Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Špela Miroševič | -- | 2149 | 2022-11-30 02:37:00 | | | |
| 2 | Vivi Li | Meta information modification | 2149 | 2022-12-01 02:16:48 | | | | |
| 3 | Vivi Li | + 3 word(s) | 2152 | 2022-12-05 05:12:37 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Miroševič, �.; Khandelwal, S.; Sušjan, P.; Žakelj, N.; Gosar, D.; Forstnerič, V.; Lainšček, D.; Jerala, R.; Osredkar, D. Correlation between Phenotype and Genotype in CTNNB1 Syndrome. Encyclopedia. Available online: https://encyclopedia.pub/entry/37180 (accessed on 28 June 2026).
Miroševič �, Khandelwal S, Sušjan P, Žakelj N, Gosar D, Forstnerič V, et al. Correlation between Phenotype and Genotype in CTNNB1 Syndrome. Encyclopedia. Available at: https://encyclopedia.pub/entry/37180. Accessed June 28, 2026.
Miroševič, Špela, Shivang Khandelwal, Petra Sušjan, Nina Žakelj, David Gosar, Vida Forstnerič, Duško Lainšček, Roman Jerala, Damjan Osredkar. "Correlation between Phenotype and Genotype in CTNNB1 Syndrome" Encyclopedia, https://encyclopedia.pub/entry/37180 (accessed June 28, 2026).
Miroševič, �., Khandelwal, S., Sušjan, P., Žakelj, N., Gosar, D., Forstnerič, V., Lainšček, D., Jerala, R., & Osredkar, D. (2022, November 30). Correlation between Phenotype and Genotype in CTNNB1 Syndrome. In Encyclopedia. https://encyclopedia.pub/entry/37180
Miroševič, Špela, et al. "Correlation between Phenotype and Genotype in CTNNB1 Syndrome." Encyclopedia. Web. 30 November, 2022.
Copy Citation
The CTNNB1 (Cadherin-associated protein, beta 1) Syndrome is a rare neurodevelopmental disorder associated with developmental delay, intellectual disability, and delayed or absent speech. Research showed wide genotypic and phenotypic variability in patients with CTNNB1 Syndrome. The most common moderate-severe phenotype manifested in facial dysmorphisms, microcephaly, various motor disabilities, language and cognitive impairments, and behavioral abnormalities (e.g., autistic-like or aggressive behavior).
beta-catenin
loss of function mutation
intellectual disability
hypotonia
microcephaly
eye movement disorders
1. Introduction
CTNNB1 Syndrome is a severe autosomal dominant neurodevelopmental disorder usually caused by de novo loss-of-function mutations in the CTNNB1 (Cadherin-associated protein, beta 1) gene [1]. CTNNB1 Syndrome manifests itself in a variety of developmental disorders including Neurodevelopmental Disorder with Spastic Diplegia and Visual Defects (NEDSDV), and visual disorders including Familial Exudative Vitreoretinopathy (FEVR). NEDSDV is a neurodevelopmental disorder characterized by global developmental delay, impaired intellectual development with absent or very limited speech, craniofacial anomalies and microencephaly, axial hypotonia, and spasticity [1][2]. FEVR is an autosomal dominant disorder characterized by incomplete development of the retinal vasculature [3]. De novo loss-of-function mutations in the CTNNB1 gene were first discovered in 2012 after diagnostic exome sequencing of individuals with severe intellectual disability [4], and since then the term CTNNB1 Syndrome has become the generic term for all disorders associated with CTNNB1 haploinsufficiency. Currently, this disorder is diagnosed in approximately 300 patients worldwide, although this number is likely an underestimation due to misdiagnosis in cerebral palsy [5][6], leading to efforts to reevaluate the diagnoses of cerebral palsy patients to enable genomics-based changes in their clinical care.
The CTNNB1 gene is located on chromosome 3 (locus 3p22.1, 41240942–41281939). It consists of 16 exons, with exons 2–15 (2346 bp) providing the coding sequence for β-catenin protein. β-catenin protein consists of 781 amino acids and belongs to the armadillo family of structural proteins involved in both embryonic development and adult homeostasis where it plays two essential roles: (1) as a transcriptional co-factor in the Wnt-signaling pathway, and (2) as an anchor in intracellular contacts and cell adhesion [7]. When the Wnt pathway is not stimulated, most of the newly expressed β-catenin is depleted from the cytoplasm by the destruction complex, while the remaining undergraded β-catenin engages with E-cadherin and α-catenin in membrane complexes that serve as cellular anchors. Within the destruction complex, which consists of Axin, Adenomatous Polyposis Coli (APC), and CK140 proteins, β-catenin undergoes a series of consecutive phosphorylations by the glycogen synthase kinase 3β (GSK3β) and CK1 kinases, which ultimately leads to β-catenin ubiquitination by β-TrCP and its proteasomal degradation. In the course of the canonical Wnt signaling pathway, Wnt ligands bind the membrane Frizzled family receptor that stimulates Dishevelled protein to sequester destruction complex proteins [8]. In this way, the degradation of β-catenin is inhibited, allowing the accumulation of free β-catenin which, transported to the nucleus, assists the T-cell factor/lymphoid enhancer factor (TCF-LEF) family of transcription factors in the transcription of various developmental genes, such as axin 1 and cyclin D. Structural and signaling roles of β-catenin are mutually exclusive, which is reflected in its protein structure. β-catenin consists of three regions with a distinct charge distribution: (1) an unstructured N-terminal region (130 amino acids), bearing amino-acid residues important for β-catenin degradation (S33, S37, Y41, S45); (2) a highly conserved central core region (550 amino acids) consisting of 12 armadillo repeats (each is a 42 amino-acid triple helix) [9][10] that form a positively charged groove [9], where β-catenin interacts with more than 20 protein partners including E-cadherin, TCF and degradation complex proteins [10][11][12]; and (3), the unstructured C-terminal region (100 amino acids), which is believed to enhance β-catenin stability by shielding the N-terminus from the destruction complex [9][10][13]. The molecular mechanism of binding exclusivity for the various β-catenin partners remains elusive—it is thought that the occlusion of ligand binding may be achieved by back-folding of termini.
