 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Frederique Tesson | -- | 3755 | 2023-02-16 16:19:45 | | | |
| 2 | Jessie Wu | Meta information modification | 3755 | 2023-02-17 02:16:30 | | | | |
| 3 | Jessie Wu | + 1 word(s) | 3756 | 2023-02-17 02:18:24 | | | | |
| 4 | Jessie Wu | Meta information modification | 3756 | 2023-02-17 02:20:03 | | |
Video Upload Options
The lamin A/C gene (LMNA) codes for nuclear intermediate filaments constitutive of the nuclear lamina. LMNA has 12 exons and alternative splicing of exon 10 results in two major isoforms of the A-type lamins - lamins A and C. Mutations found throughout LMNA cause a group of diseases collectively known as laminopathies, of which the type, diversity, penetrance, and severity of phenotypes can vary from one individual to the other, even between individuals carrying the same mutation. The majority of laminopathies affect the cardiac and/or skeletal muscles. The underlying molecular mechanisms contributing to such tissue-specific phenotypes caused by mutations in a ubiquitously expressed gene are not yet well elucidated.
1. Abnormal Nuclear Morphology, Aberrant Lamin A/C Phenotype and Lamin A/C Binding Proteins Mislocalization in Lamin A/C - Related Cardiac and Skeletal Muscle Disease
A rare lamin A/C null case in humans was discovered from a deceased newborn who was homozygous for the nonsense p.Y259X lamin A/C variant [1]. This mutation causes a complete absence of the A-type lamins in the homozygous state and half the amount of wild-type (WT) lamin A/C in the heterozygous state, resulting in limb-girdle muscular dystrophy (LGMD) type 1B [1] [2]. The homozygous child presented with severe under development and abnormalities including dystrophic muscles [1]. Skin fibroblasts from the heterozygous grandmother were comparable to WT controls while the LMNA null cells presented with misshapen nuclei, abnormal localization of nuclear proteins (e.g., B-type lamins, emerin, and syne1), and had impaired cell division capacity [2]. B-type lamins are intimately connected to A-type lamins, and appear to be involved in several cellular functions ranging from regulation of gene expression and mitotic spindle assembly to cellular senescence [3]. RNAi knockdown of LMNA in HeLa cells resulted in emerin mislocalization in the endoplasmic reticulum similar to the LMNA null cells phenotype, but it did not affect cell growth [2] [4]. To determine whether emerin was also mislocalized in the presence of other LMNA mutations, mutant lamins A and C were transfected in various cell lines including the P19 embryonic carcinoma stem cells, which lack endogenous lamin A/C [5] [6]. There was no consensus in the results, regardless of the position of the mutation along LMNA, with emerin either clearly mislocalized in the presence of some dilated cardiomyopathy (DCM) or Emery-Dreifuss muscular dystrophy (EDMD) LMNA variants or normally expressed and distributed [5] [6] [7]. Nonetheless, many LMNA mutations affecting the rod domain of the protein and causing muscular laminopathies also presented with lamin A/C aggregates, notably lamin C aggregates, in the nucleoplasm of transfected cells [6] [7] [8]. Emerin was found in these nuclear foci as well [7]. On the other hand, some amino acid substitutions affecting the globular end of the lamin A/C protein did not result in punctate lamin A structures, however, emerin remained mislocalized [7]. It has been suggested that aberrant emerin localization prevents its interaction with lamin A/C and BAF, which in turn blocks the emerin-mediated downregulation of the Signal Transducer and Activators of Transcription 3 (STAT3) signaling pathway [9]. Prolonged activation of STAT3 in muscles induces muscle wasting by activation of protein degradation pathways [9]. Analyses of fibroblasts from muscular laminopathy patients with mutations affecting the head or tail domains of the protein had overt nuclear structure aberrations [10]. Specifically, immunostaining for lamin A/C revealed defects including honeycomb staining, punctate structures, or blebbing [10]. Moreover, emerin and lamin B were also noted to be mislocalized [10]. Thus, demonstrating that lamin A/C mutations cause profound nuclear abnormalities and the mislocalization of a variety of proteins.
Transient transfection in HeLa cells of the p.E203G lamin A variant associated with DCM with conduction defects (DCM-CD), resulted in lamin aggregates and the absence of the lamin A variant in the nuclear periphery [11]. However, Ostlund and colleagues did not show this effect when they transiently transfected the same variant in C2C12 cells, a mouse skeletal myoblast cell line [12] [13]. While the difference between the observed lamin phenotypes might be attributed to the cell line used, the transient expression (lamin A only, lamin C only, or both lamins A and C to maintain the 1:1 stoichiometry of lamin A/C) of other variants associated with DCM (p.D192G, p.N195K), DCM associated with conduction disease (DCM-CD) (p.Y481X), or EDMD (p.R386K) in Cos7 (fibroblast-like cell line derived from monkey kidney tissue) and H9C2 (rat cardiomyoblast cell line) cells resulted in large and multiple lamin aggregates, which also fail to connect to the nuclear envelope unlike cells expressing WT lamin A/C or a familial partial lipodystrophy (FPLD) (p.R482W) mutant lamin A/C [14] [8]. An interesting exception is the DCM-causing lamin A/C variant p.L85R, which produced a WT lamin phenotype if mutant lamin A only was expressed but an aberrant phenotype (multiple speckles) when mutant lamin C only was expressed. Nevertheless, the abnormal mutant lamin C phenotype was rescued if co-expressed with the mutant lamin A, suggesting that mutations can affect the properties of lamins A and C differently [14]. Indeed, interactions between emerin and lamin A, and emerin and lamin C, are differently affected by LMNA mutations [15]. Moreover, fibroblasts from mice expressing only lamin C displayed normal emerin localization further illustrating that the structural abnormalities due to LMNA mutations can result in the inability of the A-type lamins to properly connect to the nuclear envelope and can negatively impact their interaction with their binding partners [16]. To better explore the effects of LMNA mutations on the interaction between A-type lamins and their binding partners, studies involving mutations found in regions known to interact with lamin A/C partners were performed.
2. Disruption of Lamin A/C Interaction with Binding Partners Can Result in Multiple Tissue Phenotypes
Mutations found throughout LMNA affect sites that are known to interact with various proteins. For instance, the C to T substitution at nucleotide position 1444 producing the p.R482W lamin A/C variant and associated with both FPLD and LGMD in some patients—is in a region known to bind actin and/or influence SREBF1 interaction [11] [17] [18] [19] [20]. Both p.R482W and the EDMD p.L530P variant, presented with decreased SREBF1 binding capacity [11]. This suggested that in some mutations causing striated muscle laminopathies, the binding capacity to a transcription factor involved in adipocyte metabolism can be affected and can result in lipodystrophic features in patients with muscular laminopathy [11]. This could also explain why the EDMD LmnaΔ8–11 mice model, which mainly exhibited striated muscle laminopathy, also notably lacked subcutaneous fat and presented with some abnormal metabolic parameters [21] [22].
