Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Aashish Soni | -- | 1864 | 2022-12-02 23:17:47 | | | |
| 2 | Catherine Yang | + 1 word(s) | 1865 | 2022-12-05 04:46:50 | | | | |
| 3 | Catherine Yang | -2 word(s) | 1863 | 2022-12-05 04:48:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Soni, A.; Lin, X.; Mladenov, E.; Mladenova, V.; Stuschke, M.; Iliakis, G. BMN673 Is a PARP Inhibitor. Encyclopedia. Available online: https://encyclopedia.pub/entry/37887 (accessed on 03 July 2026).
Soni A, Lin X, Mladenov E, Mladenova V, Stuschke M, Iliakis G. BMN673 Is a PARP Inhibitor. Encyclopedia. Available at: https://encyclopedia.pub/entry/37887. Accessed July 03, 2026.
Soni, Aashish, Xixi Lin, Emil Mladenov, Veronika Mladenova, Martin Stuschke, George Iliakis. "BMN673 Is a PARP Inhibitor" Encyclopedia, https://encyclopedia.pub/entry/37887 (accessed July 03, 2026).
Soni, A., Lin, X., Mladenov, E., Mladenova, V., Stuschke, M., & Iliakis, G. (2022, December 02). BMN673 Is a PARP Inhibitor. In Encyclopedia. https://encyclopedia.pub/entry/37887
Soni, Aashish, et al. "BMN673 Is a PARP Inhibitor." Encyclopedia. Web. 02 December, 2022.
Copy Citation
Talazoparib (BMN673) is a relatively new PARP inhibitor (PARPi) that exhibits superior efficacy in vitro compared to olaparib and other clinically relevant PARPi. BMN673, similar to most clinical PARPi, inhibits the catalytic activities of PARP-1 and PARP-2 and shows impressive anticancer potential as monotherapy in several pre-clinical and clinical studies. Tumor resistance to PARPi poses a significant challenge in the clinic and combining BMN673 with RT may offer a promising treatment modality.
PARP inhibitor
BMN673
talazoparib
olaparib
radiotherapy
1. Introduction
Radiotherapy (RT) and chemotherapy are two main pillars of cancer treatment. As the primary treatment modality, RT contributes to about 40% of curative treatments for cancer [1][2]. Therapeutically exploiting DNA repair offers a potent means to target cancer cells [3][4]. PARP inhibitors (PARPi) are widely used chemotherapeutic agents in homologous recombination (HR) deficient tumors. The seminal finding of a synthetic lethal interaction between PARP-1 and HR defects [5][6] accelerated the development of highly specific, clinically relevant PARPi, including olaparib (AZD-2281), niraparib (MK-4827), veliparib (ABT888), rucaparib (AG-014699), and talazoparib (BMN673) [7][8][9]. Such clinical PARPi exclusively target PARP-1 and PARP-2 to execute their anti-cancer effects through a synthetically lethal interaction with HR defects. The idea of synthetic lethal interactions between the PARPi and HR defects has been exploited as an Achilles heel in the management of ovarian and breast cancers and has been extensively reviewed in the past [10][11][12][13][14][15][16][17].
The PARP family of proteins regulates several biochemical processes in a living cell by mediating a post-translational modification called poly- or mono-ADP-ribosylation (PARylation or MARylation). Where PARP-1, 2, 5a, and 5b are capable of modifying their targets through PARylation, the remaining PARP family members modify their targets through MARylation [18]. Among 17 reported members of the PARP family [19], only PARP-1, PARP-2, and PARP-3 were found to regulate DNA damage response and repair (Figure 1). However, the main target of PARPi is PARP-1, which regulates DSB repair through PARylation of various signaling proteins, including itself. PARP-1 is the critical protein involved in single-strand break repair (SSB) and alternative end joining (alt-EJ) [20]. Multiple studies extend the primary functions of PARP-1 in DNA repair and suggest a broader role for PARP-1 and PARylation in the complex network of multiple DNA repair mechanisms [21][22]. This has highlighted PARP-1 as a central molecule coordinating the overall cellular responses to genotoxic stress in general and DNA damage, in particular (DDR) [23].

