisinzano, T.E.; Kivell, B.M. The 2-methoxy methyl analogue of salvinorin A attenuates cocaine-induced drug seeking and sucrose reinforcements in rats. Eur. J. Pharmacol. 2013, 720, 69–76.
- Kivell, B.M.; Ewald, A.W.M.; Prisinzano, T.E. Salvinorin A Analogs and Other Kappa-Opioid Receptor Compounds as Treatments for Cocaine Abuse. Adv. Pharmacol. 2014, 69, 481–511.
- Kivell, B.M.; Paton, K.F.; Kumar, N.; Morani, A.S.; Culverhouse, A.; Shepherd, A.; Welsh, S.A.; Biggerstaff, A.; Crowley, R.S.; Prisinzano, T.E. Kappa Opioid Receptor Agonist Mesyl Sal B Attenuates Behavioral Sensitization to Cocaine with Fewer Aversive Side-Effects than Salvinorin A in Rodents. Molecules 2018, 23, 2602.
- White, K.L.; Robinson, J.E.; Zhu, H.; DiBerto, J.F.; Polepally, P.R.; Zjawiony, J.K.; Nichols, D.E.; Malanga, C.J.; Roth, B.L. The G protein-biased κ-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J. Pharmacol. Exp. Ther. 2015, 352, 98–109.
- Bui, A.M.; Cavé, A.; Janot, M.M.; Parello, J.; Potier, P.; Scheidegger, U. Isolement et Analyse Structurale Du Collybolide, Nouveau Sesquiterpene Extrait de Collybia Maculata Alb. et Sch. Ex Fries (Basidiomycetes). Tetrahedron 1974, 30, 1327–1336.
- Shevick, S.L.; Freeman, S.M.; Tong, G.; Russo, R.J.; Bohn, L.M.; Shenvi, R.A. Asymmetric Syntheses of (+)- and (−)-Collybolide Enable Reevaluation of kappa-Opioid Receptor Agonism. ACS Cent. Sci. 2022, 8, 948–954.
- Aldrich, J.V.; McLaughlin, J.P. Peptide Kappa Opioid Receptor Ligands: Potential for Drug Development. AAPS J. 2009, 11, 312–322.
- Gentilucci, L. New Trends in the Development of Opioid Peptide Analogues as Advanced Remedies for Pain Relief. Curr. Top. Med. Chem. 2004, 4, 19–38.
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical Modifications Designed to Improve Peptide Stability: Incorporation of Non-Natural Amino Acids, Pseudo-Peptide Bonds, and Cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203.
- Chavkin, C. Dynorphin–Still an Extraordinarily Potent Opioid Peptide. Mol. Pharmacol. 2013, 83, 729–736.
- Schlechtingen, G.; DeHaven, R.N.; Daubert, J.D.; Cassel, J.A.; Chung, N.N.; Schiller, P.W.; Taulane, J.P.; Goodman, M. Structureactivity relationships of dynorphin A analogues modified in the address sequence. J. Med. Chem. 2003, 46, 2104–2109.
- Vanderah, T.W.; Schteingart, C.D.; Trojnar, J.; Junien, J.-L.; Lai, J.; Riviere, P.J.-M. FE200041 (D-Phe-D-Phe-D-Nle-D-Arg-NH2): A peripheral efficacious kappa opioid agonist with unprecedented selectivity. J. Pharmacol. Exp. Ther. 2004, 310, 326–333.
- Vanderah, T.W.; Largent-Milnes, T.; Lai, J.; Porreca, F.; Houghten, R.A.; Menzaghi, F.; Wisniewski, K.; Stalewski, J.; Sueiras-Diaz, J.; Galyean, R.; et al. Novel D-amino acid tetrapeptides produce potent antinociception by selectively acting at peripheral kappa-opioid receptors. Eur. J. Pharmacol. 2008, 583, 62–72.
- Wang, X.; Gou, X.; Yu, X.; Bai, D.; Tan, B.; Cao, P.; Qian, M.; Zheng, X.; Wang, H.; Tang, P.; et al. Antinociceptive and Antipruritic Effects of HSK21542, a Peripherally-Restricted Kappa Opioid Receptor Agonist, in Animal Models of Pain and Itch. Front. Pharmacol. 2021, 12, 773204.
- Beck, T.C.; Reichel, C.M.; Helke, K.L.; Bhadsavle, S.S.; Dix, T.A. Non-addictive orally-active kappa opioid agonists for the treatment of peripheral pain in rats. Eur. J. Pharmacol. 2019, 856, 172396.