Protein kinase C alpha (PKC-α) interacts with lamin A/C and with several lamin A/C partners [23]. One of the PKC-α targets is ERK 1/2, which is implicated in the development and progression of striated muscle laminopathies [24] [25]. PKC-α was found aberrantly localized in the nucleus of transfected cells expressing a DCM-causing LMNA mutation and in skeletal myoblasts from individuals harboring different LMNA-related congenital muscular dystrophy (L-CMD) mutations [8]. Moreover, ERK 1/2 activation was downregulated in myoblasts from L-CMD patients carrying the p.ΔK32 and p.R249W mutations and from DCM LmnaΔK32/ΔK32 and EDMD LmnaH222P/H222P mice models suggesting PKC-α involvement in striated muscle laminopathies [8].
Lamins also directly interact with specific transcriptionally repressed large genomic regions called Lamina Associated Domains (LADs) and are involved in the epigenetic regulation of chromatin [26]. The p.R482W and the EDMD p.R453W variants were shown to disrupt the appropriate formation of lamin A-associated heterochromatin domains [26]. Moreover, nucleoplasmic A-type lamins interact with the polycomb group (PcG) of proteins which are epigenetic repressors involved in muscle differentiation [27]. PcG targets genes were found to be upregulated in the muscle satellite cells of the EDMD LmnaΔ8–11 mice model leading to a lack of muscle satellite cells’ identity and senescence [28]. Furthermore, the interaction of lamin A/C with the histone acetyltransferase – PCAF is altered in HEK293 cells expressing LMNA EDMD mutations [[29]]. Taken together, these results suggest that aberrant epigenetic regulation induced by LMNA mutation contributes to the lamin-dependent tissue-specific human diseases and to the phenotypic variability observed in patients with laminopathy To further investigate the consequences of LMNA mutations on lamin A/C protein structure and the associated phenotypes, the lamin A/C protein structural stability and mobility were assessed.
3. Impaired Lamin A/C Structural Stability and Dynamics in Striated Muscle Laminopathies
To assess the effects of changes in lamin A/C structure caused by LMNA mutations, the structural stability and dynamics of various lamin A/C variants were studied. Gilchrist et al. transiently transfected epitope-tagged WT lamin A or lamin A variants in HT1080 cells, a human fibrosarcoma cell line [30]. The lamin A variants studied were the DCM-CD p.L85R and p.N195K variants, the EDMD p.L530P variant, and the p.R482W variant which is associated with FPLD with some individuals co-presenting LGMD [30]. While the transfected cells showed no significant or overt nuclear shape/size abnormalities, fluorescence recovery after photobleaching (FRAP) demonstrated that the DCM-CD and EDMD variants were more mobile and showed faster dynamics (especially the p.N195K variant) compared to WT [30]. In contrast, the predominantly lipodystrophy-causing variant p.R482W had comparable mobility to WT lamin A [30]. FRAP experiments on Cos7 cells also demonstrated increased mobility for the p.D192G (DCM-CD) and p.R386K (EDMD) lamin C only expressing variants compared to either WT lamin C or the p.L85R (DCM-CD) lamin C variant [14]. Fluorescence loss in photobleaching (FLIP) experiments further showed faster diffusion of lamin A p.N195K compared to WT [30]. Such increased mobility suggests less stability in the lamin A/C structures of the said variants [14] [30]. To determine if these unstable A-type lamins affect the mechanical properties of the nucleoskeleton and/or the cytoskeleton, studies on nuclear and cytoskeletal architecture were performed on WT and mutant LMNA-expressing cells.
4. Compromised Nuclear and Cytoskeletal Mechanics in the Presence of Lamin A/C Mutations Causing Muscular Laminopathies
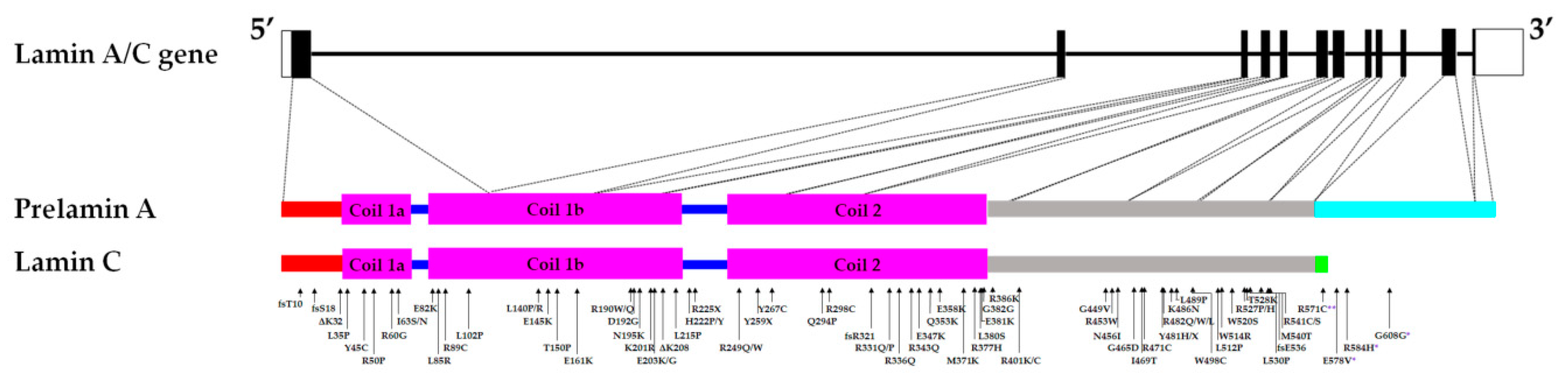
As a key component of the nuclear envelope architecture, lamin A/C has been suggested to be involved in nuclear mechanotransduction. Indeed, variations in lamin A/C expression level and spatial distribution in the nuclear envelope are associated with changes in nuclear rigidity - the nuclear stiffness increases as lamin A/C expression locally increases [31]. Conversely, nuclear envelop regions of the lowest lamin A/C expression are prone to deformation when subjected to osmotic stresses [31]. A cardiac sample from the p.D192G DCM patient presented with immense nuclear aberrations including nuclear envelope rupture, chromatin disorganization, and mitochondria mislocalization in the nucleoplasm in about 30% of the cardiomyocytes [32] [33]. Similarly, nuclear envelope breaks were observed in the DCM LmnaΔK32/+ mice model [34]. Furthermore, skin fibroblasts from DCM, EDMD, and LGMD patients carrying LMNA mutations displayed a loss of nuclear stiffness [5]. Mouse embryo fibroblasts (MEFs) and skeletal myotubes from the LmnaΔ8–11 mice also presented with reduced nuclear stiffness and a notable increase in nuclear deformity when strain was applied to the cells compared to WT [35] [36] [37]. Low- and medium-pressure micro-injection of dextran into the nucleus resulted in dextran leaking into the cytoplasm, suggesting compromised nuclear integrity of LmnaΔ8–11 mice fibroblasts [35]. Viral transfection in neonatal rat ventricular myocytes of different mutant LMNA cDNA (p.E161K, p.D192G, p.N195K variants) causing DCM or DCM-CD resulted in nuclear morphology defects including blebbing [38]. Analyses of nuclear stiffness showed that the mutant LMNA-expressing cells are stiffer, particularly the p.D192G variant, compared to the controls [38]. On the other hand, LMNA mutations that were not associated with striated muscle laminopathies did not result in nuclear deformability [5]. Nucleoplasmic localization and large aggregates of lamin A/C were also observed in transfected cells, suggesting impaired assembly of lamin A/C variants [5] [8] [14] [32] [38]. The mutations resulting in these lamin A/C protein variants are found in the rod domain of the A-type lamins, which is involved in dimerization and the formation of a higher-order lamin structure (Figure 1). Indeed, analysis of LMNA variants linked to the severe phenotype observed in LMNA-related L-CMD and the less severe phenotype associated with EDMD revealed that mutations that impair dimerization of lamin A/C led to more severe disease phenotype [39].