Given the low prevalence of CTNNB1 Syndrome and its relatively recent discovery, little is known about the effect of CTNNB1 mutation type and exonic localization on the severity of clinical phenotypes. It is also not clear whether CTNNB1 mutations are null (in which case the mutated transcripts undergo nonsense-mediated RNA decay—NMD [14], or are translated into non-functional proteins) or, on the other hand, cause a partial loss of protein function due to the presence of an incompletely functioning protein. Another type of mutation can lead to the expression of proteins that interfere with the normal function of the protein from the wild type allele. These so-called antimorphic or dominant-negative mutations (mutated transcripts escape NMD and translate into truncated variants with potentially deleterious effects on the function of the healthy allele) are rare; however, given the variability of CTNNB1 Syndrome-associated mutations in terms of type and location, a production of auto-inhibitory truncated variants cannot be ruled out.
De novo mutations of the CTNNB1 gene have been associated with neurodevelopmental disorders, with cases of intellectual disability and speech delay [4]. Addressing these open questions through phenotype–genotype correlation studies is essential in order to develop targeted interventions and focused clinical care, specific to the mutational context in the affected individuals [15]. The availability of data for such studies has been aided by genomic microarray technology, which has tremendously changed diagnostic approaches in children with neurodevelopmental disorders. Genetic testing can identify the genetic etiology in approximately 40% of cases of cerebral palsy (CP) cases, particularly those diagnosed with autism spectrum disorders (ASD) and intellectual disability (ID) with no apparent causative factor related to CP [5][16]. Access to a large number of patients who have been reliably and systematically assessed is fundamental for understanding CTNNB1 Syndrome. Researchers analyze the prevalence of clinical manifestations, and classify mutations according to their type (missense/nonsense/frameshift/splicing), exonic location, associated clinical features, and disease severity. Based on the analysis of the collected data, the genotype–phenotype correlations for CTNNB1 Syndrome are explored in detail. These may serve as a classification standard for new case studies and as a reference for researchers working to develop personalized therapeutic approaches.
2. Prevalence of Clinical Features
Clinical features are presented based on primary (>50%) and secondary criteria (20–49%) (see Table 1 and Figure 1 and Figure 2), which were established to distinguish between more common and less common features. Facial dysmorphism was one of the most commonly reported clinical features (>86.8% of cases), including small alae nasi, long and/or flat philtrum, thin upper lip vermillion, and broad nasal tip. The presence of microencephaly (occipitofrontal circumference (OFC) less than 3 SD) was noted in 73.7% of cases, while the majority of the remaining cases had an OFC smaller than average. Reported cases exhibited eye abnormalities, including strabismus (52.6%), FEVR (22.8%), hyperopia (14%), astigmatism (8.8%), myopia (5.8%), esotropia (5.3%), retinal detachment (3.8%), and optic atrophy (1.9%). Muscle tone abnormalities were found in the majority of the reported cases, including axial hypotonia (91.5%), peripheral spasticity (84.7%), and dystonia, which was reported in 11 cases and not systematically assessed.
