Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vicente Barrios Sabador | -- | 6839 | 2023-02-13 12:22:26 | | | |
| 2 | Rita Xu | -66 word(s) | 6773 | 2023-02-14 02:43:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Casado, M.E.; Collado-Pérez, R.; Frago, L.M.; Barrios, V. Leptin and Its Involvement in Pathology. Encyclopedia. Available online: https://encyclopedia.pub/entry/41161 (accessed on 27 July 2026).
Casado ME, Collado-Pérez R, Frago LM, Barrios V. Leptin and Its Involvement in Pathology. Encyclopedia. Available at: https://encyclopedia.pub/entry/41161. Accessed July 27, 2026.
Casado, María E., Roberto Collado-Pérez, Laura M. Frago, Vicente Barrios. "Leptin and Its Involvement in Pathology" Encyclopedia, https://encyclopedia.pub/entry/41161 (accessed July 27, 2026).
Casado, M.E., Collado-Pérez, R., Frago, L.M., & Barrios, V. (2023, February 13). Leptin and Its Involvement in Pathology. In Encyclopedia. https://encyclopedia.pub/entry/41161
Casado, María E., et al. "Leptin and Its Involvement in Pathology." Encyclopedia. Web. 13 February, 2023.
Copy Citation
Excess body weight is frequently associated with low-grade inflammation. Evidence indicates a relationship between obesity and cancer, as well as with other diseases, such as diabetes and non-alcoholic fatty liver disease, in which inflammation and the actions of various adipokines play a role in the pathological mechanisms involved in these disorders. Leptin is mainly produced by adipose tissue in proportion to fat stores, it is also synthesized in other organs, where leptin receptors are expressed.
adipokine
biomarkers
biochemical mechanisms
cancer
leptin
1. Introduction
Leptin, a polypeptide hormone of 167 amino acids with an N-terminal secretory-signal sequence of 21 amino acids encoded by the ob gene, was originally described as a protector against obesity, since ob/ob mice (leptin-deficient) were obese [1]. This cytokine is mainly synthesized by adipocytes from white adipose tissue in proportion to fat stores, and, since its discovery by Friedman’s group in 1994, has been essentially studied in relationship to its crucial role as a regulator of energy balance via its actions on hypothalamic nuclei [2]. Leptin modulates energy homeostasis through its potent inhibitory effect on hypothalamic orexigenic neuropeptide Y/agouti-related peptide (NPY/AgRP)-coexpressing neurons and the activation of pro-opiomelanocortin (POMC)-expressing neurons in the arcuate nucleus [3]. NPY is necessary for some acute actions of AgRP on feeding behavior, mediated through its inhibitory effect on the melanocortin 4 receptor (MC4R) [4], whereas POMC and cocaine and amphetamine-related transcript (CART) neurons trigger MC4R neurons, especially those located in the paraventricular nucleus [5]. These cells play a key role in the control of food intake and energy balance, and the disruption of this circuit by changes in leptin sensitivity or mutations in the genes encoding leptin, the leptin receptor, POMC, or MC4R, amongst other genes in this system, can lead to severe obesity [6].
After the primary recognition for its role in modulating food intake and energy balance, this adipokine has emerged as a crucial molecule with pleiotropic functions. Much evidence indicates its implication in diverse physiological processes, such as glucose metabolism, hematopoiesis, bone remodeling, neurogenesis and neuroprotection, interaction with the immune system, reproduction, angiogenesis, blood pressure, gastric emptying, and glomerular filtration rate, among other functions [7]. Leptin is not only linked to physiological processes, but it is also involved in pathological situations. It is suggested to have anti-apoptotic and pro-angiogenetic effects [8] and to promote proliferation, cell survival and migration in physiological conditions, processes that may also affect the initiation and progression of different diseases, especially cancers of the brain, breast, or lung [9].
The involvement of leptin in pathological situations is more evident in disorders that involve an increase in serum leptin levels. In this sense, data shows a close relationship between obesity and different diseases, such as cancer or cardiac hypertrophy [10][11]. Liver and renal diseases are also associated with hyperleptinemia, both in humans and experimental models of obesity [12][13]. Decreased serum levels may be also related to lipodystrophy and associated pathologies, such as metabolic syndrome and liver diseases. In addition, hypoleptinemia seems to also be associated with adaptive responses, thus the reductions in this adipokine may be a starvation signal inducing hunger, which may be more potent than its effects as a physiological satiety factor [14].
Multiple signaling pathways activated after the binding of leptin to leptin receptors (ObRs) and inter-relations with other factors that may regulate levels of the receptor and vice versa could explain the plethora of leptin actions. Leptin is a class I adipocytokine that binds to at least five OBRs isoforms, all of which share the extracellular domain; however, the ObRb isoform has a well-conserved intracellular membrane-proximal proline-rich region that is essential for Janus kinase (JAK) 2 association, given that this receptor has no intrinsic kinase activity. Activation of the ObRb starts a cascade of signal transduction pathways, with the JAK/signal transducers and activators of transcription (JAK/STAT) pathway being the best studied. Leptin also increases JAK2-dependent activation of the extracellular signal-regulated kinase 1/2 (ERK1/2) mitogen-activated protein kinase (MAPK) [15] and the insulin receptor (IR) substrate (IRS)-phosphoinositide 3-kinase (PI3K)/Akt pathway. In addition, leptin exerts a role in the adenosine monophosphate-activated protein kinase (AMPK) and the cAMP-response element binding protein (CREB)-regulated transcription coactivator (CRTC) pathway [16].
2. Functionality of Leptin and Its Involvement in Pathology
Mutations in the genes for leptin and its receptor, as well as in some of the associated signaling targets, are related to several pathological states. Likewise, changes in the transcription and translation processes can alter local and peripheral levels of this adipokine, modifying its functionality. Finally, changes in the levels of its receptors, especially ObRb, can also be modified in different diseases, most of them related to various degrees of inflammation. Consequently, its implication in diverse central and peripheral disorders is wide, given the plethora of actions of this cytokine.
2.1. Leptin-Associated Central Dysfunctions
The originally described central actions of leptin were mainly related to the regulation of food intake and energy homeostasis at the hypothalamic level [17]. Subsequently, it was reported that leptin is also involved in neurogenesis and has neuroprotective actions [18]. Thus, changes in its functionality may be related to alterations in the regulation of body weight, or different neurodegenerative processes.
2.1.1. Central Leptin Resistance and Dysregulation of Energy Homeostasis
Numerous mechanisms by which leptin resistance is caused have been described, but they are not fully clarified. Among these, a decrease in the intracellular signaling pathway of leptin in different neurons of the central nervous system (CNS) stands out, especially in the hypothalamus. This may be due to changes in the hypothalamic levels of ObRb or also to mutations in its intracellular domain that decrease the triggering of signaling after leptin binding [19]. This response is also altered by the increase in feedback inhibitors. This may be associated with an effect of leptin itself in the increase of leptin resistance, with this state called “leptin-induced leptin resistance” [20]. A prolonged augmentation of leptin in the CNS promotes an increase in STAT3 phosphorylation and translocation to the nucleus, where it activates SOCS3 transcription.
A decrease in positive regulators of the leptin cascade attenuates intracellular signaling. For example, SH2B1, a cytoplasmic adaptor protein, binds via its SH2 domain to JAK2, stimulating this leptin-related pathway. Its deletion provokes metabolic disorders in SH2B1 KO mice, including leptin resistance, obesity, and glucose intolerance whereas restoration of SH2B1 corrected these metabolic disturbances and improved JAK2-mediated leptin signaling and regulation of hypothalamic orexigenic neuropeptides [21]. In addition, pathogenic variants in SH2B1 in young adults with severe obesity corroborate the role of this adaptor protein [22]. Autophagy seems to be involved in leptin resistance as hypothalamic ablation of autophagy-related protein 7 augmented food intake, generating leptin resistance and obesity. On the other hand, the specific deletion of this factor in AgRP neurons reduced food intake and weight, with a possible increase in sensitivity to the actions of leptin [23]. Therefore, it is suggested that an endoplasmic reticulum stress-autophagy pathway regulates hypothalamic development and energy balance in leptin-deficient mice [24].
Hypothalamic inflammation plays a key role in the generation of central insulin and leptin resistance with TNF-α and IL-6 involved in the maintenance of this situation. Insulin resistance also aggravates leptin resistance, given the crosstalking between these hormones, which is essential for maintenance of normal healthy energy homeostasis [25]. C1qTNF-related protein 4 (CTRP4) acts in the hypothalamus to modulate food intake and exerts anti-inflammatory effects on several cell types. Overexpression of CTRP4 in the hypothalamus of high-fat-diet-induced obese mice reduced central TNF-α and IL-6 levels, being consistent with the suppression of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) signaling and restoring of leptin sensitivity [26].
Another key factor in the appearance of central resistance to leptin is impairment of its transport to the brain across the blood–brain barrier. This transport is dependent on the ObRb and is saturable, so if there are hyperleptinemia, the saturation of the receptor reduces its transport, and lower local levels into the brain may generate leptin resistance [27]. However, this point is controversial, since there are data that suggest an ObRb-independent transport mechanism and, in addition, astrocytes may also regulate leptin transport across the blood-brain barrier mediated by leptin receptors in a mechanism independent of reduction of paracellular permeability [28]. Hypertriglyceridemia specifically inhibits leptin transport across the blood–brain barrier [29]. Tanycytes, a specialized glial cell located in the medial eminence of the third ventricle of the hypothalamus, are involved in the transport of leptin and other hormones into the cerebrospinal fluid and play a role in the pathophysiology of central insulin resistance. These cells do not express the leptin receptor and thus, the mechanism by which this transport occurs is partially independent of the leptin receptor [30]. The recent characterization of new factors such as the cationic amino acid transporter-1, that increases leptin transport, suggests additional mechanisms for leptin transport. In this regard, the treatment of brain microvascular endothelial cell-like cells with substrates for this transporter generated a significant increase in leptin transport [31]. All of these factors mentioned above are involved in changes in the expression of the different neuropeptides that regulate energy intake and homeostasis, and in a prolonged situation, this causes metabolic disorders by disrupting the crosstalk between the hypothalamus and periphery, a situation that can be reversed with some antilipemic drugs that reduce body weight [32].
2.1.2. Leptin, Neurogenesis, and Neuroprotection
Although leptin receptors were initially identified in hypothalamic neurons, they are widely distributed in different cell types of the cerebral cortex and hippocampus, areas involved in cognition and memory processes. Expression of ObRb in neural precursor cells is necessary for preserving an adequate body weight in addition to the differentiation of these precursors into neural or glial cells after birth [33]. Leptin is an activator of transcription through triggering CREB protein, that is required for leptin-stimulated synapse formation and also augments the expression of miRNA-132, a well-known CREB target, which is necessary for leptin-induced synaptogenesis [34]. CREB regulates the expression of numerous genes in neurons, including those that modulate synaptic development.
Leptin participates in the growth of hippocampal neurons and formation of dendritic spines in the hippocampal neurons, and this results in the enhancement of glutamatergic synaptogenesis during neonatal development [35]. This action is also mediated, at least in part, by the deubiquitinase ubiquitin-specific protease 8 that is accomplished with ObRb. Acute leptin stimulation increases this activity and mRNA levels through CREB-dependent transcription [36]. Surface expression of N-methyl D-aspartate (NMDA) receptor subtype 2B (NR2B) is also regulated by leptin, and after its activation forms a complex with ObRb [37]. NMDA is the major excitatory neurotransmitter in the mammalian CNS, and it is implicated in modulating functions such as learning, memory processing, pain perception, and feeding behaviors.