Figure 1. Domain structure of human PARP-1, PARP-2, and PARP-3 proteins. The domain locations within the full-length proteins were extracted from the uniport database by using the following entries for PARP-1 (P09874), PARP-2 (Q9UGN5), and PARP-3 (Q9Y6F1). Main protein domains involved in DNA-binding, auto-modification, and catalytic functions are indicated. ZnFI: zinc finger I domain, ZnFII: zinc finger II domain, ZnFIII: zinc finger III domain, BRCT: BRCA1 C-terminal domain, WGR: Trp-Gly-Arg domain, HD: Helical domain, ART: (ADP-ribosyl) transferase domain. ZnFI and ZnFII help to recognize and bind to DNA damage sites, while ZnFIII activates the enzyme upon DNA binding. The BRCT domain of PARP-1 is required to localize XRCC1 and XRCC1-complexed DNA repair factors to DNA damage sites.
Tumors lacking HR and therefore therapeutically treated with PARPi, frequently acquire resistance when treated for a prolonged period with such inhibitors [24]. The prime mechanisms behind development of PARPi resistance in tumors are restoring HR activity, losing PARP-1 activity, or upregulating alt-EJ repair functions [24]. Combining PARPi with other chemotherapeutics or RT may help in preventing or overcoming PARPi resistance, increasing thus the tumor response to therapy.
2. BMN673 Is a Superior Radiosensitizer
A few pre-clinical studies have shown the potential of BMN673 in combination with RT on multiple cancer cell lines and xenografts. BMN673 was shown to inhibit the proliferation of glioblastoma stem cells after exposure to low or high linear energy transfer radiations [25]. In 2018, the researhcers were the first to report the strong radiosensitizing potential of BMN673 (50 nM) in a battery of cancer cell lines originating from Rhabdoid tumors, Sarcomas, lung carcinomas, osteosarcomas, and colon cancer [26]. Only a short drug treatment time (~1 h) prior to RT was sufficient to achieve robust radiosensitization. Under similar experimental conditions, olaparib (3 µM), AG14361 (0.4 µM), and PJ34 (5 µM) showed only moderate radiosensitization. A potent radiosensitizing effect of BMN673 (200 nM) but not veliparib (1600 nM) was also reported in small cell lung carcinoma (SCLC) cell lines and Xenografts [27]. The non-small cell lung carcinoma (NSCLS) cell line, H460, was strongly radiosensitized after treatment with 2 nM BMN673, which could be further enhanced by treating cells with 5-Azacytidine [28]. Colorectal cancer cells and xenografts (BRAF wild type and mutant) could also be strongly radiosensitized by BMN673 (125 nM), whereas olaparib (250nM) and rucaparib (250 nM) exerted only a moderate radiosensitizing effect [29]. Highly radio- and chemotherapy-resistant chondrosarcoma cells were also shown to be strongly radiosensitized by BMN673 (5 nM) irrespective of their IDH (isocitrate dehydrogenase) mutation status [30]. Hepatocellular carcinoma cells, HepG2 and PLC/PRF/5, were also robustly radiosensitized with BMN673 (50 nM), whereas veliparib (10 µM) was ineffective [31]. A battery of melanoma cell lines was also strongly radiosensitized by BMN673 (50 nM). In the follow-up study, niraparib could not reach similar levels of radiosensitization, even at a concentration of 2500 nM [32].