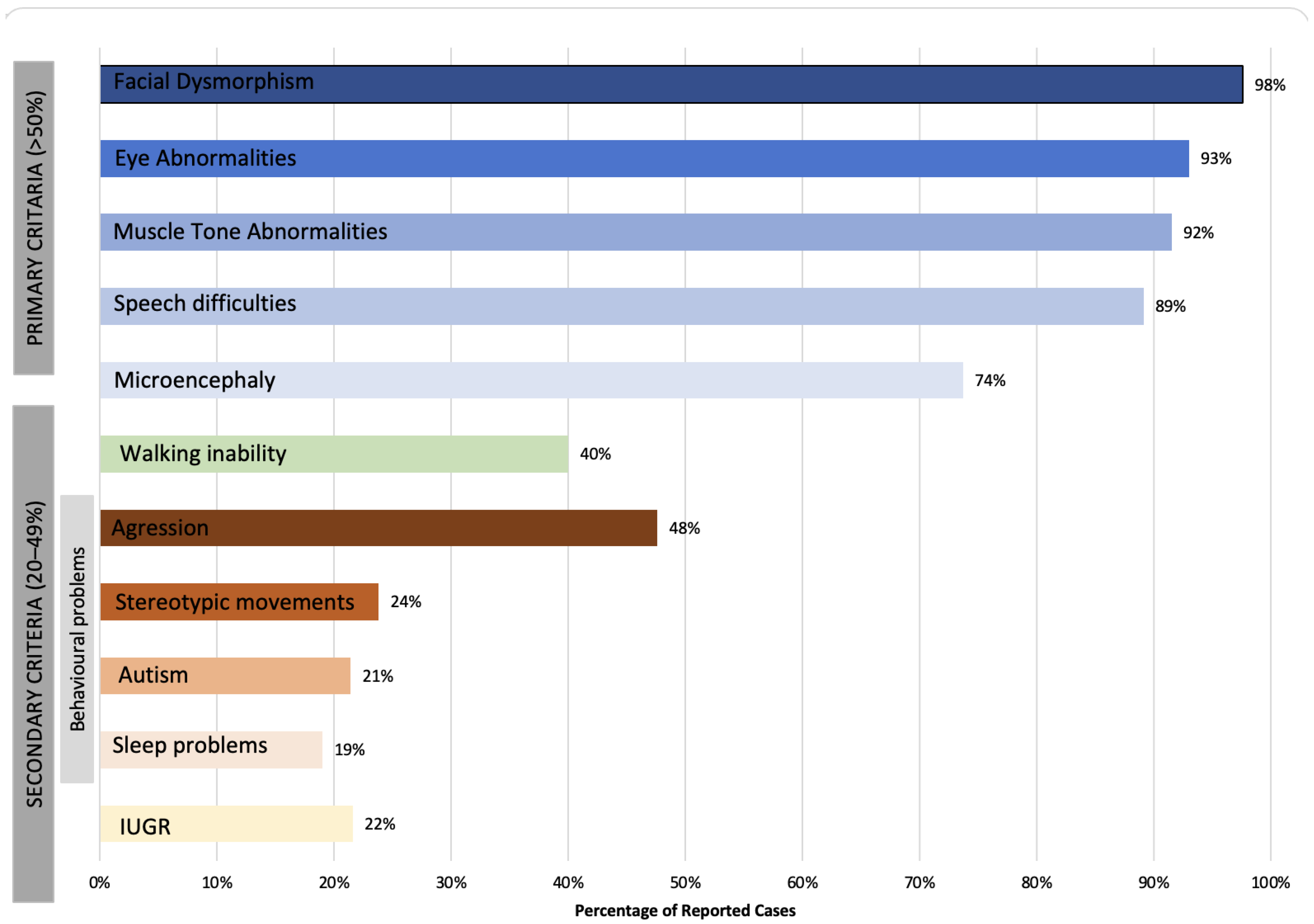
Figure 1. A 2-D bar chart representing clinical manifestations categorized based on the primary and secondary criteria.
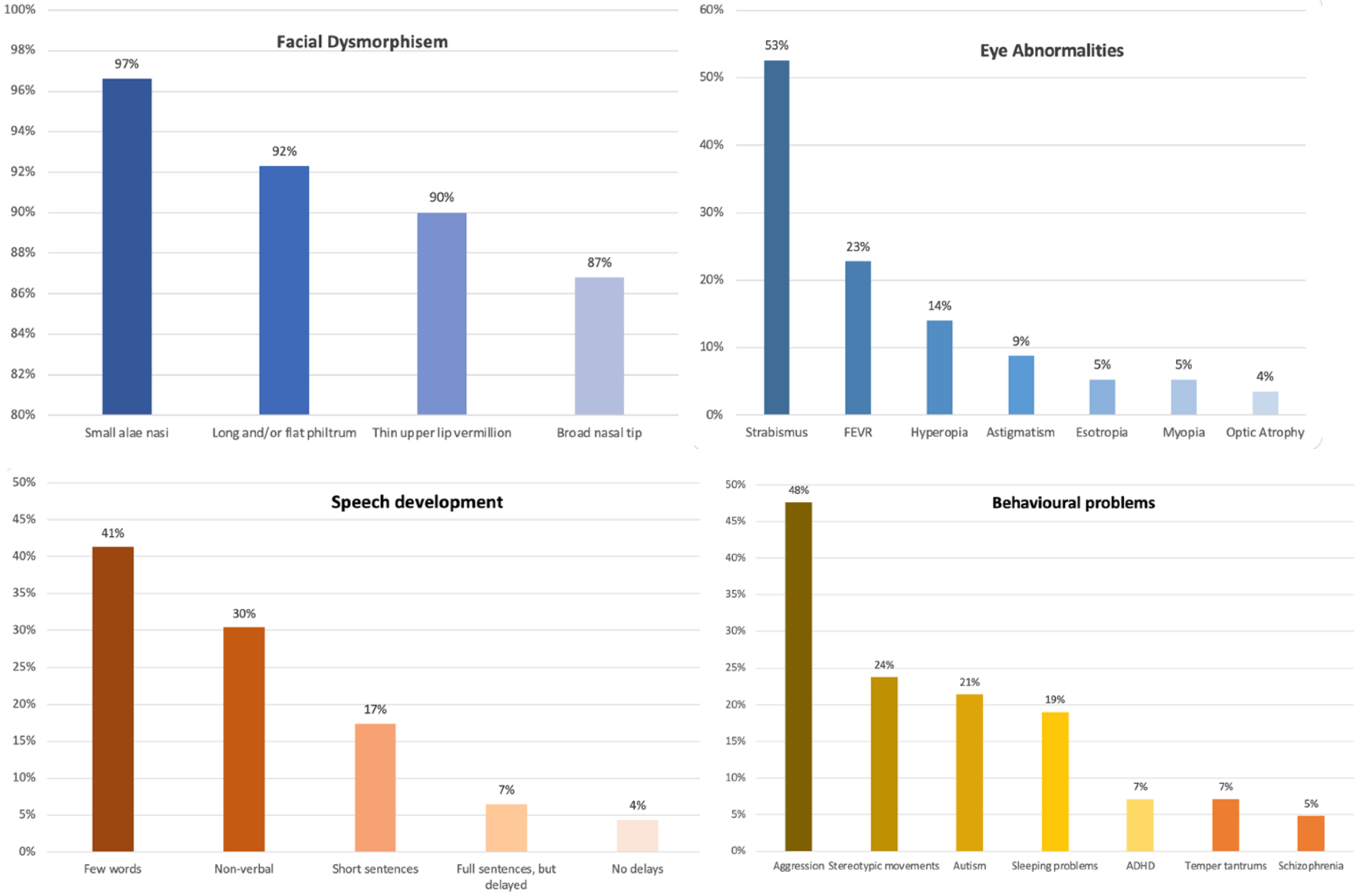
Figure 2. A column chart representing percentages of facial dysmorphism, eye abnormalities, speech development, and behavioral problems found in reported CTNNB1 patients.