Genetically obese rodents (db/db mice, fa/fa rats) exhibit impairments in hippocampal memory processes showing deficits in spatial memory tasks detected in the Morris water maze [38]. Moreover, leptin infusion improves spatial learning and behavioral performance in mice and controls excitatory synaptic transmission in the hippocampus, increasing long-term potentiation and reducing long-term depression [39]. In this manner, this adipokine increases the efficiency of excitatory synaptic transmission, and upgrades learning and memory.
Leptin also has various neuroprotective actions and has been reported to reduce damage in models of glucose deprivation and transient cerebral ischemia. Experimental data connect metabolic deterioration and Parkinson’s disease (PD) with a dysregulation of central and peripheral neuroinflammatory networks mediated by several adipokines, particularly by leptin. Leptin had protective effects in an experimental model of PD, generated by a mitochondrial neurotoxin [40], and compounds that increase leptin signaling in a 6-hydroxydopamine rat model of PD attenuate neuronal apoptosis of substantia nigra [41].
Mitochondrial dysfunction has an established role in the development of Alzheimer’s disease (AD) pathophysiology. Leptin reduces depolarization of the mitochondrial membrane and mitochondrial fragmentation induced by amyloid β (Aβ). Leptin restrains up-regulation of the mitochondrial fission protein, Fis1, and down-regulation of the fusion protein, Mfn2, and increases the expression and activity of antioxidant enzymes [42]. Chronic leptin treatment of transgenic mice overexpressing amyloid precursor protein (APP) reduced brain concentrations of Aβ and phosphorylated tau, improving memory [43], and acute central leptin infusion increased brain somatostatin [44] with a concomitant uprising of CREB, involved in memory processes. Somatostatin is a neuropeptide involved in cognitive processes and is reduced in the hippocampus of experimental models of AD [45] and in AD patients [46]. Moreover, chronic central leptin treatment reduces Aβ-mediated impairment in memory and suppression of hippocampal LTP [47].
Leptin exerts neuroprotective effects and improves chronic sleep-deprivation-induced depressive-like behaviors. Animal models of post-traumatic stress disorders present activation of NLRP3 inflammasome in astrocytes sorted from glial fibrillary acidic protein (GFAP) transgenic mice, while administration of leptin markedly suppressed the activation of astrocytic NLRP3 inflammasome. Leptin effectively improved these behavioral alterations including cognitive impairments and depressive-like conducts. These actions of leptin were facilitated by STAT3 [48]. An increase in STAT3 also mediates the effect of leptin in promoting angiogenesis after hemorrhage. Leptin infusion promoted a dose-dependent effect on vascular endothelial cell survival and proliferation after intracerebral hemorrhage in rodents [8].
2.1.3. Neurological Diseases
Clinical findings indicate that insulin and leptin resistance are related to cognitive deficits and neuropsychiatric disorders, and interestingly, these analyses propose that the associated deficits in neuroplasticity can be reversed by restoration of insulin and leptin sensitivity [49]. Parkinson’s disease, the second most common neurodegenerative disease, is characterized by gradual degeneration of dopaminergic (DA) neurons in the substantia nigra pars compacta. Leptin reversed the loss of dopaminergic neurons in an experimental model of PD that employed the dopamine cell-specific neurotoxin, 6-hydroxydopamine (6-OHDA). The neuroprotective effects occurred via ObR receptor activation and involved activation of JAK-STAT, ERK, and growth factor receptor-bound protein 2 (GRB2) [50]. Leptin augments brain-derived neurotrophic factor (BDNF) following leptin receptor activation, with this factor being a key factor for survival of dopaminergic neurons, which is also diminished in PD [51]. As leptin and BDNF activate shared signaling pathways, such as PI3K and MAPK/ERK and some intermediary molecules, including SH2 and GRB2, leptin could induce positive feedback by increasing BDNF expression. In a recent report [52], 10 PD-related hub genes, including JAK2, ObRb and IRS2, among others, are involved in AMPK and leptin signaling.
Mitochondrial dysfunction and oxidative stress are pathogenic processes involved in PD. Cells control such stress via several antioxidant factors. One family of proteins is the mitochondrial uncoupling proteins (UCPs), which are anion carriers located in the mitochondrial inner membrane. Leptin can preserve neuronal survival acting via UCP2 against mitochondrial membrane potential positive-induced toxicity, commonly used in experimental Parkinsonian models, by maintaining ATP levels and membrane potential [53]. These data indicate that leptin could have potential as a therapeutic target in PD, but research in this field is limited and generates controversies. In this regard, serum leptin levels in PD patients were non-significantly lower than controls in a meta-analysis study [54], although it was found that peripheral leptin levels in unintended weight loss PD patients were lower than those with stable weight [55].
There are data on the relationship between depression and leptin status. Animal studies of chronic stress report lower levels of leptin, and central infusion of this adipokine had antidepressant effects. Deletion of ObRb induces depression-like behaviors, suggesting that intracellular leptin signaling is implicated in the molecular mechanism of anti-depressant action of this cytokine [56]. Clinical trials examining the relationship between depressive processes and leptin levels are inconsistent. Some studies showed lower plasma levels of leptin in these patients; however, in cases of major depression disorder, serum concentrations were higher than in control subjects [57]. In another study, leptin was analyzed in depressive patients with therapy, finding a relationship between the combination of serum leptin and number of behavior disorders, suggesting that leptin levels can be a predictor of treatment responses in these patients [58].
There are data suggesting that leptin might play a role in AD. The decrease in plasma leptin levels with age has been associated with an increased risk of cognitive loss and the possible development of this disease [59]. In fact, this adipokine restrains tau phosphorylation, an essential premise for the formation of neurofibrillary tangles and studies in postmortem brains of AD patients showed a dysregulation of intracellular leptin signaling circuitry, with decreased ObRb mRNA levels in brain and colocalization of ObRb protein with neurofibrillary tangles [60], indicating a central resistance in this neurodegenerative disease.
An experimental model of AD in female rats treated chronically with Aβ showed a reduction in leptin levels and hippocampal leptin-related signaling, that was reverted with 17β-estradiol [61]. Indeed, 17β-estradiol administration is commonly related to better metabolic outcomes and neural benefits in old mice, with decreased β-amyloid burden, improved behavioral performance, and reduced microglial activation [62]. Estradiol is important for brain function; thus, 80% of postmenopausal women describe neurological symptoms including reduced cognitive performance. Preclinical evidence for neuroprotective effects of 17β-estradiol also indicate associations between menopause, cognitive aging, and AD, although there are discrepancies amongst clinical trials [63].
It has recently been reported that Aβ has a high affinity interaction with ObRb. Aβ binds allosterically to the extracellular domain of ObRb and significantly reduces different leptin signaling pathways, such as STAT3, PI3K, and ERK. In addition, the effect of leptin on POMC expression was reduced, indicating not only the disruption of the pathway, but also its effects on metabolism [64]. Chronic leptin treatment increases adult neurogenesis in transgenic mice that overexpress Aβ, with increased expression of ObRb in neurogenic niches of neural stem cells of these mice [65]. This adipokine reduced astrogliosis, microglial cell number, and the development of senile plaques, attenuating superoxide anion production and Aβ neurodegeneration.
Cytokines modulate neuronal plasticity and neurogenesis, intervening in cognitive processes. However, one must recall that leptin can promote inflammatory responses, in-creasing TNF and IL-6, which reduce synaptic plasticity and are related to the progression of AD and severity of cognitive impairment in these patients [66]. Leptin dysfunction in AD is hypothesized to be due to leptin resistance. Given that treating obesity with leptin is ineffective due to leptin resistance, it seems unlikely that AD will respond to leptin therapy as leptin resistance is thought to be an important factor in this disorder. Therefore, the use of leptin as a cognitive enhancer most likely has a reduced clinical application in these patients [67]. By itself, treatments that improve leptin sensitivity may be significantly more effective than solely replacing or supplementing this adipokine [68].
2.2. Leptin Status and Its Implication in Metabolic Pathologies
When leptin levels are in the normal range, this adipokine can exert its effects on energy intake and the numerous actions that regulate metabolism and contribute to maintaining energy homeostasis. Long-term changes in circulating leptin concentrations, either by excess or by deficiency, cause a series of disorders entangled in the pathogenesis of metabolic syndrome, diabetes mellitus, and cancer, among others.
2.2.1. Obesity and Lipodystrophy
Obesity is usually accompanied by hyperleptinemia that is directly associated with an increase in body fat mass. This leads to leptin resistance, and the underlying mechanisms are diverse, including impaired ObRb signaling, changes in hypothalamic neural wiring, reduced brain leptin transport and ObRb trafficking, endoplasmic reticulum stress, and, frequently, low-grade inflammation [69]. Through genome-wide association studies and next-generation sequencing, approximately 500 obesity-related genes were identified, with mutations in some of these genes, including leptin and its receptor genes, POMC, MC4R, BDNF, prohormone convertase 1, unique homologue 1, and neurotrophic receptor tyrosine kinase type 2, having been reported to cause obesity [70]. In this section, the first two will be described.
About 5–8% of obese patients have a monogenic form of obesity. Complete leptin deficiency causes early-onset severe obesity. These patients have undetectable serum leptin levels and other endocrine abnormalities such as hypogonadotropic hypogonadism and hypothalamic hypothyroidism, endocrine findings that are due to leptin-hypothalamic linking intracellular signaling pathways implicated in the production of sex steroids and thyroid. Mechanistically, defects in the synthesis and/or secretion of leptin have been suggested and confirmed for several of these mutations. The administration of leptin to these patients causes a loss of fat and weight, as well as a signal of adequate satiety [71].
Mutations in the leptin receptor produce a monogenic form of obesity typified by hyperphagia and weight gain, with a prevalence of around 3% of children with obesity [72]. To date, different mutations of the human leptin receptor have been reported and, in most cases, subjects present severe obesity, alterations in immune function, and delayed puberty due to hypogonadotropic hypogonadism with serum leptin levels in the range predicted by their raised fat mass [73]. However, their clinical characteristics are reported to be less pronounced than patients presenting congenital leptin deficiency due to mutations in the leptin gene. Loss of leptin receptor function has been related to infertility, although a new recent mutation described in three generations demonstrated that all these patients were able to conceive [74].
Predisposition to the development of obesity in childhood and in adulthood can be affected by the milieu in the early stages of life. In animals, dietary changes during gestation produce alterations in the metabolism and body composition of the offspring, while in humans a high body mass index and an abnormal increase in gestational weight in-crease the risk of obesity in the descendants [75]. Adiposity may involve epigenetic regulation of different genes; indeed, in murine models, variations in the maternal diet vary the methylation of key metabolic genes, with the majority of these modifications persisting throughout life, thus modifying gene expression and the metabolic processes associated with these changes. For example, Lin et al. demonstrated that high-fat diet (HFD)-exposed offspring have hypomethylation in the leptin gene in subcutaneous and visceral adipose tissue [76]. In adult mice exposed long-term to a high-fat diet, DNA methylation of leptin and PPAR-γ promoters increased in gonadal adipose tissue; the same does not occur in subcutaneous adipose tissue [77]. In humans, changes in maternal dietary composition are associated with modifications in adiposity and bone mineral content with differential methylation of several promoters in the umbilical cord [78]. Moreover, in adolescents, differentially methylated CpG loci were associated with usual adiposity and most of them positively correlated with serum leptin [79], and leptin promoter methylation at birth and 12 months predicts weight and adiposity childhood [80].