Although no overt differences were detected between the cytoskeletal architecture of WT and LmnaΔ8–11 MEFs, a significant decrease in cytoskeletal stiffness, cytoplasmic elasticity, and viscosity was observed in the LmnaΔ8–11 fibroblasts compared to WT cells, indicating that lamin A/C is involved in cytoskeleton plasticity [35] [36]. Even skin fibroblasts with EDMD mutations that did not result in nuclear deformability displayed defects in force transmission between the nucleus and the cytoskeleton [5]. Neonatal rat ventricular myocytes expressing mutant LMNA showed disorganized actin and lower levels of actin staining compared to WT and uninfected controls [38]. They also have impaired adhesion and increased plasticity, altered microtubule network organization, and present with mislocalization of Connexin 43, a plasma membrane hemichannel, suggesting a poorer capacity to handle and transmit mechanical stress due to actin defects [38] [40]. Accordingly, left ventricular LmnaΔ8–11 myocytes, as well as neonatal rat ventricular myocytes expressing the p.E161K, p.D192G, and p.N195K DCM mutations, displayed contractile dysfunction [40] [41]. Actin and the p38 MAPK, which was shown to be associated with LMNA-related DCM, are thought to interact, and the inhibition of p38 restored the mutant actin and mechanical phenotypes back to WT [38] [42] [43]. Defects of mechano-transduction in LMNA mutant cells have been further explored using L-CMD patients’ myoblasts cultured on matrix stiffness close to that of muscle (12 kPa) [44] [45]. Mutant cells displayed accumulation and disorganization of contractile actin stress fibres and increased traction forces, supporting the hypothesis that mutant cells are unable to sustain high external mechanical stretching because of impaired functional integrity of nuclear–cytoskeletal linkages [44] [45]. Furthermore, LMNA-mutated muscle cells showed aberrant activation of regulators of the mechano-response including yes-associated protein involved in cell adaptation to its microenvironment, cofilins that regulate actin-filament dynamics, and formins known to affect actin polymerisation and depolymerisation in a force-sensitive manner [44] [45] [46]. Interestingly, nuclear shape and chromatin organization can be improved in lamin A/C-depleted U2OS cells (derived from human osteosarcoma), in cells from patients with Hutchinson–Gilford progeria syndrome (premature aging laminopathy), or in aged vascular smooth muscle cells by treatment with remodelin [47] [48]. Remodelin is a small molecule that inhibits N-Acetyltransferase 10 (NAT10) activity to induce a microtubule reorganization [48]. These results highlight the significance of nuclear structural deformity in laminopathies and demonstrate that mutations in LMNA impair the mechanical capacity and integrity of both the nucleus and the whole cell by the disruption of the cytoskeleton, thus resulting in defects in mechano-transduction signalling. Experiments were then undertaken to determine if these structural changes also affect the post-translational modification status of lamin A/C.
5. Altered Lamin A/C Post-Translational Modification Status in Striated Muscle Laminopathy
As discussed in the previous paragraphs, many of the pathogenic LMNA mutations alter the lamin A/C protein structure as well as the nuclear envelope properties. In addition, studies have suggested that these changes also result in aberrant post-translational protein modifications. A-type lamins are known to be phosphorylated and the phosphorylation status may influence nuclear deformability [49]. Lamin A/C phosphorylation is involved in nuclear localization, assembly, mobility, and cytoplasmic transport of nuclear lamins during interphase [50]. The three main regions of lamin A that are phosphorylated during interphase are the N-terminal head, the proximal C-terminal region, and the lamin A specific far C-terminal region [50]. Mitsuhashi et al. noted abnormal Akt phosphorylation of S458 (located in the proximal C-terminal region) in muscle tissues and fibroblasts of patients with myopathies caused by LMNA mutations in the Ig fold in the protein’s C-terminal region [51]. Resolved structure of the lamin A/C C-terminus showed that in WT, S458 was partially buried; therefore, it was plausible that certain mutations in the Ig fold altered the protein structure and caused the exposure of S458, rendering it accessible for abnormal phosphorylation by Akt [51] [52]. Moreover, the EDMD variant p.R453W not only increased S458 phosphorylation but also decreased S390 phosphorylation compared to WT lamin A [53]. N-terminus lamin A phosphorylation was also found to be lower in myoblasts from EDMD and LGMD patients compared to the control and immunostaining for phosphorylated lamin A/C (phos-lamin A/C) in muscle fibres from these patients was markedly reduced [54]. Lower phos-lamin A/C was likewise observed in the regenerating muscle of a patient with EDMD compared to a sample from a patient with Duchenne Muscular Dystrophy, another type of muscular dystrophy [54]. Such reduction in phos-lamin A/C was only seen in myocytes but not in fibroblasts and inflammatory cells, suggesting a tissue-specific process involving kinases and lamin A/C in muscles [54]. Overall, these data showed how a mutation affecting a particular region of the protein may confer structural and chemical changes that result in a molecular hallmark common to the different types of LMNA-related myopathies.