Table 1. Summarization of the clinical features categorized according to their prevalence (n= 84).
| Clinical Feature | n (%) | Clinical Features | n (%) |
|---|---|---|---|
| Primary Criteria (>50%) | Secondary Criteria (20–49%) | ||
| Presence of microencephaly (valid cases: 57) | 42 (73.7) | Walking inability (valid cases: 40) | 16 (40) |
| Eye abnormalities (valid cases: 57) 1 | 53 (93) | Aggression | 20 (47.6) |
| Strabismus | 30 (52.6) | Stereotypic movements | 10 (23.8) |
| FEVR | 13 (22.8) | Autism | 9 (21.4) |
| Hyperopia | 8 (14) | Sleep problems | 8 (19) |
| Astigmatism | 5 (8.8) | ADHD | 3 (7.1) |
| Esotropia | 3 (5.3) | Temper tantrums | 3 (7.1) |
| Myopia | 3 (5.3) | Schizophrenia | 2 (4.8) |
| Speech difficulties (valid cases: 46) | 41 (89.1) | Abnormal MR (valid cases: 24) 2 | 4 (16.7) |
| Non-verbal | 14 (30.4) | IGR (valid cases: 37) | 8 (21.6) |
| A few words | 19 (41.3) | Additional criteria | |
| Short sentences | 8 (17.4) | Scoliosis (not systematically assessed) | 2 |
| Full sentences, but delayed | 3 (6.5) | Feeding problems (not systematically assessed) | 5 |
| No delays | 2 (4.4) |
1 Patient can have several eye abnormalities at the same time; 2 dilated ventricles, underdevelopment of the corpus callosum and brainstem, delayed myelination.
An electroencephalogram (EEG) was performed and reported in 30 patients, of whom 27 reported normal EEG (90%), whereas three patients had abnormal EEG (e.g., diffuse fast background activity, epileptiform activity with a tendency to spread). In addition, one patient’s report described focal epilepsy; however, it was not clear whether an EEG had been performed [5]. Magnetic resonance imaging (MRI) results were available for 24 cases of which 20 reports were normal (83.3%). Abnormal results were reported in four cases. These included arachnoid cysts, an enlarged Sylvian sulcus, hypoplasia of the corpus callosum, osteolytic lesions, enlarged lateral ventricles, abnormal gyration of the temporal lobe, absence of the right fornix and a hypoplastic brainstem, delayed myelination in the frontal lobes, mild dilatation of the ventricles, and mild thinning of the corpus callosum.
The gross motor milestone “sitting” was reported for 27 cases, of which 21 (77.7%) had reached this milestone at the mean age of 16 months. The remaining six had not reached it at that time, however, only three of them were older (age > 30 months) and the others still had time to reach this milestone (age ≤ 15 months). Of the 40 reported cases, 24 (60%) were able to walk independently, although most of them had difficulties (e.g., ataxic, unstable gait, use of an orthosis to stabilize the ankle). The average age for reaching this milestone was 3.8 years (range 12 months to 8 years). Of the 46 reported cases for speech development, 14 cases were nonverbal (30.4%), 19 cases used few words (41.3%), eight cases were able to speak short sentences (17.4%), and three cases were able to speak complete sentences and were only mildly delayed (6.5%). Two cases had no language delays in speech and had been achieving age-appropriate speech language milestones. In the majority of reported cases, receptive language was significantly better than expressive language. Of 25 reported cases, 13 (52%) reported “good” language comprehension, 11 (44%) reported “basic” language comprehension, and one reported “poor” language comprehension.
In most cases, the behavior was problematic. This information was available for 42 cases. Seven cases (16.6%) were described as ‘friendly and sociable’ and ‘with a generally cheerful demeanor’. In other cases, behavioral difficulties such as aggression (47.6%) were noted, ten cases showed stereotypic behavior (23.8%) and nine cases were diagnosed with autism spectrum disorder (21.4%). Three cases were diagnosed with ADHD (7.1%) and two with schizophrenia (4.8%). Data from eight cases indicated sleep problems, either in infancy (difficulty falling asleep) or in toddlerhood (night-time laughing fits).