Most studies find epigenetic variation associated with unfavorable exposures in utero, analyzing DNA methylation in genes, showing small methylation changed be-tween the compared conditions, less than 5% in case-control analysis. Both the proximal promoter of the leptin receptor and a part of its coding sequence are on a CpG island susceptible to epigenetic modifications during pregnancies with metabolic alterations. The a and b isoforms of the leptin receptor are expressed in the placenta of rodents and pregnant mice receiving diets rich in saturated fat showed a decrease in ObRa, suggesting a nutrient-availability regulatory mechanisms for the leptin receptor [81]. In humans, maternal obesity was shown to increase DNA methylation of the leptin promoter in the placenta, as well as to result in low levels of leptin receptor, although without differences in promoter DNA methylation in the fetus [82].
The different types of SOCS are negative regulators of cytokine signaling through JAK/STAT. Thus, after the binding of leptin to its receptor and the activation of STAT3, SOCS3 is synthesized, which binds to the Tyr985 of the ObRb, preventing the phosphorylation of JAK2 [83]. Deletion of SOCS3 in neurons delay the onset of central leptin resistance in mice after high caloric diet intake [84]. Leptin and insulin share many signaling targets and one of them is PTP1B, a characteristic inhibitor of the insulin pathway. This phosphatase is contained to the cytoplasmic face of the endoplasmic reticulum and constrains leptin signaling by binding and subsequent dephosphorylation of JAK2. Mice with central PTP1B-deficiency are lean, sensitive to leptin, and partially protected from diet-induced obesity [85]. Changes in leptin signaling in obesity are shown in Figure 1.
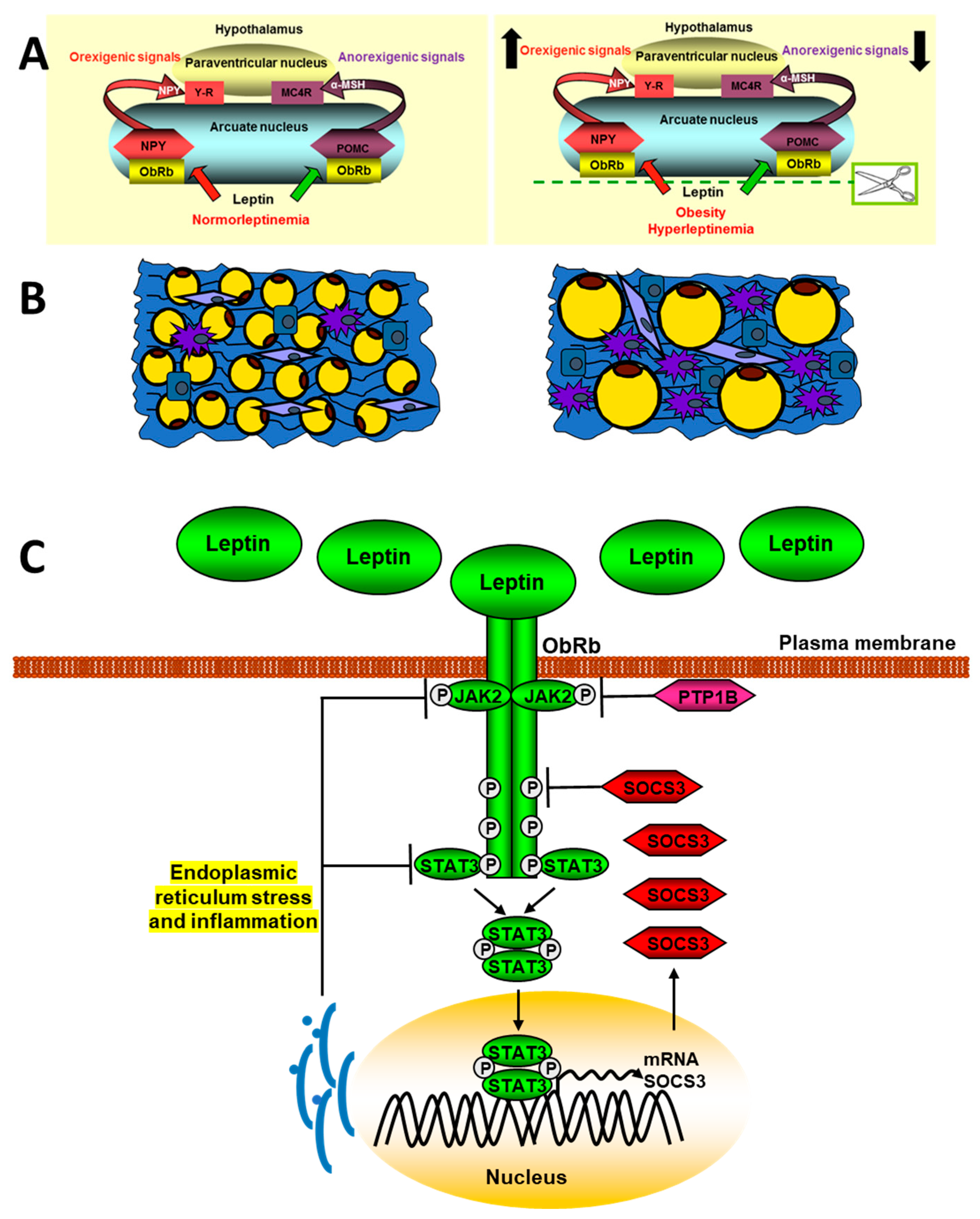
Figure 1. Schematic signaling pathways of leptin in obesity. The upper panel (A) represents a normal situation (left) and another with hyperleptinemia (right) and its effect on the expression of hypothalamic neuropeptides that regulate intake. The panel (B) shows adipose tissue in lean (left) or obese patients (right). The adipose tissue in obese subjets presents a hypertrophy of adipocytes, as well as a greater infiltration of macrophages and other inflammatory cells. In panel (C), the mechanism involved in the endoplasmic reticulum stress blocking of leptin signaling is indicated.
Proinflammatory cytokines, as IL-20, are increased in obese patients and promotes an inflammatory environment by increasing other factors, such as TNF-α, monocyte chemotactic protein 1 (MCP-1), and leptin in adipocytes. Thus, this interleukin can increase insulin resistance by reducing glucose uptake through activation of the SOCS-3 pathway [86]. Excessive intake activates hypothalamic NFκB signaling through endoplasmic reticulum stress responses, connecting this stress to central inflammation [87]. IKK inhibition reduces leptin resistance by restoring JAK2-STAT3 and PI3K signaling in the hypothalamus of mice fed high-fat diets. Likewise, the central infusion of leptin shows a peripheral inhibition of the NFκB pathway together with the activation of PI3K [88].
The changes in the levels of ObRb in the cellular membrane depend on the synthesis, transport, internalization, and recycling, among other factors. These levels regulate the efficiency of leptin action and are known to increase after fasting and decrease after food intake, with leptin increasing hypothalamic expression of leptin receptor [89] but being unable to do so after the administration of diets rich in saturated fats. There are different molecules that regulate the exposure of ObRb in the cell membrane. Among these is endospanin-1, which is codified in humans by the same gene as the leptin receptor. This protein is increased by high-fat diets and regulates ObRb trafficking and cell surface exposure [90], whereas silencing of endospanin-1 in the arcuate nucleus of obese mice completely restores leptin responsiveness [91]. Thus, defective intracellular trafficking of the leptin receptor may cause leptin resistance seen in obese patients.
Leptin crosses the blood–brain barrier actively, being transport-saturable. In situations of obesity, the ratio of leptin in serum/cerebrospinal fluid decreases, showing impaired transport. ObRa isoform seems to be involved, but mutants in this isoform have minimal effect on leptin transport [92]. The median eminence is involved in leptin transport to the hypothalamus, with tanycytes mediating this action. Leptin administration activates ObRb in these cells, through activation of the ERK pathway and subsequent delivery to hypothalamic neurons. In murine models of obesity, leptin accumulates in the median eminence and experimental activation of ERK signaling restores its transport [93].
Resistance to the action of leptin can occur due to its excess, as well as a decrease in this adipokine. Lipodystrophy syndromes, which present a complete or partial absence of fat, are forms of leptin resistance due to hypoleptinemia. Molecular bases of these syndromes include, among others, modified differentiation of adipocytes, structure and regulation of the adipocyte lipid droplet, and early cellular senescence. These patients present metabolic complications such as insulin resistance, dyslipidemia, and fatty liver disease. A recent study in lipodystrophic mice treated with leptin showed a reduction in inflammation [94] and given the evidence of leptin deficiency in lipodystrophy syndromes; leptin replacement therapy has been contemplated as a treatment option. Long-term studies in children with lipodystrophy on the use of therapy with a methionylated human leptin analogue, metreleptin, have showed great improvements in the clinical situation and metabolic circumstances of these patients [95]. However, there are studies in obese patients with diabetes that established that metreleptin develops leptin tolerance; thus, this circumstance may limit the efficiency of this therapy [96].
MicroRNAs may be implicated in obesity and in modifications in leptin signaling activation. For example, miR-200a is overexpressed in the hypothalamus of ob/ob mice and its blockade restores both leptin and insulin signaling in the hypothalamus and liver [97]. In addition, leptin treatment significantly reduced hypothalamic miR-200a expression. Overexpression of pre-miR-200a causes a significant increase in miR-200a expression in SH-SY5Y human neuroblastoma cells, leading to considerable down-regulation of ObRb at both the mRNA and protein levels. In addition, miR-200a overexpression affects STAT-3 phosphorylation and Erk 1/2 phosphorylation in response to leptin. POMC expression is decreased in ob/ob mice, whereas miR-200a blockade in the hypothalamus increases its expression [97]. Moreover, Zhang et al. [98] found that leptin strongly up-regulates the expression of miR-21 in human mature adipocytes. It is known that miR-21 expression is upregulated by TNF-α, IL-6, leptin, resistin, and FFAs, but not by glucose. Leptin may affect obesity-associated insulin resistance by promoting miR-21 expression in adipocytes. Furthermore, miR-21 enhances the adipogenesis of human adipose tissue stromal cells by modulating TGF-β [99] and targeting STAT3 signaling [100]. These two signaling pathways interact with PPAR-γ signaling [101]. The miRNA let-7 is also related to obesity, with let-7 deficiency shown to prevent obesity induced by HFD feeding in mice, as well as hepatic steatosis accompanied by inhibition of the PPAR-α signaling [102]. A specific mouse line with hepatocyte-specific let-7b/c2 knockout shows that the direct target of let-7 ring finger protein 8 (Rnf8) is increased in let7b/c2ΔHep mouse liver and identified as a E3 ubiquitin ligase for obligate PPARα heterodimer partner retinoid X receptor α (RXRα). The study highlights a role for let-7 in the RNF8-RXRα regulatory axis, which regulates hepatic lipid catabolism [102].
2.2.2. Diabetes
Serum adipokine levels exhibit similar bivariate relations with anthropometric variables in patients with type 1 diabetes mellitus (T1DM) to those in normal weight subjects. Circulating leptin is higher in overweight and obese patient with T1DM [103]. Leptin and soluble leptin receptor levels are increased in T1DM children and related to anthropometrics parameters, metabolic control, age of debut of diabetes, and kind of insulin therapy [104].