One of the prominent features of BMN673, evident in the above-mentioned studies, is that it can be used at low nanomolar concentrations to achieve strong radiosensitization. Such concentrations are achievable in the blood of treated patients also ensuring in vivo radiosensitization [33]. No other clinical PARPi showed such efficacy and similar pharmacological properties, as pointed out earlier in this section. Not surprisingly, therefore, in pre-clinical and clinical settings, BMN673 showed robust antitumor activities at relatively low concentrations. For instance, olaparib was administered at 50 mg/kg, whereas BMN673 was used at a much lower dose of 0.3 mg/kg in pre-clinical studies (8). Similar trends were also noted in the clinical setting (protocols: NCT02032823, NCT02184195, NCT01945775, NCT02326844) [34][35][36]. Importantly, BMN673 as a monotherapy showed a manageable tolerability profile in breast cancer patients who participated in EMBRACA and ABRAZO studies [37][38].
Another critical property of BMN673 is the flexible drug treatment time and administration schedule required to achieve strong radiosensitization, at least in certain cell systems. A one-hour treatment with BMN673 prior to RT exerted equally strong radiosensitization as continuous treatment during the course of incubation for colony formation, and that even administration several hours after IR radiosensitized cells with impressive efficacy [26]. This property, if proven general, will be very useful in the clinic, as it simplifies the effective combination of BMN673 treatment with radiation exposure in the hectic environment of a radiation therapy facility. Such administration-flexibility has not been reported for other radiosensitizers and requires in-depth analysis of its mechanistic underpinnings.
In addition, BMN673 specifically sensitized cancer cells to RT, whereas normal human cell lines were not sensitized [26], again a favorable parameter in the clinic, promising reduced normal tissue toxicity. The mechanistic underpinnings of this interesting observation require further in-depth investigations as well.
3. Speculations on the Mechanism of BMN673-Induced Radiosensitization
Retention of inhibited PARP on DNA lesions (the so-called PARP trapping) has been proposed as a key parameter of PAPRi-induced cytotoxicity underpinning the effects of monotherapy applications. PARPi-mediated inhibition of Parp1 auto-PARylation prevents its removal from DNA lesions and leads to replication fork collapse in S-phase cells, or to the suppression of other aspects of the DNA metabolism [39]. BMN63 has been reported as the most potent inducer of PARP-1 trapping, compared to other clinically relevant PARPi, and this property is often causally correlated to its increased cytotoxicity [39].
When analyzing the effects of PAPRi, it is important to address separately effects on cytotoxicity that is enhanced in HR deficient cells, and effects on radiosensitivity and DSB processing reviewed here, as they may be caused by different mechanisms. Thus, while it is possible that the enhanced trapping ability of BMN673 explains its toxicity in HR deficient cancers [8][11], it may not underpin, in its non-specific form, the notable radiosensitizing potential. It is relevant in this regard that the PARP trapping ability of clinical PARPi has been mostly demonstrated using DNA alkylating agents such as MMS that are likely to generate lesions strongly inhibiting DNA replication [27][39][40]. Similar forms of PARP1 trapping have not been reported for irradiated cells, and indeed the experiments failed to generate unequivocal evidence for trapping.
Can Parp1 trapping explain the above observed effects of BMN673 on DSB processing? The shift of DSB repair from c-NHEJ to HR and alt-EJ that requires extensive processing of DNA ends rules out general Parp1 trapping stopping all processing at the DSB end as a mechanism. Indeed, BMN673 mediated excessive resection and accretion of RPA70 and Rad51 after IR [26] is plausible only when DSB ends are not blocked by trapped PARP-1. Therefore, if Parp1 trapping underpins the above discussed DSB repair effects, it must be happening in a highly specific manner centering on c-NHEJ. The researchers hypothesize that after BMN673 treatment, Parp1 inhibits the binding of Ku to DNA ends. Indeed, there is evidence that Parp1 precedes Ku at the DSB end [41], and it is therefore possible that the engagement of Ku is changed by trapped Parp1. Existing evidence suggests that this effect is observed both at high as well as at low doses of IR. The researchers further hypothesize that the interaction of inhibited Parp1 is highly specific for Ku and that it leaves unchanged, or even facilitates all processes associated with DNA end resection.