Additional clinical features were considered to be rare (two or fewer cases): Scoliosis, osteogenesis imperfecta, blue sclera, sacral dimple, left clubfoot, increased dermatoglyphic whorls, type 1 diabetes, dysplastic bicuspid pulmonary valve, delayed bone age, absent left testis, brachydactyly, Achilles tendon contracture, abnormal lung growth, pulmonary hypertension, mild thumb adduction, eczema, bicoronal craniosynostosis, single supernumerary maxillary incisor, bilateral orchidopexy, syringomyelia, hypermobile joints, and glue ear.
3. Genotype
The CTNNB1 gene is located on chromosome 3 and spans 40.94 kb wherein the coding region is 2345 nt in length encoding a protein of 781 amino acids. β-catenin belongs to the armadillo family of structural proteins and is composed of three regions: an unstructured N-terminal region, bearing amino-acid residues important for β-catenin degradation; a highly conserved central core region consisting of 12 armadillo repeats, where several important interaction regions of β-catenin with many different protein partners reside; and the unstructured C-terminal region (Figure 3 and Figure 4).
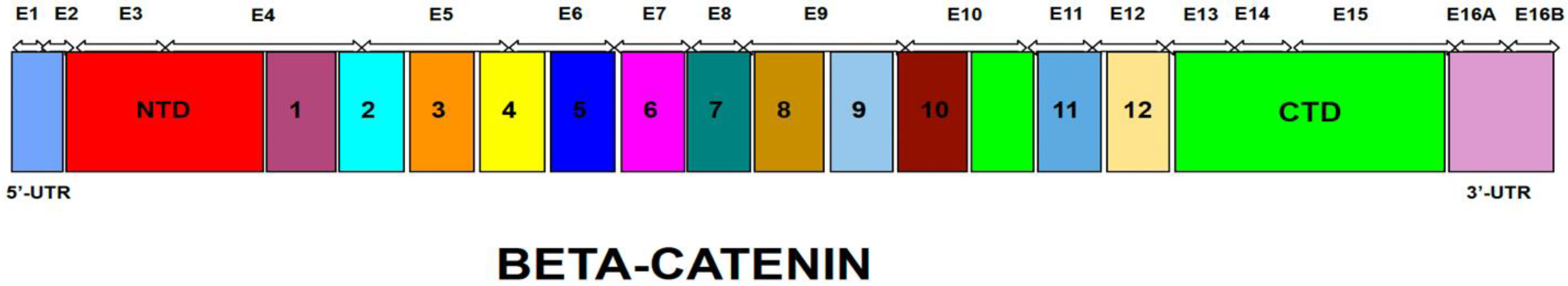
Figure 3. Schematic representation of the β catenin coding region; exon structure in correspondence to encoded protein domains.
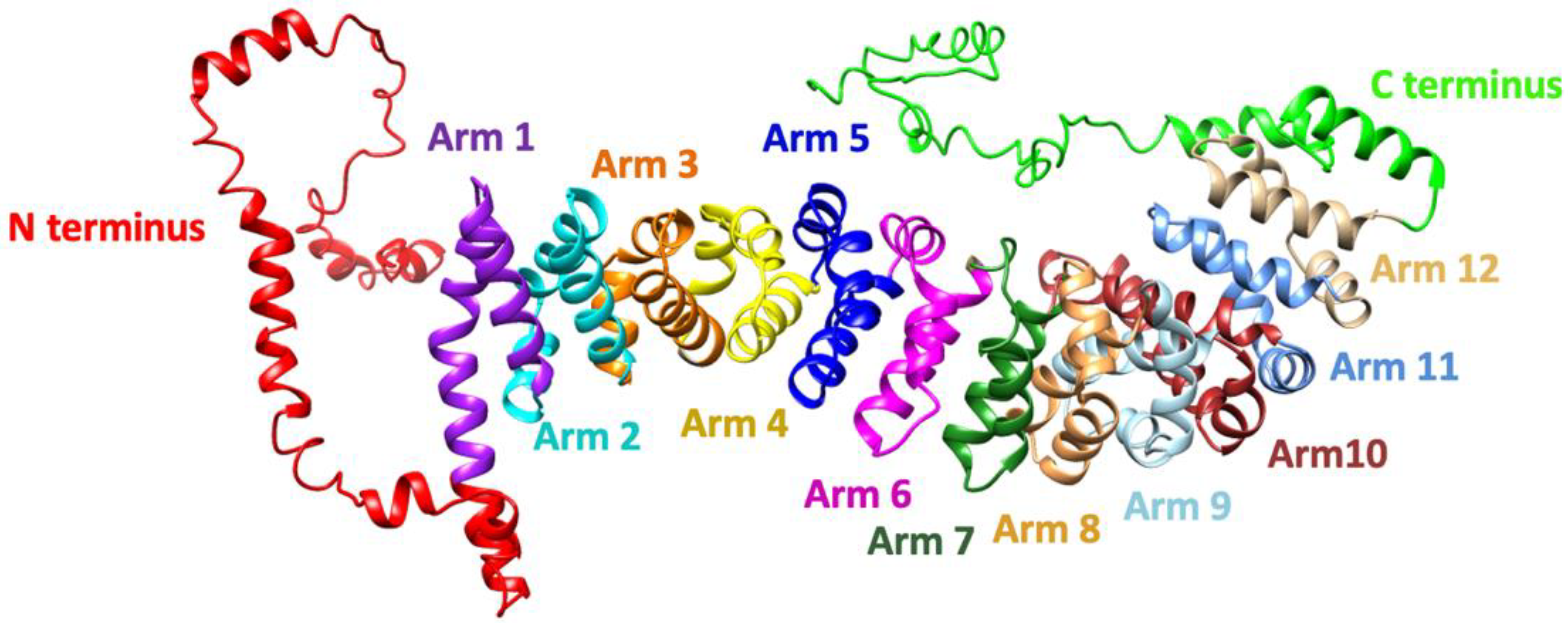
Figure 4. Three-dimensional model of human β-catenin protein generated by I-Tasser. Annotation was performed according to Huber et al., 1997. The model shows the N-terminus (red), armadillo repeat arms 1–12, and a helix and unstructured region of the C-terminal domain.