Animal studies show that leptin reduces hepatic glucose production by decreasing the MAPK phosphatase-3 (MKP-3) protein level via STAT3-enhanced MKP-3 and ERK combination [105], increases glucose uptake into tissues, modulates IGF-binding protein 2, and lowers blood glucose independent of hepatic leptin signaling [106]. This adipokine can also improve the diabetes of lipodystrophic mice independently of insulin [107], suggesting that leptin has therapeutic potential for the treatment of T1DM. PTP1B inhibits leptin signaling, and mice with T1DM induced by streptozotocin show hyperglycemia and reduced glucose metabolism, and leptin administration improves glucose metabolism in these mice. These effects were enhanced in mice with PTP1B deficiency [108]. Thus, inhibition of this leptin receptor inhibitor improves energy homeostasis in this metabolic disorder.
Central leptin action is sufficient to restore euglycemia in T1DM and is dependent on STAT3 activation, but not the release of fast-acting neurotransmitters such as glutamate and γ-aminobutyric acid [109]. In fact, central leptin can rescue T1DM hyperglycemia and recently, another mechanism has been proposed for this action. It was found that neurons expressing leptin receptor in the arcuate nucleus are selectively activated in this disease. These neurons have defective nutrient sensing and signs of energy deprivation that may be restored by leptin. The abnormal activation of these neurons due to energy deficiency as the neural basis for T1DM hyperglycemia and leptin action is mediated by inhibiting these neurons across withdrawing energy deprivation [110].
Hypoglycemia is the most serious complication of insulin therapy; therefore, adjunctive leptin treatment could reduce the severity of these episodes; indeed, the combination of insulin and leptin in T1DM mice was shown to improve glycemic stability. In addition, leptin may also have insulin-independent effects to reverse hyperglycemia and ketoacidosis in poorly controlled T1DM animal models. Leptin reduces hyperphagia and insulin resistance in liver and muscle in patients with lipodystrophy [111], and this fact together the data mentioned above increases the interest in possible treatment of T1DM patients with this cytokine, as these patients often present with weight loss and hyperphagia as a consequence of complete or partial insulin deficiency. However, differences between T1DM mice and humans may affect the efficacy of leptin treatment. T1DM mice are leptin deficient due to decreased fat mass resulting from uncontrolled diabetes unlike humans with T1DM that receive insulin therapy. In addition, leptin treatment in T1DM patients with normal leptin levels may have a different effect on blood glucose than treatment in T1DM mice deficient in this hormone [112]. On the other hand, leptin therapy might also minimize the weight gain that is associated with increased doses of insulin. Consequently, additional studies will be needed to evaluate these possibilities.
Diabetes mellitus may also result primarily from a state of insulin resistance (type 2 diabetes mellitus, T2DM). More than 80% of subjects with T2DM are overweight [113]. In fact, an underlying pathophysiological condition of obesity and T2DM is the reduced biological response to insulin in peripheral tissues such as the liver, adipose tissue, and skeletal muscle. These organs, together with the pancreas, regulate glycemia. Nevertheless, this is a simplified view, as the brain is a crucial insulin target and plays a key role in glucose homeostasis. Above all, the arcuate nucleus is of critical importance for sensing of adiposity signals, not only insulin, but also leptin and circulating nutrients [114]. There is a partial intersection between neuronal populations that regulate energy balance and glucose homeostasis; hence, obesity and T2DM may have similar origins that are related to dysfunctions in the central nervous system [115].
The genetically induced hyperphagia and obesity in leptin- and leptin receptor-knockout mice, as well as in Zucker obese rats, are associated with the development of insulin resistance, T2DM, and abnormal leptin signaling. Studies in a new model of mice, BTBR. Cg-Lepob/WiscJ ob/ob (BTBR ob/ob) mice with sustained hyperglycemia at less for 20 weeks, present anatomical alterations in the brain–blood barrier that provokes an ab-normal entry of leptin into the brain, with subsequent reduced central leptin signaling, indicating again the role of the brain in this metabolic disease [116].
The Zucker Diabetic Sprague–Dawley (ZDSD) rat is another model for experimental studies of T2DM, with a characteristic phenotype that is independent of leptin receptor signaling [117]. This rat does not carry the leptin receptor mutation (fa/fa) and develops a prediabetic state that progresses to overt diabetes with age, with common morbidities, such as cardiomyopathy, that are associated with an increased risk occurrence in prediabetic patients. Clinical deteriorations associated with human T2DM and observed in the ZDSD rat include delayed wound healing, nephropathy, and neuropathy [118]. Accordingly, ZDSD rats may be an advantageous model for understanding molecular mechanisms and discovering potential new treatments for T2DM.
Changes in leptin-signaling-related molecules play a role in T2DM patients. The analysis of the association of SNPs with T2DM demonstrated that SOCS3 and JAK2 genes may be associated with T2DM, whereas interaction between the SOCS3, JAK2, and STAT3 are related to metabolic syndrome features [119]. T2DM is associated with central and peripheral inflammation [120][121] and the JAK/STAT signaling pathway mediates the effects of multiple cytokines, with abnormal signaling closely related to diabetic complications. In this context, aberrant leptin signaling can be implicated in the pathogenesis of this disease. Increased activation of JAK2 and STAT3, accompanied by elevated levels of SOCS3 contribute to the development of vascular complications by mediating inflammation associated with vascular endothelial cells [122]. It is important to highlight the growing evidence that indicates that subjects with type 2 diabetes are at higher risk for some common tumors, including cancers of the prostate, colon, breast, endometrium, pancreas, and liver [123]. Increased insulin levels may trigger the cancer associations of several additional risk factors [124], comprising high waist circumference, visceral fat, body mass index, sedentary lifestyle, and an inadequate food intake [125], among others.
MicroRNAs may be involved in changes in leptin-signaling activation in diabetes. For example, studies in experimental models of diabetes showed that miR-125a-5p may be a modulator of glycolipid metabolism in T2DM, by restraining lipogenesis and gluconeogenesis in the liver and increasing glycogen synthesis by targeting STAT3 [126]. Furthermore, several adipokines increase leptin concentrations and reduce concentrations of adiponectin, influencing insulin sensitivity and its related diseases. This process modifies expression of miRNAs, including let-7 [127]. Let-7 functions with RNA-binding proteins Lin28a/b that when is overexpressed in mice, provoking an insulin-sensitized state able to resist high-fat-diet-induced diabetes [128]. Overexpression of let-7 causes insulin resistance and impaired glucose tolerance in mice by repressing some components of the insulin-PI3K-mTOR pathway, such as IR, IRS2, PI3K interacting protein 1, Akt2, tuberous sclerosis complex 1, and rapamycin-insensitive companion of mTOR. In addition, these authors found that some targets of let-7 presented SNPs that were related to changes in the control of fasting glucose and T2DM [128]. Moreover, miR-26b is downregulated by leptin [129] and this miRNA was found to be reduced in visceral adipose tissue (VAT) in insulin-resistant adipocytes, obese rodent models, and human obesity [130]. Insulin-stimulated glucose uptake is promoted by miR-26b, as well as the translocation of insulin-stimulated glucose transporter type 4 to the plasma membrane in mature human adipocytes. miR-26b inhibits phosphatase and tensin homolog deleted on chromosome 10 (PTEN), its target gene altering Akt activation in response to insulin, which increases insulin sensitivity via PI3K/Akt pathway [131].
Epigenetic mechanisms such as the methylation of DNA are reported to be involved in the prenatal development of leptin and insulin resistance and these mechanisms may explain the risk of T2DM for subsequent generations. On this way, South Asians present an abnormal burden of type 2 diabetes that may be attributed to a “thin-fat-phenotype”. Asian ethnicity and gestational diabetes were associated with higher placental leptin gene methylation, with maternal glucose and lipid metabolism associated with placental methylation of the gene for this cytokine [132].
Different strategies have been used in the management of T2DM, including metformin and related drugs. Aerobic exercise is suggested as an effective non-pharmacological strategy to reduce cognitive disorders that have an increased risk in this disease [133]. In experimental models of T2DM, exercise results in an improvement in the cognitive disorders and this is mediated by an in increase in the AMPK/sirtuin 1 pathway, while AMPK inhibitors suppress the beneficial effect of this exercise [134]. Different phosphatases exert feedback inhibition on insulin signaling. Among these, PTP1B stands out, which also exerts an inhibition on leptin signaling. Drugs targeting PTP1B have given promising results in T2DM animal models [135], but due to limited information on their safety, it has not been possible to use them in patients. There is a close relationship between obesity and leptin resistance, which may precede or co-occur with T2DM. Therefore, leptin sensitivity may be a possible therapeutic target in this disease. Insulin sensitivity modulators in combination with amylin and/or GLP-1 analogues may be of interest due to their central activity in the hypothalamus [136].
2.2.3. Cancer
More than twenty years ago, it was reported that both leptin mRNA and protein levels were increased in several breast cancer cell lines and breast tumors [137]. Afterwards, other studies showed changes in circulating leptin levels, together with a decrease in one or more isoforms of its receptor in different cancer types [138]. Subsequent findings established that leptin signaling was implicated in promotion of endometrial cancer cell proliferation by activating STAT3 and ERK2 pathways and that leptin-induced phosphorylation of ERK2 and Akt was determined by STAT3 activation [139].
Evidence links obesity to cancer, which may contribute to the increased morbidity and mortality associated with obesity. An excess of food intake causes an increase in fat depots, altering the balance between pro-inflammatory and anti-inflammatory cytokines and thus, increased levels of some proinflammatory cytokines associated with predicted cancer development [140]. Changes in macrophage composition can be an additional mechanism linking obesity with the initiation and progression of cancer. Proinflammatory macrophages have often been associated with a classically activated phenotype, which enhances the immune response and opposes tumorigenesis whereas anti-inflammatory macrophages alternatively adopt a phenotype that lessens immunity and promotes tumorigenesis. Leptin in obese patients may generate an alternative phenotype (M2) [141], but obesity is also related to a proinflammatory “metabolically activated” phenotype that is both mechanistically and functionally distinct from the classic (M1) phenotype. This metabolically activated phenotype is induced by saturated fatty acids released by insulin-resistant fat cells in obesity that are not only in fat but also in mammary fat and in other tissues, and secrete proinflammatory cytokines involved in tumorigenesis [142].
Leptin and different isoforms of its receptor are abnormally expressed in cancer cells and tumor-adjacent areas. Methylation of the leptin receptor gene is lower in tumor samples than in adjacent non-tumor samples and its mRNA levels are directly related to the appearance of metastatic cells. The over-activation of ObRb leads to an increase in signaling in the JAK2/STAT3, PI3K, and ERKs pathways, which modulate the expression of genes related to cancer such as vascular endothelial growth factor (VEGF), cyclin D1, and cyclooxygenase-2 that favor angiogenesis, cell proliferation, and migration processes [143]. Leptin can also promote changes in the inflammatory environment by increasing the expression and secretion of different cytokines, which leads to increased migration and invasion of cancer cells in different organs [144]. Leptin may have synergistic actions with other cytokines in different types of cancers. Thus, these actions activate NFkB, increasing the synthesis of VEGF and MCP-1 by cancer cells leading to recruitment and activation of infiltrating tumor-associated macrophages, as well as the synthesis of other cytokines related to inflammation processes [145] (Figure 2).