It has been shown recently that p97 ATPase/segregase extracts BMN673-induced trapped PARP-1 from chromatin, a process that requires post-translational modification of PARP-1 by PIAS4-mediated SUMOylation and RNF4-mediated ubiquitination [42]. The chromatin remodeler ALC1 (amplified in liver cancer 1) could also remove inactive PARP1 through binding to readily PARylated chromatin prior to PARPi treatment. Moreover, ALC1 is implicated in the recruitment of Rad51, Ku70, RPA and MRE11 to DNA damage sites, thus acting upstream of DSB repair pathway choice after removing trapped PARP [43].
The above evidence in aggregate shows that BMN673 indeed traps Parp1 at the DSBs and provides clues on possible mechanisms to undo this trapping. It leaves however open the mechanism of how BMN673 specifically inhibits c-NHEJ and enhances in a directly compensating manner, resection-dependent DSB processing. The elucidation of the mechanisms underpinning these effects should be given high priority.
Figure 2 summarizes the postulated altered balance in DSB repair utilization after treatment of irradiated cells with BMN673, based on the assumption that it inhibits c-NHEJ. The color zones in the bars show the fraction of DSBs processed by each DSB repair pathway, except SSA that has not been sufficiently analyzed for BMN673 effects. It is drawn on the assumption that only very few DSBs remain unprocessed and that this fraction slightly increases with dose. It is also considering G2-phase cells to include effects on HR.
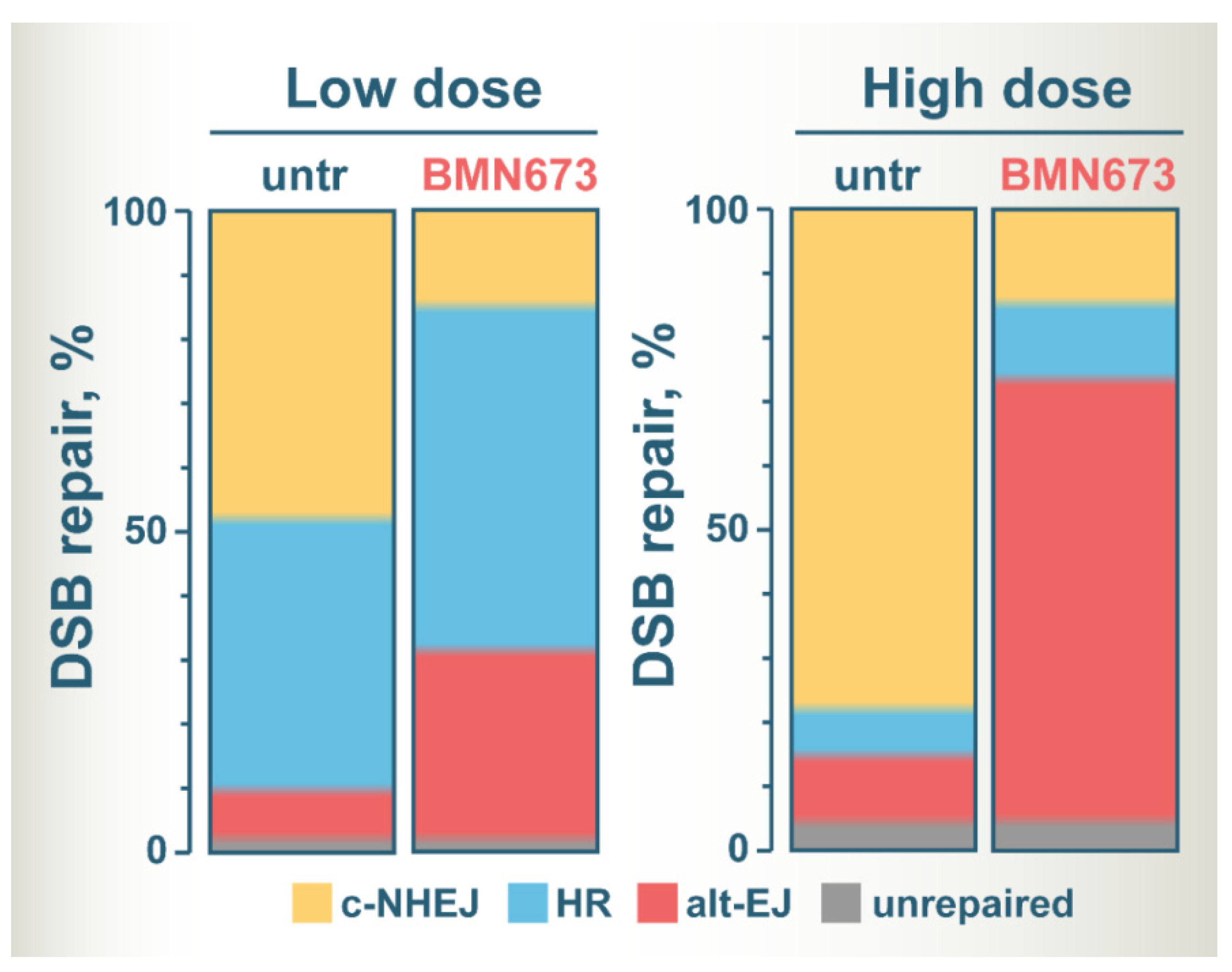
Figure 2. A model of altered DSB repair pathway balance at low and high IR doses in BMN673-treated cells. See text for details.
At low IR doses, c-NHEJ and HR repair similar fractions of DSBs, with alt-EJ having a small contribution [44][45][46]. BMN673 treatment at low IR doses inhibits c-NHEJ and shifts the balance towards HR and to alt-EJ that shows robust increases [26]. At high IR doses, HR is inherently suppressed and the majority of DSBs are processed by c-NHEJ, and to a much lesser extent by alt-EJ [44][46]. BMN673 treatment after exposure to high IR doses also inhibits c-NHEJ, but leaves now the majority of DSBs to be processed by alt-EJ [26].