Researchers' dataset of 84 patients diagnosed with CTNNB1 Syndrome shows that CTNNB1 genetic mutations are scattered throughout the gene with the majority of mutations located in the central armadillo repeat region (75.3%). The remaining mutations are roughly equally distributed between the N-terminal domain (10.6%) and the C-terminal domain (14.1%) (Figure 5).
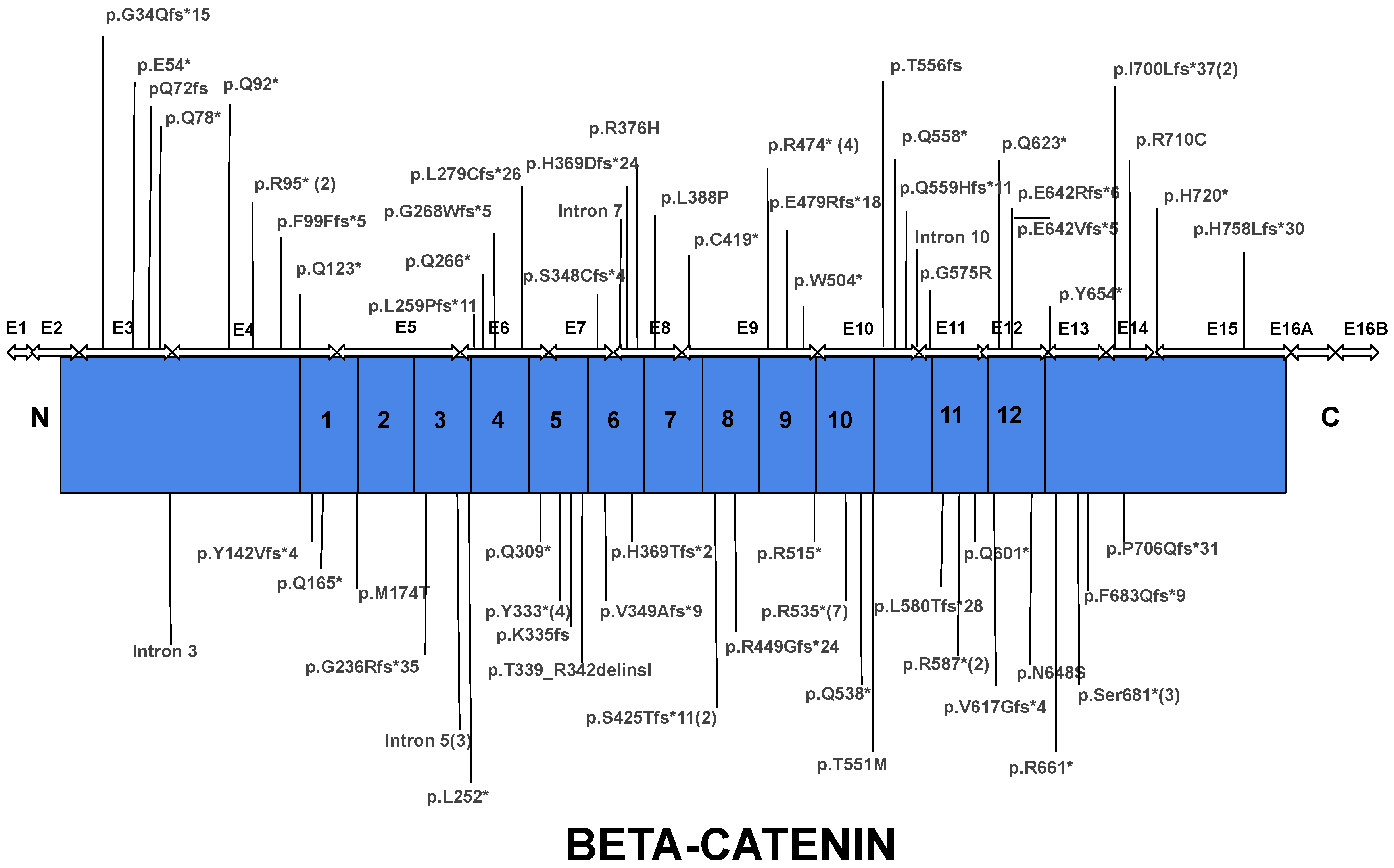
Figure 5. Distribution of mutation causative of CTNNB1 Syndrome throughout the protein coding region in accordance with exon location and subsequent encoded protein domains. Most mutations reside within the armadillo repeat region of the protein. Asterisk indicates a nonsense mutation and number in parenthesis indicates the number of cases reported the mutation.