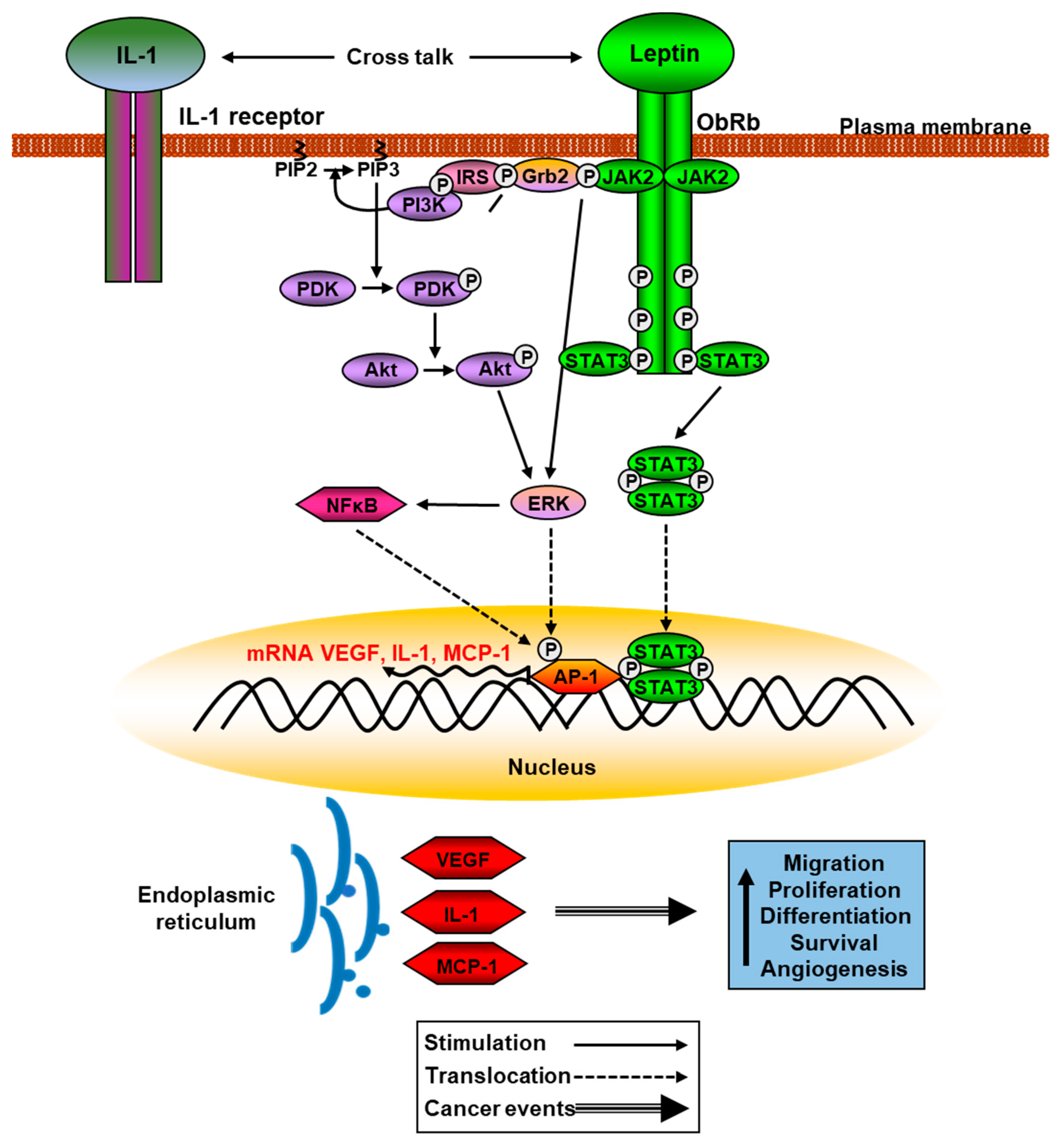
Figure 2. Schematic leptin signaling pathways involved in cancer. Phosphorylation of JAK2 and Akt activate ERK pathway that phosphorylates AP-1, increasing translation of VEGF, IL-1, and MCP-1, among other proinflammatory cytokines.
References
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546.
- Schwartz, M.W.; Seeley, R.J.; Campfield, L.A.; Burn, P.; Baskin, D.G. Identification of targets of leptin action in rat hypothalamus. J. Clin. Investig. 1996, 98, 1101–1106.
- Kalra, S.P.; Dube, M.G.; Pu, S.; Xu, B.; Horvath, T.L.; Kalra, P.S. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr. Rev. 1999, 20, 68–100.
- King, P.J.; Widdowson, P.S.; Doods, H.; Williams, G. Regulation of neuropeptide Y release from hypothalamic slices by melanocortin-4 agonists and leptin. Peptides 2000, 21, 45–48.
- Mountjoy, K.G.; Mortrud, M.T.; Low, M.J.; Simerly, R.B.; Cone, R.D. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol. Endocrinol. 1994, 8, 1298–1308.
- Rajcsanyi, L.S.; Zheng, Y.; Fischer-Posovszky, P.; Wabitsch, M.; Hebebrand, J.; Hinney, A. Prevalence estimates of putatively pathogenic leptin variants in the gnomAD database. PLoS ONE 2022, 17, e0266642.
- de Candia, P.; Prattichizzo, F.; Garavelli, S.; Alviggi, C.; La Cava, A.; Matarese, G. The pleiotropic roles of leptin in metabolism, immunity, and cancer. J. Exp. Med. 2021, 218, e20191593.
- Cui, Q.; Zhang, Y.; Tian, N.; Yang, J.; Ya, D.; Xiang, W.; Zhou, Z.; Jiang, Y.; Deng, J.; Yang, B.; et al. Leptin promotes angiogenesis via pericyte STAT3 pathway upon intracerebral hemorrhage. Cells 2022, 11, 2755.
- Cairat, M.; Rinaldi, S.; Navionis, A.S.; Romieu, I.; Biessy, C.; Viallon, V.; Olsen, A.; Tjønneland, A.; Fournier, A.; Severi, G.; et al. Circulating inflammatory biomarkers, adipokines and breast cancer risk-a case-control study nested within the EPIC cohort. BMC Med. 2022, 20, 118.
- Kinoshita, Y.; Arita, S.; Ogawa, T.; Takenouchi, A.; Inagaki-Ohara, K. Augmented leptin-induced trefoil factor 3 expression and epidermal growth factor receptor transactivation differentially influences neoplasia progression in the stomach and colorectum of dietary fat-induced obese mice. Arch. Biochem. Biophys. 2022, 729, 109379.
- Leifheit-Nestler, M.; Wagner, N.M.; Gogiraju, R.; Didié, M.; Konstantinides, S.; Hasenfuss, G.; Schäfer, K. Importance of leptin signaling and signal transducer and activator of transcription-3 activation in mediating the cardiac hypertrophy associated with obesity. J. Transl. Med. 2013, 11, 170.
- Özkan, E.A.; Sadigov, A.; Öztürk, O. Evaluation of serum omentin-1, vaspin, leptin, adiponectin levels in obese/overweight children and their relationship with non-alcoholic fatty liver disease. Clin. Nutr. Res. 2022, 11, 194–203.
- Bukosza, E.N.; Kaucsár, T.; Godó, M.; Lajtár, E.; Tod, P.; Koncsos, G.; Varga, Z.V.; Baranyai, T.; Nguyen, M.T.; Schachner, H.; et al. Glomerular collagen deposition and lipocalin-2 expression are early signs of renal injury in prediabetic obese rats. Int. J. Mol. Sci. 2019, 20, 4266.
- Ranea-Robles, P.; Lund, J.; Clemmensen, C. The physiology of experimental overfeeding in animals. Mol. Metab. 2022, 64, 101573.
- Huang, X.; He, Q.; Zhu, H.; Fang, Z.; Che, L.; Lin, Y.; Xu, S.; Zhuo, Y.; Hua, L.; Wang, J.; et al. Hepatic leptin signaling improves hyperglycemia by stimulating mapk phosphatase-3 protein degradation via STAT3. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 983–1001.
- Kim, G.H.; Szabo, A.; King, E.M.; Ayala, J.; Ayala, J.E.; Altarejos, J.Y. Leptin recruits Creb-regulated transcriptional coactivator 1 to improve hyperglycemia in insulin-deficient diabetes. Mol. Metab. 2014, 4, 227–236.
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdán, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484.
- Zhang, Y.; Cheng, D.; Jie, C.; Liu, T.; Huang, S.; Hu, S. Leptin alleviates endoplasmic reticulum stress induced by cerebral ischemia/reperfusion injury via the PI3K/Akt signaling pathway. Biosci. Rep. 2022, 42, BSR20221443.
- Bjørbaek, C. Central leptin receptor action and resistance in obesity. J. Investig. Med. 2009, 57, 789–794.
- Liu, H.; Du, T.; Li, C.; Yang, G. STAT3 phosphorylation in central leptin resistance. Nutr. Metab. 2021, 18, 39.
- Ren, D.; Zhou, Y.; Morris, D.; Li, M.; Li, Z.; Rui, L. Neuronal SH2B1 is essential for controlling energy and glucose homeostasis. J. Clin. Investig. 2007, 117, 397–406.
- Saeed, S.; Janjua, Q.M.; Haseeb, A.; Khanam, R.; Durand, E.; Vaillant, E.; Ning, L.; Badreddine, A.; Berberian, L.; Boissel, M.; et al. Rare variant analysis of obesity-associated genes in young adults with severe obesity from a consanguineous population of Pakistan. Diabetes 2022, 71, 694–705.
- Kaushik, S.; Rodriguez-Navarro, J.A.; Arias, E.; Kiffin, R.; Sahu, S.; Schwartz, G.J.; Cuervo, A.M.; Singh, R. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011, 14, 173–183.
- Park, S.; Aintablian, A.; Coupe, B.; Bouret, S.G. The endoplasmic reticulum stress-autophagy pathway controls hypothalamic development and energy balance regulation in leptin-deficient neonates. Nat. Commun. 2020, 11, 1914.
- Boucsein, A.; Kamstra, K.; Tups, A. Central signalling cross-talk between insulin and leptin in glucose and energy homeostasis. J. Neuroendocrinol. 2021, 33, e12944.
- Ye, L.; Jia, G.; Li, Y.; Wang, Y.; Chen, H.; Yu, L.; Wu, D. C1q/TNF-related protein 4 restores leptin sensitivity by downregulating NF-κB signaling and microglial activation. J. Neuroinflamm. 2021, 18, 159.
- Crujeiras, A.B.; Carreira, M.C.; Cabia, B.; Andrade, S.; Amil, M.; Casanueva, F.F. Leptin resistance in obesity: An epigenetic landscape. Life Sci. 2015, 140, 57–63.
- Hsuchou, H.; Kastin, A.J.; Tu, H.; Joan Abbott, N.; Couraud, P.O.; Pan, W. Role of astrocytic leptin receptor subtypes on leptin permeation across hCMEC/D3 human brain endothelial cells. J. Neurochem. 2010, 115, 1288–1298.
- Banks, W.A.; Coon, A.B.; Robinson, S.M.; Moinuddin, A.; Shultz, J.M.; Nakaoke, R.; Morley, J.E. Triglycerides induce leptin resistance at the blood-brain barrier. Diabetes 2004, 53, 1253–1260.
- Yoo, S.; Cha, D.; Kim, D.W.; Hoang, T.V.; Blackshaw, S. Tanycyte-independent control of hypothalamic leptin signaling. Front. Neurosci. 2019, 13, 240.
- Shi, Y.; Kim, H.; Hamann, C.A.; Rhea, E.M.; Brunger, J.M.; Lippmann, E.S. Nuclear receptor ligand screening in an iPSC-derived in vitro blood-brain barrier model identifies new contributors to leptin transport. Fluids Barriers CNS 2022, 19, 77.