Sumoylation is another post-translation modification observed on lamin A [55] [56] [57]. Small ubiquitin-like modifier (SUMO) addition is a post-translational modification that regulates several cellular processes, such as chromatin organization, transcription, nuclear transport, and DNA repair [58]. Fibroblasts from a p.E203K DCM patient and Cos 7 cells transfected with FPLD-causing mutations showed decreased lamin A sumoylation and increased cell death [55] [56]. Moreover, transient transfection of p.D192G lamin C only or both p.D192G lamins A and C in Cos7, C2C12, and H9C2 cells resulted in large lamin foci which co-localized with SUMO1 [32] [59]. Mislocalization of SUMO1 was also noted in the primary myoblasts and skeletal muscle tissue of the LmnaH222P/H222P mice [59], thus suggesting that lamin A sumoylation impairment is involved in the disease mechanism underlying striated muscle laminopathies.
These results from patient cells and immortalized cell lines demonstrate the wide breadth of the effects caused by lamin A/C mutations. Cellular phenotypic analyses can be further characterized using derived cells from patients’ induced pluripotent stem cells (iPSCs). These iPSCs can be of immense benefit, particularly in cases when multiple tissue phenotypes are co-present, as there will be no need to collect multiple cell types from the patients.
6. Myogenic or Cardiac Cells from Patients’ Induced Pluripotent Stem Cells Provide a Promising Model to Identify and Characterize Subtle Morphological Defects in Mutant Cells
In primary culture, dermal fibroblasts from a DCM-CD patient carrying p.R225X lamin A/C presented with decreased lamin A/C and profound nuclear defects including condensed heterochromatin clumps, nuclear blebs, nuclear pore complexes (NPCs) clustering, and mitochondria around the nuclear envelope [60]. Electrical induction of these fibroblasts resulted in a significant increase in senescence and apoptosis in the mutant cells, the inhibition of the ERK 1/2 branch of the MAPK pathway ameliorating this phenotype [60]. Surprisingly, cardiomyocytes derived from the iPSCs obtained from the fibroblasts of the p.R225X patient and of other DCM patients with LMNA mutations were morphologically comparable to WT [60] [61] [62] [63] [64]. This discrepant result might be due to the limited amounts of A-type lamins in human embryonic and induced pluripotent stem cells as lamin A/C expression only becomes detectable upon differentiation [65] [66]. However, it was recently shown that when DCM-associated mutations were introduced into human iPSCs, iPSC-derived cardiomyocytes, in contrast to hepatocytes or adipocytes, exhibit aberrant nuclear morphology and disruptions in lamina-chromatin interactions similar to the defects seen in the patient’s fibroblasts [67]. Upon electrical stimulation, mutant cardiomyocytes presented with marked electrophysiological and contractile alterations, nuclear senescence, and/or an increase in apoptosis [60] [64]. The electrical susceptibility of these cardiomyocytes seems to be caused by lamin A/C haploinsufficiency as electrical stimulation of LMNA knocked-down control cells resulted in the same phenotype as the mutant cells [60] [64]. Defects in the sarcomere organization and abnormal activation of ERK1/2 signaling were reported in iPSC-derived cardiomyocytes carrying the p.R190W lamin A/C variant [68]. After hypoxia, sarcomere damage and increased sensitivity to cellular stress with bradyarrhythmia and more frequent arrhythmias in mutant iPSC-derived cardiomyocytes on β-adrenergic stimulation were also observed [62].
A few studies investigated the role of epigenetic regulation mechanisms in cardiac-relevant cells. It has been shown that cardiomyocytes derived from the iPSCs obtained from human cells carrying a DCM variant displayed conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy suggesting a new mechanism underlying the conduction abnormalities associated with LMNA-cardiomyopathy [69].
A major application of iPSC technology is drug testing [70]. iPSC-derived cardiomyocytes carrying the DCM-CD p.R225X variant treated with PTC124 (ataluren), a compound that acts by promoting read-through of premature stop codons, showed reduced morphological defects at the nuclear level and improvement of the functional performance [71]. Lysine-specific demethylase 1 (LSD1) is another promising compound that targets methylated histones H3K4 and H3K9 as well as non-histone substrates to regulate gene expression. LSD1 appeared to be a therapeutic target for lamin A/C-associated DCM in the LmnaH222P mouse model [72].
Finally, the relevance of 3D culturing was highlighted in a study using derived myogenic cells from the iPSCs of patients with various striated muscle laminopathies (p.K32del, p.L35P, and p.R249W) that presented with deformed nuclei and aberrant localization of emerin (localized in one pole of the cells) and lamin B1 [73]. As expected, lamins A and C also formed aggregates or displayed a honeycomb pattern [73]. Although an abnormal nuclear shape (observed in p.L35P and p.R249W cells) and mislocalized lamin A/C (observed in p.R249W cells) were noted in the myotubes (differentiated myoblasts), the phenotype tended to be less pronounced in the myotubes than in the myoblasts [73]. However, 3D culture of these mutant myotubes, which better simulated the cellular environment in patients, enhanced the phenotypes observed compared to what was seen in the monolayer-cultured myotubes [73].
These data, therefore, highlight the immense potential of iPSCs to model striated laminopathies and the promising use of 3D culturing as a means to detect, identify and more accurately characterize cellular changes that might not be readily observable in 2D culturing. Altogether, the use of cellular models of striated laminopathy may help to better understand the cellular consequences of lamin A/C variants and open up new avenues for laminopathies’ treatment.
References
- Engelen, B.G.M. van; Muchir, A.; Hutchison, C.J.; Kooi, A.J. van der; Bonne, G.; Lammens, M. The Lethal Phenotype of a Homozygous Nonsense Mutation in the Lamin A/C Gene. Neurology 2005, 64, 374–376, 10.1212/01.WNL.0000149763.15180.00.
- Muchir, A.; van Engelen, B.G.; Lammens, M.; Mislow, J.M.; McNally, E.; Schwartz, K.; Bonne, G. Nuclear Envelope Alterations in Fibroblasts from LGMD1B Patients Carrying Nonsense Y259X Heterozygous or Homozygous Mutation in Lamin A/C Gene. Experimental Cell Research 2003, 291, 352–362, 10.1016/j.yexcr.2003.07.002.
- Hutchison, C.J. B-Type Lamins in Health and Disease. Seminars in Cell & Developmental Biology 2014, 29, 158–163, 10.1016/j.semcdb.2013.12.012.