Most of the mutations leading to CTNNB1 Syndrome were nonsense mutations (47.6%), followed by frameshift mutations (34.5%), missense mutations (8.3%), splice mutations (7.1%), and complete gene deletions (2.4%) (Figure 4). For all six missense mutations, American College of Medical Genetics and Genomics (ACMG) scores were extracted from both ClinVar and the Human Gene Mutation Database (HGMD). This information was available for three of six cases. Mutations c.1163T > C and c.2128C > T are classified as mutations of ‘Uncertain Significance’ and mutation c.1723G > A is classified as ‘Pathogenic/Likely Pathogenic’. The occurrence of mutations was found in all exons, except exon 1 and 2. The most frequently reported mutations include mutations in exon 10 (c.1603C > T, p.R535*), exon 9 (c.1420C > T, p.R474*), exon 7 (c.998dupA, c.999C > G, c.999del causing p.Y333*), intron 5 (c.734 + 1G > T, c.734 + 1G > A, causing splice mutation), and exon 13 (c.2038_2041dup, p.S681*) (Figure 5). Specific mutations in exon 9 (c.1272_1275del, p.Ser425Thrfs*11, and p.Glu479Argfs*18) and exon 4 (c.283C > T, p.R95*) occurred twice. Notably, there were two patients with reported gross deletion of the entire gene (Figure 6 and Figure 7). All but six cases found in three articles were reported as de novo mutations [17][18][19].
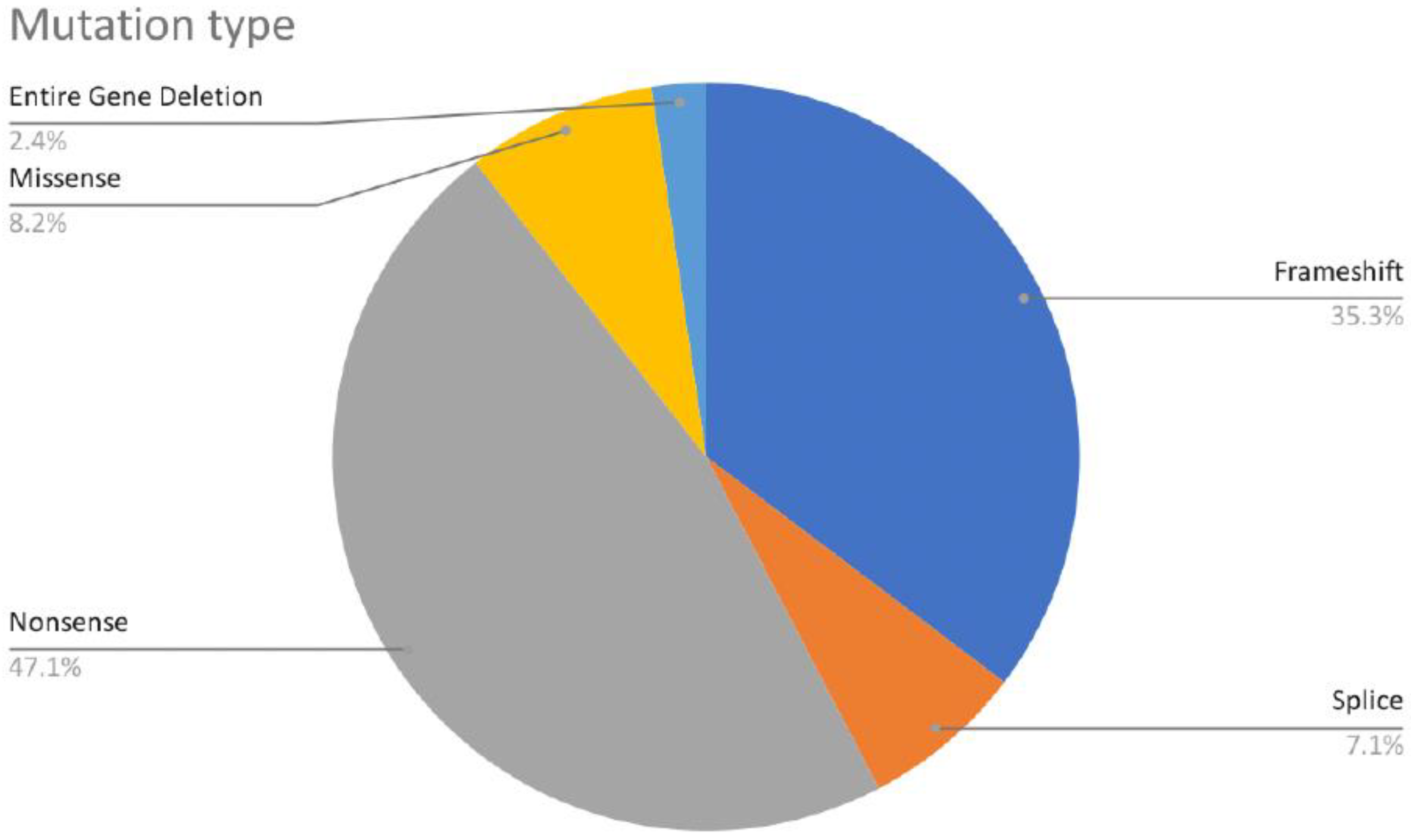
Figure 6. Distribution of CTNNB1 mutation types.