- Nabil, M.; El Demellawy, M.A.; Mahmoud, M.F.; Mahmoud, A.A.A. Prolonged overnutrition with fructose or fat induces metabolic derangements in rats by disrupting the crosstalk between the hypothalamus and periphery: Possible amelioration with fenofibrate. Eur. J. Pharmacol. 2020, 879, 173136.
- Ren, Z.; Liu, Y.; Hong, W.; Pan, X.; Gong, P.; Liu, Q.; Zhou, G.; Qin, S. Conditional knockout of leptin receptor in neural stem cells leads to obesity in mice and affects neuronal differentiation in the hypothalamus early after birth. Mol. Brain. 2020, 13, 109.
- Dhar, M.; Zhu, M.; Impey, S.; Lambert, T.J.; Bland, T.; Karatsoreos, I.N.; Nakazawa, T.; Appleyard, S.M.; Wayman, G.A. Leptin induces hippocampal synaptogenesis via CREB-regulated microRNA-132 suppression of p250GAP. Mol. Endocrinol. 2014, 28, 1073–1087.
- Sahin, G.S.; Dhar, M.; Dillon, C.; Zhu, M.; Shiina, H.; Winters, B.D.; Lambert, T.J.; Impey, S.; Appleyard, S.M.; Wayman, G.A. Leptin stimulates synaptogenesis in hippocampal neurons via KLF4 and SOCS3 inhibition of STAT3 signaling. Mol. Cell. Neurosci. 2020, 106, 103500.
- Bland, T.; Sahin, G.S.; Zhu, M.; Dillon, C.; Impey, S.; Appleyard, S.M.; Wayman, G.A. USP8 deubiquitinates the leptin receptor and is necessary for leptin-mediated synapse formation. Endocrinology 2019, 160, 1982–1998.
- Bland, T.; Zhu, M.; Dillon, C.; Sahin, G.S.; Rodriguez-Llamas, J.L.; Appleyard, S.M.; Wayman, G.A. Leptin controls glutamatergic synaptogenesis and NMDA-receptor trafficking via fyn kinase regulation of NR2B. Endocrinology 2020, 161, bqz030.
- McGregor, G.; Harvey, J. Leptin regulation of synaptic function at hippocampal TA-CA1 and SC-CA1 synapses: Implications for health and disease. Neurochem. Res. 2019, 44, 650–660.
- Luo, X.; McGregor, G.; Irving, A.J.; Harvey, J. Leptin induces a novel form of NMDA receptor-dependent LTP at hippocampal temporoammonic-CA1 synapses. eNeuro 2015, 2, ENEURO.0007-15.2015.
- Ho, P.W.; Ho, J.W.; Liu, H.F.; So, D.H.; Tse, Z.H.; Chan, K.H.; Ramsden, D.B.; Ho, S.L. Mitochondrial neuronal uncoupling proteins: A target for potential disease-modification in Parkinson’s disease. Transl. Neurodegener. 2012, 1, 3.
- Jiang, W.; Zou, W.; Hu, M.; Tian, Q.; Xiao, F.; Li, M.; Zhang, P.; Chen, Y.J.; Jiang, J.M. Hydrogen sulphide attenuates neuronal apoptosis of substantia nigra by re-establishing autophagic flux via promoting leptin signalling in a 6-hydroxydopamine rat model of Parkinson’s disease. Clin. Exp. Pharmacol. Physiol. 2022, 49, 122–133.
- Cheng, Y.; Buchan, M.; Vitanova, K.; Aitken, L.; Gunn-Moore, F.J.; Ramsay, R.R.; Doherty, G. Neuroprotective actions of leptin facilitated through balancing mitochondrial morphology and improving mitochondrial function. J. Neurochem. 2020, 155, 191–206.
- Greco, S.J.; Bryan, K.J.; Sakar, S.; Zhu, X.; Smith, M.A.; Ashford, J.W.; Johnston, J.M.; Tezapsidis, N.; Casadesus, G. Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 1155–1167.
- Perianes-Cachero, A.; Burgos-Ramos, E.; Puebla-Jiménez, L.; Canelles, S.; Frago, L.M.; Hervás-Aguilar, A.; de Frutos, S.; Toledo-Lobo, M.V.; Mela, V.; Viveros, M.P.; et al. Acute up-regulation of the rat brain somatostatin receptor-effector system by leptin is related to activation of insulin signaling and may counteract central leptin actions. Neuroscience 2013, 252, 289–301.
- Aguado-Llera, D.; Canelles, S.; Fernández-Mendívil, C.; Frago, L.M.; Argente, J.; Arilla-Ferreiro, E.; López, M.G.; Barrios, V. Improvement in inflammation is associated with the protective effect of Gly-Pro-Glu and cycloprolylglycine against Aβ-induced depletion of the hippocampal somatostatinergic system. Neuropharmacology 2019, 151, 112–126.
- Gonzalez-Rodriguez, M.; Astillero-Lopez, V.; Villanueva-Anguita, P.; Paya-Rodriguez, M.E.; Flores-Cuadrado, A.; Villar-Conde, S.; Ubeda-Banon, I.; Martinez-Marcos, A.; Saiz-Sanchez, D. Somatostatin and astroglial involvement in the human limbic system in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 8434.
- Tong, J.Q.; Zhang, J.; Hao, M.; Yang, J.; Han, Y.F.; Liu, X.J.; Shi, H.; Wu, M.N.; Liu, Q.S.; Qi, J.S. Leptin attenuates the detrimental effects of β-amyloid on spatial memory and hippocampal later-phase long term potentiation in rats. Horm. Behav. 2015, 73, 125–130.
- Ji, M.; Gong, W.; Wang, S.; Zhang, D.; Chen, B.; Li, X.; Wu, X.; Cui, L.; Feng, Y.; Verkhratsky, A.; et al. Leptin attenuates fear memory by inhibiting astrocytic NLRP3 inflammasome in post-traumatic stress disorder model. Neurochem. Res. 2022.
- Erichsen, J.M.; Fadel, J.R.; Reagan, L.P. Peripheral versus central insulin and leptin resistance: Role in metabolic disorders, cognition, and neuropsychiatric diseases. Neuropharmacology 2022, 203, 108877.
- Weng, Z.; Signore, A.P.; Gao, Y.; Wang, S.; Zhang, F.; Hastings, T.; Yin, X.M.; Chen, J. Leptin protects against 6-hydroxydopamine-induced dopaminergic cell death via mitogen-activated protein kinase signaling. J. Biol. Chem. 2007, 282, 34479–34491.
- Sharma, V.; Singh, T.G.; Kaur, A.; Mannan, A.; Dhiman, S. Brain-derived neurotrophic factor: A novel dynamically regulated therapeutic modulator in neurological disorders. Neurochem. Res. 2022.
- Yin, X.; Wang, M.; Wang, W.; Chen, T.; Song, G.; Niu, Y.; Jiang, Z.; Gao, Z.; Wang, Z. Identification of potential miRNA-mRNA regulatory network contributing to Parkinson’s Disease. Parkinsons Dis. 2022, 2022, 2877728.
- Ho, P.W.; Liu, H.F.; Ho, J.W.; Zhang, W.Y.; Chu, A.C.; Kwok, K.H.; Ge, X.; Chan, K.H.; Ramsden, D.B.; Ho, S.L. Mitochondrial uncoupling protein-2 (UCP2) mediates leptin protection against MPP+ toxicity in neuronal cells. Neurotox. Res. 2010, 17, 332–343.
- Rahnemayan, S.; Mirghafourvand, M.; Fathalizadeh, A.; Faramarzi, E.; Reyhanifard, A.; Mahmoodpoor, A.; Sanaie, S. Leptin levels in patients with Parkinson’s disease: A systematic review and meta-analysis. Clin. Nutr. ESPEN 2021, 41, 104–109.
- Zou, X.; Zhong, L.; Zhu, C.; Zhao, H.; Zhao, F.; Cui, R.; Gao, S.; Li, B. Role of leptin in mood disorder and neurodegenerative disease. Front. Neurosci. 2019, 13, 378.
- Guo, M.; Huang, T.Y.; Garza, J.C.; Chua, S.C.; Lu, X.Y. Selective deletion of leptin receptors in adult hippocampus induces depression-related behaviours. Int. J. Neuropsychopharmacol. 2013, 16, 857–867.
- Milaneschi, Y.; Lamers, F.; Bot, M.; Drent, M.L.; Penninx, B.W. Leptin dysregulation is specifically associated with major depression with atypical features: Evidence for a mechanism connecting obesity and depression. Biol. Psychiatry 2017, 81, 807–814.
- Choi, W.; Kim, J.W.; Kang, H.J.; Kim, H.K.; Kang, H.C.; Lee, J.Y.; Kim, S.W.; Stewart, R.; Kim, J.M. Interactive effects of serum leptin levels and physical comorbidity on the pharmacotherapeutic response of depressive disorders. Clin. Psychopharmacol. Neurosci. 2022, 20, 662–674.
- Tian, J.; Wang, T.; Jia, K.; Guo, L.; Swerdlow, R.H.; Du, H. Nonobese male patients with Alzheimer’s Disease are vulnerable to decrease in plasma leptin. J. Alzheimers Dis. 2022, 88, 1017–1027.
- Bonda, D.J.; Stone, J.G.; Torres, S.L.; Siedlak, S.L.; Perry, G.; Kryscio, R.; Jicha, G.; Casadesus, G.; Smith, M.A.; Zhu, X.; et al. Dysregulation of leptin signaling in Alzheimer disease: Evidence for neuronal leptin resistance. J. Neurochem. 2014, 128, 162–172.
- Perianes-Cachero, A.; Canelles, S.; Aguado-Llera, D.; Frago, L.M.; Toledo-Lobo, M.V.; Carrera, I.; Cacabelos, R.; Chowen, J.A.; Argente, J.; Arilla-Ferreiro, E.; et al. Reduction in Aβ-induced cell death in the hippocampus of 17β-estradiol-treated female rats is associated with an increase in IGF-I signaling and somatostatinergic tone. J. Neurochem. 2015, 135, 1257–1271.
- Christensen, A.; Liu, J.; Pike, C.J. Aging reduces estradiol protection against neural but not metabolic effects of obesity in female 3xTg-AD mice. Front. Aging Neurosci. 2020, 12, 113.
- Conley, A.C.; Albert, K.M.; McDonald, B.C.; Saykin, A.J.; Dumas, J.A.; Newhouse, P.A. Estradiol treatment in young postmenopausal women with self-reported cognitive complaints: Effects on cholinergic-mediated cognitive performance. Hum. Psychopharmacol. 2022, 37, e2838.
- Cecon, E.; Lhomme, T.; Maurice, T.; Luka, M.; Chen, M.; Silva, A.; Wauman, J.; Zabeau, L.; Tavernier, J.; Prévot, V.; et al. Amyloid beta peptide is an endogenous negative allosteric modulator of leptin receptor. Neuroendocrinology 2021, 111, 370–387.
- Calió, M.L.; Mosini, A.C.; Marinho, D.S.; Salles, G.N.; Massinhani, F.H.; Ko, G.M.; Porcionatto, M.A. Leptin enhances adult neurogenesis and reduces pathological features in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2021, 148, 105219.
- Lai, K.S.P.; Liu, C.S.; Rau, A.; Lanctôt, K.L.; Köhler, C.A.; Pakosh, M.; Carvalho, A.F.; Herrmann, N. Peripheral inflammatory markers in Alzheimer’s disease: A systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 876–882.
- Mejido, D.C.P.; Peny, J.A.; Vieira, M.N.N.; Ferreira, S.T.; De Felice, F.G. Insulin and leptin as potential cognitive enhancers in metabolic disorders and Alzheimer’s disease. Neuropharmacology 2020, 171, 108115.