- Harborth, J.; Elbashir, S.M.; Bechert, K.; Tuschl, T.; Weber, K. Identification of Essential Genes in Cultured Mammalian Cells Using Small Interfering RNAs. Journal of Cell Science 2001, 114, 4557–4565, 10.1242/jcs.114.24.4557.
- Zwerger, M.; Jaalouk, D.E.; Lombardi, M.L.; Isermann, P.; Mauermann, M.; Dialynas, G.; Herrmann, H.; Wallrath, L.L.; Lammerding, J. Myopathic Lamin Mutations Impair Nuclear Stability in Cells and Tissue and Disrupt Nucleo-Cytoskeletal Coupling. Human Molecular Genetics 2013, 22, 2335–2349, 10.1093/hmg/ddt079.
- Raharjo, W.H.; Enarson, P.; Sullivan, T.; Stewart, C.L.; Burke, B. Nuclear Envelope Defects Associated with LMNA Mutations Cause Dilated Cardiomyopathy and Emery-Dreifuss Muscular Dystrophy. Journal of Cell Science 2001, 114, 4447–4457, 10.1242/jcs.114.24.4447.
- Holt, I.; Östlund, C.; Stewart, C.L.; Man, N. thi; Worman, H.J.; Morris, G.E. Effect of Pathogenic Mis-Sense Mutations in Lamin A on Its Interaction with Emerin in Vivo. Journal of Cell Science 2003, 116, 3027–3035, 10.1242/jcs.00599.
- Nicolas, H.A.; Bertrand, A.T.; Labib, S.; Mohamed-Uvaize, M.; Bolongo, P.M.; Wu, W.Y.; Bilińska, Z.T.; Bonne, G.; Akimenko, M.-A.; Tesson, F..; et al. Protein Kinase C Alpha Cellular Distribution, Activity, and Proximity with Lamin A/C in Striated Muscle Laminopathies. Cells 2020, 9, 2388, 10.3390/cells9112388.
- Lee, B.; Lee, S.; Lee, Y.; Park, Y.; Shim, J. Emerin Represses STAT3 Signaling through Nuclear Membrane-Based Spatial Control. International Journal of Molecular Sciences 2021, 22, 6669, 10.3390/ijms22136669.
- Muchir, A.; Medioni, J.; Laluc, M.; Massart, C.; Arimura, T.; Kooi, A.J.V.D.; Desguerre, I.; Mayer, M.; Ferrer, X.; Briault, S.; et al.et al. Nuclear Envelope Alterations in Fibroblasts from Patients with Muscular Dystrophy, Cardiomyopathy, and Partial Lipodystrophy Carrying Lamin A/C Gene Mutations. Muscle & Nerve 2004, 30, 444–450, 10.1002/mus.20122.
- Lloyd, D.J.; Trembath, R.C.; Shackleton, S. A Novel Interaction between Lamin A and SREBP1: Implications for Partial Lipodystrophy and Other Laminopathies. Human Molecular Genetics 2002, 11, 769–777, 10.1093/hmg/11.7.769.
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J.; Spudich, S.; De Girolami, U.; et al.et al. Missense Mutations in the Rod Domain of the Lamin A/C Gene as Causes of Dilated Cardiomyopathy and Conduction-System Disease. New England Journal of Medicine 1999, 341, 1715–1724, 10.1056/NEJM199912023412302.
- Östlund, C.; Bonne, G.; Schwartz, K.; Worman, H.J. Properties of Lamin A Mutants Found in Emery-Dreifuss Muscular Dystrophy, Cardiomyopathy and Dunnigan-Type Partial Lipodystrophy. Journal of Cell Science 2001, 114, 4435–4445, 10.1242/jcs.114.24.4435.
- Sylvius, N.; Hathaway, A.; Boudreau, E.; Gupta, P.; Labib, S.; Bolongo, P.M.; Rippstein, P.; McBride, H.; Bilinska, Z.T.; Tesson, F.; et al. Specific Contribution of Lamin A and Lamin C in the Development of Laminopathies. Experimental Cell Research 2008, 314, 2362–2375, 10.1016/j.yexcr.2008.04.017.
- Motsch, I.; Kaluarachchi, M.; Emerson, L.J.; Brown, C.A.; Brown, S.C.; Dabauvalle, M.-C.; Ellis, J.A. Lamins A and C Are Differentially Dysfunctional in Autosomal Dominant Emery-Dreifuss Muscular Dystrophy. European Journal of Cell Biology 2005, 84, 765–781, 10.1016/j.ejcb.2005.04.004.
- Fong, L.G.; Ng, J.K.; Lammerding, J.; Vickers, T.A.; Meta, M.; Coté, N.; Gavino, B.; Qiao, X.; Chang, S.Y.; Young, S.R.; et al.et al. Prelamin A and Lamin A Appear to Be Dispensable in the Nuclear Lamina. J Clin Invest 2006, 116, 743–752, 10.1172/JCI27125.
- Vigouroux, C.; Magré, J.; Vantyghem, M.C.; Bourut, C.; Lascols, O.; Shackleton, S.; Lloyd, D.J.; Guerci, B.; Padova, G.; Valensi, P.; et al.et al. Lamin A/C Gene: Sex-Determined Expression of Mutations in Dunnigan-Type Familial Partial Lipodystrophy and Absence of Coding Mutations in Congenital and Acquired Generalized Lipoatrophy. Diabetes 2000, 49, 1958–1962, 10.2337/diabetes.49.11.1958.
- Simon, D.N.; Zastrow, M.S.; Wilson, K.L. Direct Actin Binding to A- and B-Type Lamin Tails and Actin Filament Bundling by the Lamin A Tail. Nucleus 2010, 1, 264–272, 10.4161/nucl.11799.
- Buendia, B. LMNA p.R482W Mutation Related to FPLD2 Alters SREBP1-A Type Lamin Interactions in Human Fibroblasts and Adipose Stem Cells. Orphanet Journal of Rare Diseases 2015, 10, O13, 10.1186/1750-1172-10-S2-O13.
- Vadrot, N.; Duband-Goulet, I.; Cabet, E.; Attanda, W.; Barateau, A.; Vicart, P.; Gerbal, F.; Briand, N.; Vigouroux, C.; Oldenburg, A.R.; et al.et al. The p.R482W Substitution in A-Type Lamins Deregulates SREBP1 Activity in Dunnigan-Type Familial Partial Lipodystrophy. Human Molecular Genetics 2015, 24, 2096–2109, 10.1093/hmg/ddu728.