References
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253.
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and Radiation Therapy: Current Advances and Future Directions. Int. J. Med. Sci. 2012, 9, 193–199.
- Jackson, S.P.; Helleday, T. DNA REPAIR. Drugging DNA repair. Science 2016, 352, 1178–1179.
- Nickoloff, J.A.; Jones, D.; Lee, S.-H.; Williamson, E.A.; Hromas, R. Drugging the Cancers Addicted to DNA Repair. JNCI J. Natl. Cancer Inst. 2017, 109, djx059.
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917.
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921.
- Wang, B.; Chu, D.; Feng, Y.; Shen, Y.; Aoyagi-Scharber, M.; Post, L.E. Discovery and Characterization of (8S,9R)-5-Fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-tetrahydro-3H-pyridophthalazin-3-one (BMN 673, Talazoparib), a Novel, Highly Potent, and Orally Efficacious Poly(ADP-ribose) Polymerase-1/2 Inhibitor, as an Anticancer Agent. J. Med. Chem. 2016, 59, 335–357.
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. BMN 673, a Novel and Highly Potent PARP1/2 Inhibitor for the Treatment of Human Cancers with DNA Repair Deficiency. Clin. Cancer Res. 2013, 19, 5003–5015.
- Hoy, S.M. Talazoparib: First Global Approval. Drugs 2018, 78, 1939–1946.
- Thomas, H. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393.
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158.
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576.
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 1–22.
- Xie, T.; Dickson, K.-A.; Yee, C.; Ma, Y.; Ford, C.E.; Bowden, N.A.; Marsh, D.J. Targeting Homologous Recombination Deficiency in Ovarian Cancer with PARP Inhibitors: Synthetic Lethal Strategies That Impact Overall Survival. Cancers 2022, 14, 4621.
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623.
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2019, 19, 23–38.
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic Lethality and Cancer Therapy: Lessons Learned from the Development of PARP Inhibitors. Annu. Rev. Med. 2015, 66, 455–470.