- Fugal, J.; Serpa, S.M. Difelikefalin: A New κ-Opioid Receptor Agonist for the Treatment of Hemodialysis-Dependent Chronic Kidney Disease–Associated Pruritus. Ann. Pharmacother. 2022; Online First.
- Kutlu Yalcin, E.; Araujo-Duran, J.; Turan, A. Emerging drugs for the treatment of postsurgical pain. Expert Opin. Emerg. Drugs 2021, 26, 371–384.
- Schmidtko, A.; Loetsch, J.; Freynhagen, R.; Geisslinger, G. Ziconotide for treatment of severe chronic pain. Lancet 2010, 375, 1569–1577.
- Brust, A.; Croker, D.E.; Colless, B.; Ragnarsson, L.; Andersson, Å.; Jain, K.; Garcia-Caraballo, S.; Castro, J.; Brierley, S.M.; Alewood, P.F.; et al. Conopeptide-derived κ opioid agonists (Conorphins): Potent, selective, and metabolic stable dynorphin A mimetics with antinociceptive properties. J. Med. Chem. 2016, 59, 2381–2395.
- Muratspahić, E.; Tomašević, N.; Koehbach, J.; Duerrauer, L.; Hadžić, S.; Castro, J.; Schober, G.; Sideromenos, S.; Clark, R.J.; Brierley, S.M.; et al. Design of a Stable Cyclic Peptide Analgesic Derived from Sunflower Seeds that Targets the κ-Opioid Receptor for the Treatment of Chronic Abdominal Pain. J. Med. Chem. 2021, 64, 9042–9055.
- Zadina, J.E.; Hackler, L.; Ge, L.J.; Kastin, A.J. A potent and selective endogenous agonist for the mu-opiate receptor. Nature 1997, 386, 499–502.
- Harrison, C.; McNulty, S.; Smart, D.; Rowbotham, D.J.; Grandy, D.K.; Devi, L.A.; Lambert, D.G. The effects of endomorphin-1 and endomorphin-2 in CHO cells expressing recombinant mu-opioid receptors and SH-SY5Y cells. Br. J. Pharmacol. 1999, 128, 472–478.
- Fichna, J.; Janecka, A.; Costentin, J.; Do Rego, J.C. The endomorphin system and its evolving neurophysiological role. Pharmacol. Rev. 2007, 59, 88–123.
- De Marco, R.; Bedini, A.; Spampinato, S.; Comellini, L.; Zhao, J.; Artali, R.; Gentilucci, L. Constraining endomorphin-1 by beta,alpha-hybrid dipeptide/heterocycle scaffolds: Identification of a novel kappa-opioid receptor selective partial agonist. J. Med. Chem. 2018, 61, 5751–5757.
- Greco, A.; Tani, S.; De Marco, R.; Gentilucci, L. Synthesis and analysis of the conformational preferences of 5-aminomethyloxazolidine-2,4-dione scaffolds: First examples of β2- and β2,2-homo-Freidinger lactam analogues. Chem. Eur. J. 2014, 20, 13390–13404.
- Saito, T.; Hirai, H.; Kim, Y.J.; Kojima, Y.; Matsunaga, Y.; Nishida, H.; Sakakibara, T.; Suga, O.; Sujaku, T.; Kojima, N. CJ-15,208, a novel kappa opioid receptor antagonist from a fungus, Ctenomyces serratus ATCC15502. J. Antibiot. 2002, 55, 847–854.
- Dolle, R.E.; Michaut, M.; Martinez-Teipel, B.; Seida, P.R.; Ajello, C.W.; Muller, A.L.; DeHaven, R.N.; Carroll, P.J. Nascent structure-activity relationship study of a diastereomeric series of kappa opioid receptor antagonists derived from CJ-15,208. Bioorg. Med. Chem. Lett. 2009, 19, 3647–3650.
- Aldrich, J.V.; Kulkarni, S.S.; Senadheera, S.N.; Ross, N.C.; Reilley, K.J.; Eans, S.O.; Ganno, M.L.; Murray, T.F.; McLaughlin, J.P. Unexpected opioid activity profiles of analogues of the novel peptide kappa opioid receptor ligand CJ-15,208. ChemMedChem 2011, 6, 1739–1745.
- Ross, N.C.; Reilley, K.J.; Murray, T.F.; Aldrich, J.V.; McLaughlin, J.P. Novel opioid cyclic tetrapeptides: Trp isomers of CJ-15,208 exhibit distinct opioid receptor agonism and short-acting k opioid receptor antagonism. Br. J. Pharmacol. 2012, 165, 1097–1108.