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-Type Lamin Expression Compromises Nuclear Envelope Integrity Leading to Muscular Dystrophy. Journal of Cell Biology 1999, 147, 913–920, 10.1083/jcb.147.5.913.
- Ruan, J.; Liu, X.G.; Zheng, H.L.; Li, J.B.; Xiong, X.D.; Zhang, C.L.; Luo, C.Y.; Zhou, Z.J.; Shi, Q.; Weng, Y.G.; et al. Deletion of the Lmna Gene Induces Growth Delay and Serum Biochemical Changes in C57BL/6 Mice. Asian-Australas J Anim Sci 2014, 27, 123–130, 10.5713/ajas.2013.13246.
- Martelli, A.M.; Bortul, R.; Tabellini, G.; Faenza, I.; Cappellini, A.; Bareggi, R.; Manzoli, L.; Cocco, L. Molecular characterization of protein kinase C-alpha binding to lamin A. J Cell Biochem 2002, 86, 320–330, 10.1002/jcb.10227.
- Mauro, A.; Ciccarelli, C.; Cesaris, P.D.; Scoglio, A.; Bouche, M.; Molinaro, M.; Aquino, A.; Zani, B.M. PKCalpha-mediated ERK, JNK and p38 activation regulates the myogenic program in human rhabdomyosarcoma cells. J Cell Sci 2002, 115, 3587–3599, 10.1242/jcs.00037.
- Muchir, A.; Kim, Y.J.; Reilly, S.A.; Wu, W.; Choi, J.C.; Worman, H.J. Inhibition of extracellular signal-regulated kinase 1/2 signaling has beneficial effects on skeletal muscle in a mouse model of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutation. Skelet Muscle 2013, 3, 17, 10.1186/2044-5040-3-17.
- Bianchi, A.; Manti, P.G.; Lucini, F.; Lanzuolo, C. Mechanotransduction, Nuclear Architecture and Epigenetics in Emery Dreifuss Muscular Dystrophy: Tous Pour Un, Un Pour Tous. Nucleus 2018, 9, 276–290, 10.1080/19491034.2018.1460044.
- Marullo, F.; Cesarini, E.; Antonelli, L.; Gregoretti, F.; Oliva, G.; Lanzuolo, C. Nucleoplasmic Lamin A/C and Polycomb Group of Proteins: An Evolutionarily Conserved Interplay. Nucleus 2016, 7, 103–111, 10.1080/19491034.2016.1157675.
- Bianchi, A.; Mozzetta, C.; Pegoli, G.; Lucini, F.; Valsoni, S.; Rosti, V.; Petrini, C.; Cortesi, A.; Gregoretti, F.; Antonelli, L.; et al.et al. Dysfunctional Polycomb Transcriptional Repression Contributes to Lamin A/C–Dependent Muscular Dystrophy. J Clin Invest 2020, 130, 2408–2421, 10.1172/JCI128161.
- Santi, S.; Cenni, V.; Capanni, C.; Lattanzi, G.; Mattioli, E. PCAF Involvement in Lamin A/C-HDAC2 Interplay during the Early Phase of Muscle Differentiation. Cells 2020, 9, 1735, 10.3390/cells9071735.
- Gilchrist, S.; Gilbert, N.; Perry, P.; Östlund, C.; Worman, H.J.; Bickmore, W.A. Altered Protein Dynamics of Disease-Associated Lamin A Mutants. BMC Cell Biology 2004, 5, 46, 10.1186/1471-2121-5-46.
- Srivastava, L.K.; Ju, Z.; Ghagre, A.; Ehrlicher, A.J. Spatial Distribution of Lamin A/C Determines Nuclear Stiffness and Stress-Mediated Deformation. Journal of Cell Science 2021, 134, jcs248559, 10.1242/jcs.248559.
- Sylvius, N.; Bilinska, Z.T.; Veinot, J.P.; Fidzianska, A.; Bolongo, P.M.; Poon, S.; McKeown, P.; Davies, R.A.; Chan, K.-L.; Tang, A.S.L.; et al.et al. In Vivo and in Vitro Examination of the Functional Significances of Novel Lamin Gene Mutations in Heart Failure Patients. Journal of Medical Genetics 2005, 42, 639–647, 10.1136/jmg.2004.023283.
- Gupta, P.; Bilinska, Z.T.; Sylvius, N.; Boudreau, E.; Veinot, J.P.; Labib, S.; Bolongo, P.M.; Hamza, A.; Jackson, T.; Ploski, R.; et al.et al. Genetic and Ultrastructural Studies in Dilated Cardiomyopathy Patients: A Large Deletion in the Lamin A/C Gene Is Associated with Cardiomyocyte Nuclear Envelope Disruption. Basic Res Cardiol 2010, 105, 365–377, 10.1007/s00395-010-0085-4.
- Cattin, M.-E.; Bertrand, A.T.; Schlossarek, S.; Le Bihan, M.-C.; Skov Jensen, S.; Neuber, C.; Crocini, C.; Maron, S.; Lainé, J.; Mougenot, N.; et al.et al. Heterozygous LmnadelK32 Mice Develop Dilated Cardiomyopathy through a Combined Pathomechanism of Haploinsufficiency and Peptide Toxicity. Human Molecular Genetics 2013, 22, 3152–3164, 10.1093/hmg/ddt172.
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T. Lamin A/C Deficiency Causes Defective Nuclear Mechanics and Mechanotransduction. J Clin Invest 2004, 113, 370–378, 10.1172/JCI19670.
- Lee, J.S.H.; Hale, C.M.; Panorchan, P.; Khatau, S.B.; George, J.P.; Tseng, Y.; Stewart, C.L.; Hodzic, D.; Wirtz, D. Nuclear Lamin A/C Deficiency Induces Defects in Cell Mechanics, Polarization, and Migration. Biophysical Journal 2007, 93, 2542–2552, 10.1529/biophysj.106.102426.
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins A and C but Not Lamin B1 Regulate Nuclear Mechanics. Journal of Biological Chemistry 2006, 281, 25768–25780, 10.1074/jbc.M513511200.
- Laurini, E.; Martinelli, V.; Lanzicher, T.; Puzzi, L.; Borin, D.; Chen, S.N.; Long, C.S.; Lee, P.; Mestroni, L.; Taylor, M.R.G.; et al.et al. Biomechanical Defects and Rescue of Cardiomyocytes Expressing Pathologic Nuclear Lamins. Cardiovascular Research 2018, 114, 846–857, 10.1093/cvr/cvy040.