- Corrigan, R.R.; Piontkivska, H.; Casadesus, G. Amylin pharmacology in Alzheimer’s disease pathogenesis and treatment. Curr. Neuropharmacol. 2022, 20, 1894–1907.
- Menendez, A.; Wanczyk, H.; Walker, J.; Zhou, B.; Santos, M.; Finck, C. Obesity and adipose tissue dysfunction: From pediatrics to adults. Genes 2022, 13, 1866.
- Mahmoud, R.; Kimonis, V.; Butler, M.G. Genetics of obesity in humans: A clinical review. Int. J. Mol. Sci. 2022, 23, 11005.
- Baxter, J.; Armijo, P.R.; Flores, L.; Krause, C.; Samreen, S.; Tanner, T. Updates on monogenic obesity in a multifactorial disease. Obes. Surg. 2019, 29, 4077–4083.
- Funcke, J.B.; von Schnurbein, J.; Lennerz, B.; Lahr, G.; Debatin, K.M.; Fischer-Posovszky, P.; Wabitsch, M. Monogenic forms of childhood obesity due to mutations in the leptin gene. Mol. Cell. Pediatr. 2014, 1, 3.
- Farooqi, I.S.; Wangensteen, T.; Collins, S.; Kimber, W.; Matarese, G.; Keogh, J.M.; Lank, E.; Bottomley, B.; Lopez-Fernandez, J.; Ferraz-Amaro, I.; et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N. Engl. J. Med. 2007, 356, 237–247.
- Chaves, C.; Kay, T.; Anselmo, J. Early onset obesity due to a mutation in the human leptin receptor gene. Endocrinol. Diabetes Metab. Case Rep. 2022, 2022, 21-0124.
- Yang, J.; Wang, M.; Tobias, D.K.; Rich-Edwards, J.W.; Darling, A.M.; Abioye, A.I.; Pembe, A.B.; Madzorera, I.; Fawzi, W.W. Gestational weight gain during the second and third trimesters and adverse pregnancy outcomes, results from a prospective pregnancy cohort in urban Tanzania. Reprod. Health 2022, 19, 140.
- Lin, X.H.; Gao, L.; Tian, S.; Klausen, C.; Guo, M.X.; Gao, Q.; Liu, M.E.; Wang, H.; Wu, D.D.; Zhou, C.L.; et al. Maternal high-fat-diet exposure is associated with elevated blood pressure and sustained increased leptin levels through epigenetic memory in offspring. Sci. Rep. 2021, 11, 316.
- Zwamborn, R.A.; Slieker, R.C.; Mulder, P.C.; Zoetemelk, I.; Verschuren, L.; Suchiman, H.E.; Toet, K.H.; Droog, S.; Slagboom, P.E.; Kooistra, T.; et al. Prolonged high-fat diet induces gradual and fat depot-specific DNA methylation changes in adult mice. Sci. Rep. 2017, 7, 43261.
- Harvey, N.C.; Sheppard, A.; Godfrey, K.M.; McLean, C.; Garratt, E.; Ntani, G.; Davies, L.; Murray, R.; Inskip, H.M.; Gluckman, P.D.; et al. Childhood bone mineral content is associated with methylation status of the RXRA promoter at birth. J. Bone Miner. Res. 2014, 29, 600–607.
- Huang, R.C.; Melton, P.E.; Burton, M.A.; Beilin, L.J.; Clarke-Harris, R.; Cook, E.; Godfrey, K.M.; Burdge, G.C.; Mori, T.A.; Anderson, D.; et al. Adiposity associated DNA methylation signatures in adolescents are related to leptin and perinatal factors. Epigenetics 2022, 17, 819–836.
- Mansell, T.; Ponsonby, A.L.; Collier, F.; Burgner, D.; Pezic, A.; Vuillermin, P.; Ryan, J.; Saffery, R. Methylation of the LEP gene promoter in blood at 12 months and BMI at 4 years of age-a population-based cohort study. Int. J. Obes. 2020, 44, 842–847.
- Mazzucco, M.B.; Higa, R.; Capobianco, E.; Kurtz, M.; Jawerbaum, A.; White, V. Saturated fat-rich diet increases fetal lipids and modulates LPL and leptin receptor expression in rat placentas. J. Endocrinol. 2013, 217, 303–315.
- Nogues, P.; Dos Santos, E.; Jammes, H.; Berveiller, P.; Arnould, L.; Vialard, F.; Dieudonné, M.N. Maternal obesity influences expression and DNA methylation of the adiponectin and leptin systems in human third-trimester placenta. Clin. Epigenetics 2019, 11, 20.
- Eyckerman, S.; Broekaert, D.; Verhee, A.; Vandekerckhove, J.; Tavernier, J. Identification of the Y985 and Y1077 motifs as SOCS3 recruitment sites in the murine leptin receptor. FEBS Lett. 2000, 486, 33–37.
- McEwen, H.J.; Inglis, M.A.; Quennell, J.H.; Grattan, D.R.; Anderson, G.M. Deletion of suppressor of cytokine signaling 3 from forebrain neurons delays infertility and onset of hypothalamic leptin resistance in response to a high caloric diet. J. Neurosci. 2016, 36, 7142–7153.
- Ancel, C.M.; Evans, M.C.; Kerbus, R.I.; Wallace, E.G.; Anderson, G.M. Deletion of PTP1B from brain neurons partly protects mice from diet-induced obesity and minimally improves fertility. Endocrinology 2022, 163, bqab266.
- Hsu, Y.H.; Wu, C.H.; Chiu, C.J.; Chen, W.T.; Chang, Y.C.; Wabitsch, M.; Chang, M.S. IL-20 is involved in obesity by modulation of adipogenesis and macrophage dysregulation. Immunology 2021, 164, 817–833.
- Cakir, I.; Nillni, E.A. Endoplasmic reticulum stress, the hypothalamus, and energy balance. Trends Endocrinol. Metab. 2019, 30, 163–176.
- Barrios, V.; Campillo-Calatayud, A.; Guerra-Cantera, S.; Canelles, S.; Martín-Rivada, Á.; Frago, L.M.; Chowen, J.A.; Argente, J. Opposite effects of chronic central leptin infusion on activation of insulin signaling pathways in adipose tissue and liver are related to changes in the inflammatory environment. Biomolecules 2021, 11, 1734.
- Ahi, E.P.; Brunel, M.; Tsakoumis, E.; Chen, J.; Schmitz, M. Appetite regulating genes in zebrafish gut; a gene expression study. PLoS ONE 2022, 17, e0255201.
- Roujeau, C.; Jockers, R.; Dam, J. Endospanin 1 determines the balance of leptin-regulated hypothalamic functions. Neuroendocrinology 2019, 108, 132–141.
- Vauthier, V.; Swartz, T.D.; Chen, P.; Roujeau, C.; Pagnon, M.; Mallet, J.; Sarkis, C.; Jockers, R.; Dam, J. Endospanin 1 silencing in the hypothalamic arcuate nucleus contributes to sustained weight loss of high fat diet obese mice. Gene Ther. 2014, 21, 638–644.
- Li, Z.; Ceccarini, G.; Eisenstein, M.; Tan, K.; Friedman, J.M. Phenotypic effects of an induced mutation of the ObRa isoform of the leptin receptor. Mol. Metab. 2013, 2, 364–375.
- Guillebaud, F.; Duquenne, M.; Djelloul, M.; Pierre, C.; Poirot, K.; Roussel, G.; Riad, S.; Lanfray, D.; Morin, F.; Jean, A.; et al. Glial endozepines reverse high-fat diet-induced obesity by enhancing hypothalamic response to peripheral leptin. Mol. Neurobiol. 2020, 57, 3307–3333.
- Stürzebecher, P.E.; Kralisch, S.; Schubert, M.R.; Filipova, V.; Hoffmann, A.; Oliveira, F.; Sheikh, B.N.; Blüher, M.; Kogel, A.; Scholz, M.; et al. Leptin treatment has vasculo-protective effects in lipodystrophic mice. Proc. Natl. Acad. Sci. USA 2022, 119, e2110374119.
- Mainieri, F.; Tagi, V.M.; Chiarelli, F. Treatment options for lipodystrophy in children. Front. Endocrinol. 2022, 13, 879979.
- Moon, H.S.; Matarese, G.; Brennan, A.M.; Chamberland, J.P.; Liu, X.; Fiorenza, C.G.; Mylvaganam, G.H.; Abanni, L.; Carbone, F.; Williams, C.J.; et al. Efficacy of metreleptin in obese patients with type 2 diabetes: Cellular and molecular pathways underlying leptin tolerance. Diabetes 2011, 60, 1647–1656.
- Crépin, D.; Benomar, Y.; Riffault, L.; Amine, H.; Gertler, A.; Taouis, M. The over-expression of miR-200a in the hypothalamus of ob/ob mice is linked to leptin and insulin signaling impairment. Mol. Cell. Endocrinol. 2014, 384, 1–11.
- Zhang, N.; Zhang, N.; Song, L.; Xie, H.; Zhao, C.; Li, S.; Zhao, W.; Zhao, Y.; Gao, C.; Xu, G. Adipokines and free fatty acids regulate insulin sensitivity by increasing microRNA-21 expression in human mature adipocytes. Mol. Med. Rep. 2017, 16, 2254–2258.
- Kim, Y.J.; Hwang, S.J.; Bae, Y.C.; Jung, J.S. MiR-21 regulates adipogenic differentiation through the modulation of TGF-beta signaling in mesenchymal stem cells derived from human adipose tissue. Stem Cells 2009, 27, 3093–3102.
- Kim, Y.J.; Hwang, S.H.; Cho, H.H.; Shin, K.K.; Bae, Y.C.; Jung, J.S. MicroRNA 21 regulates the proliferation of human adipose tissue derived mesenchymal stem cells and high-fat diet-induced obesity alters microRNA 21 expression in white adipose tissues. J. Cell. Physiol. 2012, 227, 183–193.
- Dong, T. Anticancer activities of PPARγ in breast cancer are context dependent. Am. J. Pathol. 2013, 182, 1–4.
- Yagai, T.; Yan, T.; Luo, Y.; Takahashi, S.; Aibara, D.; Kim, D.; Brocker, C.N.; Levi, M.; Motohashi, H.; Gonzalez, F.J. Feedback repression of PPARα signaling by Let-7 microRNA. Cell. Rep. 2021, 36, 109506.
- Kollari, E.; Zografou, I.; Sampanis, C.; Athyros, V.G.; Didangelos, T.; Mantzoros, C.S.; Karagiannis, A. Serum adipokine levels in patients with type 1 diabetes are associated with degree of obesity but only resistin is independently associated with atherosclerosis markers. Hormones 2022, 21, 91–101.
- Szalecki, M.; Pańkowska, E.; Wysocka-Mincewicz, M.; Klupa, T.; Janas, R. Leptin and soluble leptin receptor in children with type 1 diabetes mellitus. Pediatr. Endocrinol. Diabetes Metab. 2010, 16, 262–269.
- Gamarra, J.R.; Haeusler, R.A. Hepatocentric leptin signaling modulates gluconeogenesis via MKP-3. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 1166–1167.
- Denroche, H.C.; Levi, J.; Wideman, R.D.; Sequeira, R.M.; Huynh, F.K.; Covey, S.D.; Kieffer, T.J. Leptin therapy reverses hyperglycemia in mice with streptozotocin-induced diabetes, independent of hepatic leptin signaling. Diabetes 2011, 60, 1414–1423.