- Aldrich, J.V.; Senadheera, S.N.; Ross, N.C.; Reilley, K.A.; Ganno, M.L.; Eans, S.E.; Murray, T.F.; McLaughlin, J.P. Alanine analogues of CJ-15,208: Novel opioid activity profiles and prevention of drug- and stress-induced reinstatement of cocaine seeking behavior. Br. J. Pharmacol. 2014, 171, 3212–3222.
- Aldrich, J.V.; Senadheera, S.N.; Ross, N.C.; Ganno, M.L.; Eans, S.O.; McLaughlin, J.P. The macrocyclic peptide natural product CJ-15,208 is orally active and prevents reinstatement of extinguished cocaine-seeking behavior. J. Nat. Prod. 2013, 76, 433–438.
- Eans, S.O.; Ganno, M.L.; Reilley, K.J.; Patkar, K.A.; Senadheera, S.N.; Aldrich, J.V.; McLaughlin, J.P. The macrocyclic tetrapeptide CJ-15,208 produces short-acting k opioid receptor antagonism in the CNS after oral administration. Br. J. Pharmacol. 2013, 169, 426–436.
- Brice-Tutt, A.C.; Senadheera, S.N.; Ganno, M.L.; Eans, S.O.; Khaliq, T.; Murray, T.F.; McLaughlin, J.P.; Aldrich, J.V. Phenylalanine Stereoisomers of CJ-15,208 and CJ-15,208 Exhibit Distinctly Different Opioid Activity Profiles. Molecules 2020, 25, 3999.
- Cardillo, G.; Gentilucci, L.; Tolomelli, A.; Spinosa, R.; Calienni, M.; Qasem, A.R.; Spampinato, S. Synthesis and evaluation of the affinity toward μ-opioid receptors of atypical, lipophilic ligands based on the sequence c. J. Med. Chem. 2004, 47, 5198–5203.
- Bedini, A.; Baiula, M.; Gentilucci, L.; Tolomelli, A.; De Marco, R.; Spampinato, S. Peripheral antinociceptive effects of the cyclic endomorphin-1 analog c in a mouse visceral pain model. Peptides 2010, 31, 2135–2140.
- Gentilucci, L.; Tolomelli, A.; De Marco, R.; Spampinato, S.; Bedini, A.; Artali, R. The inverse type II beta-turn on D-Trp-Phe, a pharmacophoric motif for MOR agonists. ChemMedChem 2011, 6, 1640–1653.
- De Marco, R.; Tolomelli, A.; Spampinato, S.; Bedini, A.; Gentilucci, L. Opioid activity profiles of oversimplified peptides lacking in the protonable N-terminus. J. Med. Chem. 2012, 55, 10292–10296.
- De Marco, R.; Bedini, A.; Spampinato, S.; Gentilucci, L. Synthesis of tripeptides containing D-Trp substituted at the indole ring, assessment of opioid receptor binding and in vivo central antinociception. J. Med. Chem. 2014, 57, 6861–6866.
- Gentilucci, L.; Squassabia, F.; Artali, R. Re-discussion of the importance of ionic interactions in stabilizing ligand-opioid receptor complex and in activating signal transduction. Curr. Drug Targets 2007, 8, 185–196.
- Gentilucci, L.; Tolomelli, A.; De Marco, R.; Artali, R. Molecular docking of opiates and opioid peptides, a tool for the design of selective agonists and antagonists, and for the investigation of atypical ligand-receptor interactions. Curr. Med. Chem. 2012, 19, 1587–1601.
- De Marco, R.; Bedini, A.; Spampinato, S.; Cavina, L.; Pirazzoli, E.; Gentilucci, L. Versatile Picklocks To Access All Opioid Receptors: Tuning the Selectivity and Functional Profile of the Cyclotetrapeptide c (CJ-15,208). J. Med. Chem. 2016, 59, 9255–9261.
- Bedini, A.; Di Cesare Mannelli, L.; Micheli, L.; Baiula, M.; Vaca, G.; De Marco, R.; Gentilucci, L.; Ghelardini, C.; Spampinato, S. Functional selectivity and antinociceptive effects of a novel KOR agonist. Front. Pharmacol. 2020, 11, 188.
- De Marco, R.; Gentilucci, L. Tryptophan-Containing Non-Cationizable Opioid Peptides—A new chemotype with unusual structure and in vivo activity. Future Med. Chem. 2017, 9, 2099–2115.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
20 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No