- Bertrand, A.T.; Brull, A.; Azibani, F.; Benarroch, L.; Chikhaoui, K.; Stewart, C.L.; Medalia, O.; Ben Yaou, R.; Bonne, G. Lamin A/C Assembly Defects in LMNA-Congenital Muscular Dystrophy Is Responsible for the Increased Severity of the Disease Compared with Emery–Dreifuss Muscular Dystrophy. Cells 2020, 9, 844, 10.3390/cells9040844.
- Borin, D.; Peña, B.; Chen, S.N.; Long, C.S.; Taylor, M.R.G.; Mestroni, L.; Sbaizero, O. Altered Microtubule Structure, Hemichannel Localization and Beating Activity in Cardiomyocytes Expressing Pathologic Nuclear Lamin A/C. Heliyon 2020, 6, e03175, 10.1016/j.heliyon.2020.e03175.
- Nikolova, V.; Leimena, C.; McMahon, A.C.; Tan, J.C.; Chandar, S.; Jogia, D.; Kesteven, S.H.; Michalicek, J.; Otway, R.; Verheyen, F.; et al.et al. Defects in Nuclear Structure and Function Promote Dilated Cardiomyopathy in Lamin A/C–Deficient Mice. J Clin Invest 2004, 113, 357–369, 10.1172/JCI19448.
- Muchir, A.; Wu, W.; Choi, J.C.; Iwata, S.; Morrow, J.; Homma, S.; Worman, H.J. Abnormal P38α Mitogen-Activated Protein Kinase Signaling in Dilated Cardiomyopathy Caused by Lamin A/C Gene Mutation. Human Molecular Genetics 2012, 21, 4325–4333, 10.1093/hmg/dds265.
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiology and Molecular Biology Reviews 2011, 75, 50–83, 10.1128/MMBR.00031-10.
- Schwartz, C.; Fischer, M.; Mamchaoui, K.; Bigot, A.; Lok, T.; Verdier, C.; Duperray, A.; Michel, R.; Holt, I.; Voit, T.; et al.et al. Lamins and Nesprin-1 Mediate inside-out Mechanical Coupling in Muscle Cell Precursors through FHOD1. Sci Rep 2017, 7, 1253, 10.1038/s41598-017-01324-z.
- Bertrand, A.T.; Ziaei, S.; Ehret, C.; Duchemin, H.; Mamchaoui, K.; Bigot, A.; Mayer, M.; Quijano-Roy, S.; Desguerre, I.; Lainé, J.; et al.et al. Cellular Microenvironments Reveal Defective Mechanosensing Responses and Elevated YAP Signaling in LMNA-Mutated Muscle Precursors. Journal of Cell Science 2014, 127, 2873–2884, 10.1242/jcs.144907.
- Vignier, N.; Chatzifrangkeskou, M.; Pinton, L.; Wioland, H.; Marais, T.; Lemaitre, M.; Le Dour, C.; Peccate, C.; Cardoso, D.; Schmitt, A.; et al.et al. The Non-Muscle ADF/Cofilin-1 Controls Sarcomeric Actin Filament Integrity and Force Production in Striated Muscle Laminopathies. Cell Reports 2021, 36, 109601, 10.1016/j.celrep.2021.109601.
- Cobb, A.M.; Larrieu, D.; Warren, D.T.; Liu, Y.; Srivastava, S.; Smith, A.J.O.; Bowater, R.P.; Jackson, S.P.; Shanahan, C.M. Prelamin A Impairs 53BP1 Nuclear Entry by Mislocalizing NUP153 and Disrupting the Ran Gradient. Aging Cell 2016, 15, 1039–1050, 10.1111/acel.12506.
- Larrieu, D.; Britton, S.; Demir, M.; Rodriguez, R.; Jackson, S.P. Chemical Inhibition of NAT10 Corrects Defects of Laminopathic Cells. Science 2014, 344, 527–532, 10.1126/science.1252651.
- Davidson, P.M.; Lammerding, J. Broken Nuclei – Lamins, Nuclear Mechanics, and Disease. Trends in Cell Biology 2014, 24, 247–256, 10.1016/j.tcb.2013.11.004.
- Kochin, V.; Shimi, T.; Torvaldson, E.; Adam, S.A.; Goldman, A.; Pack, C.-G.; Melo-Cardenas, J.; Imanishi, S.Y.; Goldman, R.D.; Eriksson, J.E.; et al. Interphase Phosphorylation of Lamin A. Journal of Cell Science 2014, 127, 2683–2696, 10.1242/jcs.141820.
- Mitsuhashi, H.; Hayashi, Y.K.; Matsuda, C.; Noguchi, S.; Wakatsuki, S.; Araki, T.; Nishino, I. Specific Phosphorylation of Ser458 of A-Type Lamins in LMNA-Associated Myopathy Patients. Journal of Cell Science 2010, 123, 3893–3900, 10.1242/jcs.072157.
- Krimm, I.; Östlund, C.; Gilquin, B.; Couprie, J.; Hossenlopp, P.; Mornon, J.-P.; Bonne, G.; Courvalin, J.-C.; Worman, H.J.; Zinn-Justin, S.; et al. The Ig-like Structure of the C-Terminal Domain of Lamin A/C, Mutated in Muscular Dystrophies, Cardiomyopathy, and Partial Lipodystrophy. Structure 2002, 10, 811–823, 10.1016/S0969-2126(02)00777-3.
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.D.P.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.-W.; Tewari, M.; et al.et al. Nuclear Lamin-A Scales with Tissue Stiffness and Enhances Matrix-Directed Differentiation. Science 2013, 341, 1240104, 10.1126/science.1240104.
- Cenni, V.; Sabatelli, P.; Mattioli, E.; Marmiroli, S.; Capanni, C.; Ognibene, A.; Squarzoni, S.; Maraldi, N.M.; Bonne, G.; Columbaro, M.; et al.et al. Lamin A N-Terminal Phosphorylation Is Associated with Myoblast Activation: Impairment in Emery–Dreifuss Muscular Dystrophy. Journal of Medical Genetics 2005, 42, 214–220, 10.1136/jmg.2004.026112.
- Zhang, Y.-Q.; Sarge, K.D. Sumoylation Regulates Lamin A Function and Is Lost in Lamin A Mutants Associated with Familial Cardiomyopathies. Journal of Cell Biology 2008, 182, 35–39, 10.1083/jcb.200712124.
- Simon, D.N.; Domaradzki, T.; Hofmann, W.A.; Wilson, K.L. Lamin A Tail Modification by SUMO1 Is Disrupted by Familial Partial Lipodystrophy–Causing Mutations. Mol Biol Cell 2013, 24, 342–350, 10.1091/mbc.E12-07-0527.