- Ebihara, K.; Nakao, K. Translational research of leptin in lipodystrophy and its related diseases. In Innovative Medicine: Basic Research and Development ; Nakao, K., Minato, N., Uemoto, S., Eds.; Springer: Tokyo, Japan, 2015.
- Ito, Y.; Sun, R.; Yagimuma, H.; Taki, K.; Mizoguchi, A.; Kobayashi, T.; Sugiyama, M.; Onoue, T.; Tsunekawa, T.; Takagi, H.; et al. Protein tyrosine phosphatase 1B deficiency improves glucose homeostasis in type 1 diabetes treated with leptin. Diabetes 2022, 71, 1902–1914.
- Xu, Y.; Chang, J.T.; Myers, M.G., Jr.; Xu, Y.; Tong, Q. Euglycemia restoration by central leptin in type 1 diabetes requires STAT3 signaling but not fast-acting neurotransmitter release. Diabetes 2016, 65, 1040–1049.
- Fan, S.; Xu, Y.; Lu, Y.; Jiang, Z.; Li, H.; Morrill, J.C.; Cai, J.; Wu, Q.; Xu, Y.; Xue, M.; et al. A neural basis for brain leptin action on reducing type 1 diabetic hyperglycemia. Nat. Commun. 2021, 12, 2662.
- Bonnefond, A.; Semple, R.K. Achievements, prospects and challenges in precision care for monogenic insulin-deficient and insulin-resistant diabetes. Diabetologia 2022, 65, 1782–1795.
- Perry, R.J.; Petersen, K.F.; Shulman, G.I. Pleotropic effects of leptin to reverse insulin resistance and diabetic ketoacidosis. Diabetologia 2016, 59, 933–937.
- Zhang, Q.; Wang, Y.; Huang, E.S. Changes in racial/ethnic disparities in the prevalence of Type 2 diabetes by obesity level among US adults. Ethn. Health 2009, 14, 439–457.
- Jais, A.; Brüning, J.C. Arcuate nucleus-dependent regulation of metabolism-pathways to obesity and diabetes mellitus. Endocr. Rev. 2022, 43, 314–328.
- Agrawal, R.; Reno, C.M.; Sharma, S.; Christensen, C.; Huang, Y.; Fisher, S.J. Insulin action in the brain regulates both central and peripheral functions. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E156–E163.
- Hayden, M.R.; Banks, W.A. Deficient leptin cellular signaling plays a key role in brain ultrastructural remodeling in obesity and type 2 diabetes mellitus. Int. J. Mol. Sci. 2021, 22, 5427.
- Wang, A.N.; Carlos, J.; Fraser, G.M.; McGuire, J.J. Zucker Diabetic-Sprague Dawley (ZDSD) rat: Type 2 diabetes translational research model. Exp. Physiol. 2022, 107, 265–282.
- Suckow, M.A.; Gobbett, T.A.; Peterson, R.G. Wound healing delay in the ZDSD rat. In Vivo 2017, 31, 55–60.
- Zhang, Y.; Lin, C.; Chen, R.; Luo, L.; Huang, J.; Liu, H.; Chen, W.; Xu, J.; Yu, H.; Ding, Y. Association analysis of SOCS3, JAK2 and STAT3 gene polymorphisms and genetic susceptibility to type 2 diabetes mellitus in Chinese population. Diabetol. Metab. Syndr. 2022, 14, 4.
- van Doorn, M.; Kemme, M.; Ouwens, M.; van Hoogdalem, E.J.; Jones, R.; Romijn, H.; de Kam, M.; Schoemaker, R.; Burggraaf, K.; Cohen, A. Evaluation of proinflammatory cytokines and inflammation markers as biomarkers for the action of thiazolidinediones in Type 2 diabetes mellitus patients and healthy volunteers. Br. J. Clin. Pharmacol. 2006, 62, 391–402.
- Li, Y.; Chen, S.; Zhao, T.; Li, M. Serum IL-36 cytokines levels in type 2 diabetes mellitus patients and their association with obesity, insulin resistance, and inflammation. J. Clin. Lab. Anal. 2021, 35, e23611.
- Yang, M.; Tian, M.; Zhang, X.; Xu, J.; Yang, B.; Yu, J.; Li, F.; Li, Y.; Li, S.; Li, X. Role of the JAK2/STAT3 signaling pathway in the pathogenesis of type 2 diabetes mellitus with macrovascular complications. Oncotarget 2017, 8, 96958–96969.
- Maskarinec, G.; Fontaine, A.; Torfadottir, J.E.; Lipscombe, L.L.; Lega, I.C.; Figueroa, J.; Wild, S. The relation of type 2 diabetes and breast cancer incidence in Asian, Hispanic and African American populations—A review. Can. J. Diabetes 2018, 42, 100–105.
- Karra, P.; Winn, M.; Pauleck, S.; Bulsiewicz-Jacobsen, A.; Peterson, L.; Coletta, A.; Doherty, J.; Ulrich, C.M.; Summers, S.A.; Gunter, M.; et al. Metabolic dysfunction and obesity-related cancer: Beyond obesity and metabolic syndrome. Obesity 2022, 30, 1323–1334.
- Rossi, M.; Turati, F.; Lagiou, P.; Trichopoulos, D.; Augustin, L.S.; La Vecchia, C.; Trichopoulou, A. Mediterranean diet and glycaemic load in relation to incidence of type 2 diabetes: Results from the Greek cohort of the population-based European Prospective Investigation into Cancer and Nutrition (EPIC). Diabetologia 2013, 56, 2405–2413.
- Xu, L.; Li, Y.; Yin, L.; Qi, Y.; Sun, H.; Sun, P.; Xu, M.; Tang, Z.; Peng, J. miR-125a-5p ameliorates hepatic glycolipid metabolism disorder in type 2 diabetes mellitus through targeting of STAT3. Theranostics 2018, 8, 5593–5609.
- Arner, P.; Kulyte, A. MicroRNA regulatory networks in human adipose tissue and obesity. Nat. Rev. Endocrinol. 2015, 11, 276–288.
- Zhu, H.; Shyh-Chang, N.; Segrè, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011, 147, 81–94.
- Al Saqri, A.; Malgundkar, S.H.; Al Kindi, F.; Gupta, I.; Al Moundhri, M.; Tamimi, Y. SOCS3 gene silencing does not occur through methylation and mutations in gastric cancer. Hum. Cell. 2022, 35, 1114–1125.
- Xu, G.; Ji, C.; Shi, C.; Fu, H.; Zhu, L.; Xu, L.; Xu, L.; Chen, L.; Feng, Y.; Zhao, Y.; et al. Modulation of hsa-mir-26b levels following adipokine stimulation. Mol. Biol. Rep. 2013, 40, 3577–3582.
- Xu, G.; Ji, C.; Song, G.; Zhao, C.; Shi, C.; Song, L.; Chen, L.; Yang, L.; Huang, F.; Pang, L.; et al. MiR-26b modulates insulin sensitivity in adipocytes by interrupting the PTEN/PI3K/AKT pathway. Int. J. Obes. 2015, 39, 1523–1530.
- Sletner, L.; Moen, A.E.F.; Yajnik, C.S.; Lekanova, N.; Sommer, C.; Birkeland, K.I.; Jenum, A.K.; Böttcher, Y. Maternal glucose and LDL-cholesterol levels are related to placental leptin gene methylation, and, together with nutritional factors, largely explain a higher methylation level among ethnic South Asians. Front. Endocrinol. 2021, 12, 809916.
- Fanelli, G.; Mota, N.R.; Salas-Salvadó, J.; Bulló, M.; Fernandez-Aranda, F.; Camacho-Barcia, L.; Testa, G.; Jiménez-Murcia, S.; Bertaina-Anglade, V.; Franke, B.; et al. The link between cognition and somatic conditions related to insulin resistance in the UK Biobank study cohort: A systematic review. Neurosci. Biobehav. Rev. 2022, 143, 104927.
- Lin, L.; Wang, Y.; Xu, W.; Huang, C.; Hu, J.; Chen, X.; Lv, X.; Qin, Y.; Zhao, X.; Li, H. Aerobic exercise improves type 2 diabetes mellitus-related cognitive impairment by inhibiting JAK2/STAT3 and enhancing AMPK/SIRT1 pathways in mice. Dis. Markers 2022, 2022, 6010504.
- Behl, T.; Gupta, A.; Sehgal, A.; Albarrati, A.; Albratty, M.; Meraya, A.M.; Najmi, A.; Bhatia, S.; Bungau, S. Exploring protein tyrosine phosphatases (PTP) and PTP-1B inhibitors in management of diabetes mellitus. Biomed. Pharmacother. 2022, 153, 113405.
- Salazar, J.; Chávez-Castillo, M.; Rojas, J.; Ortega, A.; Nava, M.; Pérez, J.; Rojas, M.; Espinoza, C.; Chacin, M.; Herazo, Y.; et al. Is “leptin resistance” another key resistance to manage type 2 diabetes? Curr. Diabetes Rev. 2020, 16, 733–749.
- O’Brien, S.N.; Welter, B.H.; Price, T.M. Presence of leptin in breast cell lines and breast tumors. Biochem. Biophys. Res. Commun. 1999, 259, 695–698.
- Yuan, S.S.; Tsai, K.B.; Chung, Y.F.; Chan, T.F.; Yeh, Y.T.; Tsai, L.Y.; Su, J.H. Aberrant expression and possible involvement of the leptin receptor in endometrial cancer. Gynecol. Oncol. 2004, 92, 769–775.
- Sharma, D.; Saxena, N.K.; Vertino, P.M.; Anania, F.A. Leptin promotes the proliferative response and invasiveness in human endometrial cancer cells by activating multiple signal-transduction pathways. Endocr. Relat. Cancer 2006, 13, 629–640.
- Yeung, C.Y.; Tso, A.W.; Xu, A.; Wang, Y.; Woo, Y.C.; Lam, T.H.; Lo, S.V.; Fong, C.H.; Wat, N.M.; Woo, J.; et al. Pro-inflammatory adipokines as predictors of incident cancers in a Chinese cohort of low obesity prevalence in Hong Kong. PLoS ONE 2013, 8, e78594.
- Acedo, S.C.; Gambero, S.; Pereira Cunha, F.G.; Lorand-Metze, I.; Gambero, A. Participation of leptin in the determination of the macrophage phenotype: An additional role in adipocyte and macrophage crosstalk. In Vitro Cell. Dev. Biol. Anim. 2013, 49, 473–478.
- Tiwari, P.; Blank, A.; Cui, C.; Schoenfelt, K.Q.; Zhou, G.; Xu, Y.; Khramtsova, G.; Olopade, F.; Shah, A.M.; Khan, S.A.; et al. Metabolically activated adipose tissue macrophages link obesity to triple-negative breast cancer. J. Exp. Med. 2019, 216, 1345–1358.
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; Sánchez-Margalet, V. Role of leptin in inflammation and vice versa. Int. J. Mol. Sci. 2020, 21, 5887.
- García-Miranda, A.; Garcia-Hernandez, A.; Castañeda-Saucedo, E.; Navarro-Tito, N.; Maycotte, P. Adipokines as regulators of autophagy in obesity-linked cancer. Cells 2022, 11, 3230.
- Lipsey, C.C.; Harbuzariu, A.; Daley-Brown, D.; Gonzalez-Perez, R.R. Oncogenic role of leptin and Notch interleukin-1 leptin crosstalk outcome in cancer. World, J. Methodol. 2016, 6, 43–55.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
14 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No