- Zhu, H.; Tang, Y.-D.; Zhan, G.; Su, C.; Zheng, C. The Critical Role of PARPs in Regulating Innate Immune Responses. Front. Immunol. 2021, 12, 712556.
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424.
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. Mol. Mech. Mutagen. 2011, 711, 61–72.
- Pazzaglia, S.; Pioli, C. Multifaceted Role of PARP-1 in DNA Repair and Inflammation: Pathological and Therapeutic Implications in Cancer and Non-Cancer Diseases. Cells 2019, 9, 41.
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621.
- Hall, M.; Benafif, S. An update on PARP inhibitors for the treatment of cancer. Onco Targets Ther. 2015, 8, 519–528.
- Du, Y.; Yamaguchi, H.; Hsu, J.L.; Hung, M.-C. PARP inhibitors as precision medicine for cancer treatment. Natl. Sci. Rev. 2017, 4, 576–592.
- Lesueur, P.; Chevalier, F.; El-Habr, E.A.; Junier, M.-P.; Chneiweiss, H.; Castera, L.; Müller, E.; Stefan, D.; Saintigny, Y. Radiosensitization Effect of Talazoparib, a Parp Inhibitor, on Glioblastoma Stem Cells Exposed to Low and High Linear Energy Transfer Radiation. Sci. Rep. 2018, 8, 1–12.
- Soni, A.; Li, F.; Wang, Y.; Grabos, M.; Krieger, L.M.; Chaudhary, S.; Hasan, M.S.M.; Ahmed, M.; Coleman, C.N.; Teicher, B.A.; et al. Inhibition of Parp1 by BMN673 Effectively Sensitizes Cells to Radiotherapy by Upsetting the Balance of Repair Pathways Processing DNA Double-Strand Breaks. Mol. Cancer Ther. 2018, 17, 2206–2216.
- Laird, J.H.; Lok, B.H.; Ma, J.; Bell, A.; de Stanchina, E.; Poirier, J.T.; Rudin, C.M. Talazoparib Is a Potent Radiosensitizer in Small Cell Lung Cancer Cell Lines and Xenografts. Clin. Cancer Res. 2018, 24, 5143–5152.
- Abbotts, R.; Topper, M.J.; Biondi, C.; Fontaine, D.; Goswami, R.; Stojanovic, L.; Choi, E.Y.; McLaughlin, L.; Kogan, A.A.; Xia, L.; et al. DNA methyltransferase inhibitors induce a BRCAness phenotype that sensitizes NSCLC to PARP inhibitor and ionizing radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 22609–22618.
- Carter, R.; Cheraghchi-Bashi, A.; Westhorpe, A.; Yu, S.; Shanneik, Y.; Seraia, E.; Ouaret, D.; Inoue, Y.; Koch, C.; Wilding, J.; et al. Identification of anticancer drugs to radiosensitise BRAF-wild-type and mutant colorectal cancer. Cancer Biol. Med. 2019, 16, 234–246.
- Venneker, S.; Kruisselbrink, A.B.; Bruijn, I.H.B.-D.; de Jong, Y.; van Wijnen, A.J.; Danen, E.H.; Bovée, J.V. Inhibition of PARP Sensitizes Chondrosarcoma Cell Lines to Chemo- and Radiotherapy Irrespective of the IDH1 or IDH2 Mutation Status. Cancers 2019, 11, 1918.
- Gerossier, L.; Dubois, A.; Paturel, A.; Fares, N.; Cohen, D.; Merle, P.; Lachuer, J.; Wierinckx, A.; Saintigny, P.; Bancel, B.; et al. PARP inhibitors and radiation potentiate liver cell death in vitro. Do hepatocellular carcinomas have an achilles’ heel? Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101553.
- Jonuscheit, S.; Jost, T.; Gajdošová, F.; Wrobel, M.; Hecht, M.; Fietkau, R.; Distel, L. PARP Inhibitors Talazoparib and Niraparib Sensitize Melanoma Cells to Ionizing Radiation. Genes 2021, 12, 849.