- Sharma, P.; Kuehn, M.R. SENP1-Modulated Sumoylation Regulates Retinoblastoma Protein (RB) and Lamin A/C Interaction and Stabilization. Oncogene 2016, 35, 6429–6438, 10.1038/onc.2016.177.
- Gill, G. SUMO and Ubiquitin in the Nucleus: Different Functions, Similar Mechanisms?. Genes Dev 2004, 18, 2046–2059, 10.1101/gad.1214604.
- Boudreau, É.; Labib, S.; Bertrand, A.T.; Decostre, V.; Bolongo, P.M.; Sylvius, N.; Bonne, G.; Tesson, F. Lamin A/C Mutants Disturb Sumo1 Localization and Sumoylation in Vitro and in Vivo. PLOS ONE 2012, 7, e45918, 10.1371/journal.pone.0045918.
- Siu, C.-W.; Lee, Y.-K.; Ho, J.C.-Y.; Lai, W.-H.; Chan, Y.-C.; Ng, K.-M.; Wong, L.-Y.; Au, K.-W.; Lau, Y.-M.; Zhang, J.; et al.et al. Modeling of Lamin A/C Mutation Premature Cardiac Aging Using Patient-Specific Induced Pluripotent Stem Cells. Aging 2012, 4, 803–822, 10.18632/aging.100503.
- Ho, J.C.; Zhou, T.; Lai, W.-H.; Huang, Y.; Chan, Y.-C.; Li, X.; Wong, N.L.; Li, Y.; Au, K.-W.; Guo, D.; et al.et al. Generation of Induced Pluripotent Stem Cell Lines from 3 Distinct Laminopathies Bearing Heterogeneous Mutations in Lamin A/C. Aging 2011, 3, 380–390, 10.18632/aging.100277.
- Shah, D.; Virtanen, L.; Prajapati, C.; Kiamehr, M.; Gullmets, J.; West, G.; Kreutzer, J.; Pekkanen-Mattila, M.; Heliö, T.; Kallio, P.; et al.et al. Modeling of LMNA-Related Dilated Cardiomyopathy Using Human Induced Pluripotent Stem Cells. Cells 2019, 8, 594, 10.3390/cells8060594.
- Mehrabi, M.; Morris, T.A.; Cang, Z.; Nguyen, C.H.H.; Sha, Y.; Asad, M.N.; Khachikyan, N.; Greene, T.L.; Becker, D.M.; Nie, Q.; et al.et al. A Study of Gene Expression, Structure, and Contractility of IPSC-Derived Cardiac Myocytes from a Family with Heart Disease Due to LMNA Mutation. Ann Biomed Eng 2021, 49, 3524–3539, 10.1007/s10439-021-02850-8.
- Yang, J.; Argenziano, M.A.; Burgos Angulo, M.; Bertalovitz, A.; Beidokhti, M.N.; McDonald, T.V. Phenotypic Variability in IPSC-Induced Cardiomyocytes and Cardiac Fibroblasts Carrying Diverse LMNA Mutations. Frontiers in Physiology 2021, 12, 778982, 10.3389/fphys.2021.778982.
- Rober, R.A.; Weber, K.; Osborn, M. Differential Timing of Nuclear Lamin A/C Expression in the Various Organs of the Mouse Embryo and the Young Animal: A Developmental Study. Development 1989, 105, 365–378, 10.1242/dev.105.2.365.
- Constantinescu, D.; Gray, H.L.; Sammak, P.J.; Schatten, G.P.; Csoka, A.B. Lamin A/C Expression Is a Marker of Mouse and Human Embryonic Stem Cell Differentiation. Stem Cells 2006, 24, 177–185, 10.1634/stemcells.2004-0159.
- Shah, P.P.; Lv, W.; Rhoades, J.H.; Poleshko, A.; Abbey, D.; Caporizzo, M.A.; Linares-Saldana, R.; Heffler, J.G.; Sayed, N.; Thomas, D.; et al.et al. Pathogenic LMNA Variants Disrupt Cardiac Lamina-Chromatin Interactions and de-Repress Alternative Fate Genes. Cell Stem Cell 2021, 28, 938-954, 10.1016/j.stem.2020.12.016.
- Chatzifrangkeskou, M.; Yadin, D.; Marais, T.; Chardonnet, S.; Cohen-Tannoudji, M.; Mougenot, N.; Schmitt, A.; Crasto, S.; Di Pasquale, E.; Macquart, C.; et al.et al. Cofilin-1 Phosphorylation Catalyzed by ERK1/2 Alters Cardiac Actin Dynamics in Dilated Cardiomyopathy Caused by Lamin A/C Gene Mutation. Human Molecular Genetics 2018, 27, 3060–3078, 10.1093/hmg/ddy215.
- Salvarani, N.; Crasto, S.; Miragoli, M.; Bertero, A.; Paulis, M.; Kunderfranco, P.; Serio, S.; Forni, A.; Lucarelli, C.; Dal Ferro, M.; et al.et al. The K219T-Lamin Mutation Induces Conduction Defects through Epigenetic Inhibition of SCN5A in Human Cardiac Laminopathy. Nat Commun 2019, 10, 2267, 10.1038/s41467-019-09929-w.
- Takahashi, K.; Yamanaka, S. A Decade of Transcription Factor-Mediated Reprogramming to Pluripotency. Nat Rev Mol Cell Biol 2016, 17, 183–193, 10.1038/nrm.2016.8.
- Lee, Y.; Lau, Y.; Cai, Z.; Lai, W.; Wong, L.; Tse, H.; Ng, K.; Siu, C. Modeling Treatment Response for Lamin A/C Related Dilated Cardiomyopathy in Human Induced Pluripotent Stem Cells. J Am Heart Assoc 2017 , 6, e005677, 10.1161/JAHA.117.005677.
- Guénantin, A.-C.; Jebeniani, I.; Leschik, J.; Watrin, E.; Bonne, G.; Vignier, N.; Pucéat, M. Targeting the Histone Demethylase LSD1 Prevents Cardiomyopathy in a Mouse Model of Laminopathy. J Clin Invest 2021, 131, e136488, 10.1172/JCI136488.
- Steele-Stallard, H.B.; Pinton, L.; Sarcar, S.; Ozdemir, T.; Maffioletti, S.M.; Zammit, P.S.; Tedesco, F.S. Modeling Skeletal Muscle Laminopathies Using Human Induced Pluripotent Stem Cells Carrying Pathogenic LMNA Mutations. Front Physiol 2018, 9, 1332, 10.3389/fphys.2018.01332.