- de Bono, J.S.; Mina, L.A.; Gonzalez, M.; Curtin, N.J.; Wang, E.; Henshaw, J.W.; Chadha, M.; Sachdev, J.C.; Matei, D.; Jameson, G.S.; et al. First-in-human trial of novel oral PARP inhibtior BMN673 in patients with solid tumors. J. Clin. Oncol. 2013, 31, 2580.
- Carney, B.; Kossatz, S.; Lok, B.H.; Schneeberger, V.; Gangangari, K.K.; Pillarsetty, N.V.K.; Weber, W.A.; Rudin, C.M.; Poirier, J.T.; Reiner, T. Target engagement imaging of PARP inhibitors in small-cell lung cancer. Nat. Commun. 2018, 9, 1–13.
- Loap, P.; Loirat, D.; Berger, F.; Ricci, F.; Vincent-Salomon, A.; Ezzili, C.; Mosseri, V.; Fourquet, A.; Ezzalfani, M.; Kirova, Y. Combination of Olaparib and Radiation Therapy for Triple Negative Breast Cancer: Preliminary Results of the RADIOPARP Phase 1 Trial. Int. J. Radiat. Oncol. 2020, 109, 436–440.
- Boussios, S.; Abson, C.; Moschetta, M.; Rassy, E.; Karathanasi, A.; Bhat, T.; Ghumman, F.; Sheriff, M.; Pavlidis, N. Poly (ADP-Ribose) Polymerase Inhibitors: Talazoparib in Ovarian Cancer and Beyond. Drugs RD 2020, 20, 55–73.
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763.
- Ettl, J.; Quek, R.G.W.; Lee, K.H.; Rugo, H.S.; Hurvitz, S.; Gonçalves, A.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Quality of life with talazoparib versus physician’s choice of chemotherapy in patients with advanced breast cancer and germline BRCA1/2 mutation: Patient-reported outcomes from the EMBRACA phase III trial. Ann. Oncol. 2018, 29, 1939–1947.
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443.
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477.
- Yang, G.; Liu, C.; Chen, S.-H.; Kassab, M.A.; Hoff, J.D.; Walter, N.G.; Yu, X. Super-resolution imaging identifies PARP1 and the Ku complex acting as DNA double-strand break sensors. Nucleic Acids Res. 2018, 46, 3446–3457.
- Krastev, D.B.; Li, S.; Sun, Y.; Wicks, A.J.; Hoslett, G.; Weekes, D.; Badder, L.M.; Knight, E.G.; Marlow, R.; Pardo, M.C.; et al. The ubiquitin-dependent ATPase p97 removes cytotoxic trapped PARP1 from chromatin. Nature 2022, 24, 62–73.
- Juhász, S.; Smith, R.; Schauer, T.; Spekhardt, D.; Mamar, H.; Zentout, S.; Chapuis, C.; Huet, S.; Timinszky, G. The chromatin remodeler ALC1 underlies resistance to PARP inhibitor treatment. Sci. Adv. 2020, 6, eabb8626.
- Mladenov, E.; Staudt, C.; Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Iliakis, G. Strong suppression of gene conversion with increasing DNA double-strand break load delimited by 53BP1 and RAD52. Nucleic Acids Res. 2019, 48, 1905–1924.
- Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Pantelias, G.E.; Iliakis, G. Chromosome breaks generated by low doses of ionizing radiation in G2-phase are processed exclusively by gene conversion. DNA Repair 2020, 89, 102828.
- Murmann-Konda, T.; Soni, A.; Stuschke, M.; Iliakis, G. Analysis of chromatid-break-repair detects a homologous recombination to non-homologous end-joining switch with increasing load of DNA double-strand breaks. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2021, 867, 503372.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
744
Revisions:
3 times
(View History)
Update Date:
05 Dec 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No