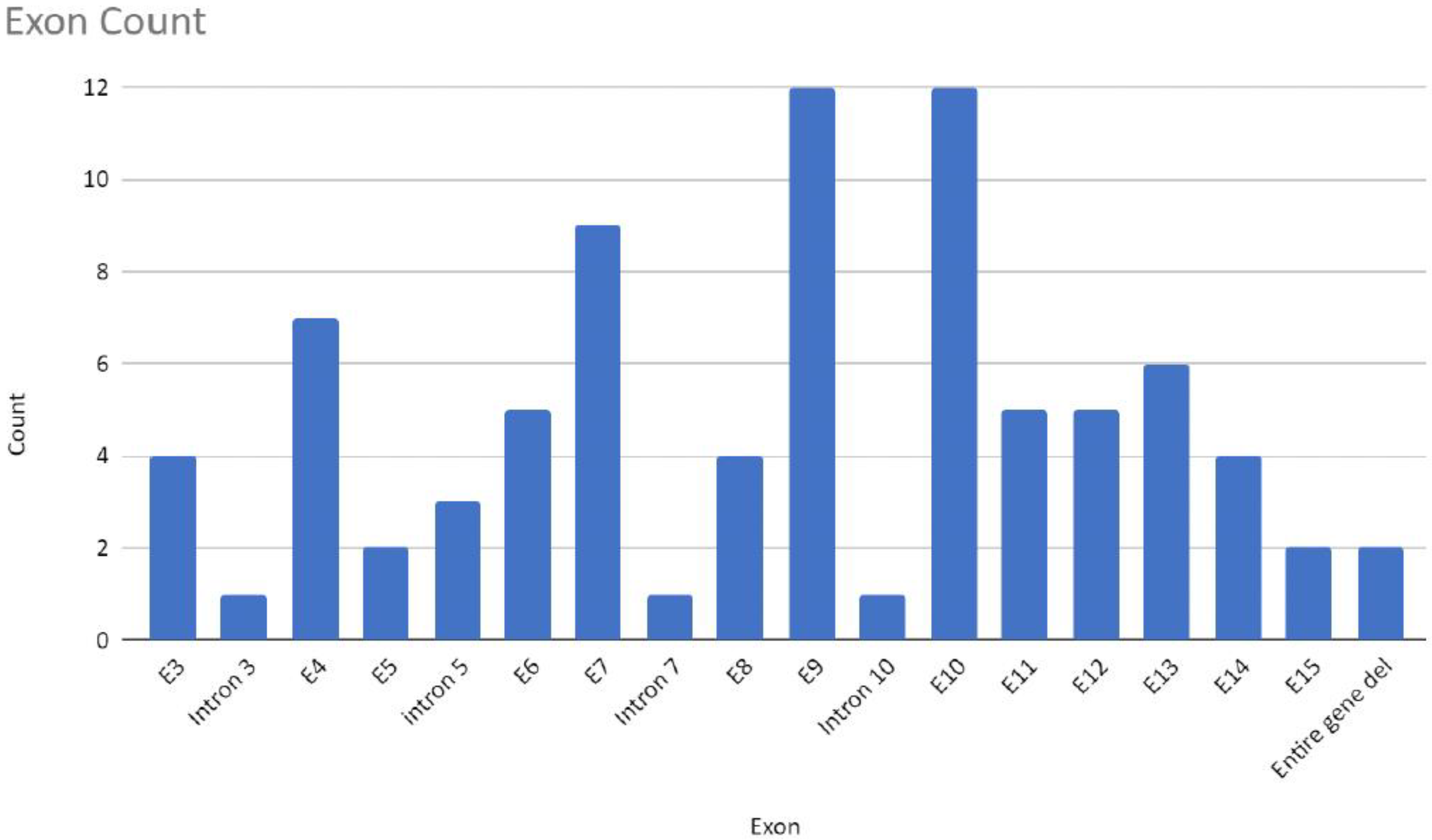
Figure 7. Number of mutations detected in individual intron or exon regions or whole gene deletions of analyzed samples in this entry (n = 84).
References
- Verhoeven, W.M.A.; Egger, J.I.M.; Jongbloed, R.E.; van Putten, M.M.; de Bruin-van Zandwijk, M.; Zwemer, A.S.; Pfundt, R.; Willemsen, M.H. A de novo CTNNB1 Novel Splice Variant in an Adult Female with Severe Intellectual Disability. Int. Med. Case Rep. J. 2020, 13, 487–492.
- Kuechler, A.; Willemsen, M.H.; Albrecht, B.; Bacino, C.A.; Bartholomew, D.W.; van Bokhoven, H.; van den Boogaard, M.J.; Bramswig, N.; Buttner, C.; Cremer, K.; et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: Expanding the mutational and clinical spectrum. Hum. Genet. 2015, 134, 97–109.
- Gilmour, D.F. Familial exudative vitreoretinopathy and related retinopathies. Eye 2015, 29, 1–14.
- de Ligt, J.; Willemsen, M.H.; van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929.
- Jin, S.C.; Lewis, S.A.; Bakhtiari, S.; Zeng, X.; Sierant, M.C.; Shetty, S.; Nordlie, S.M.; Elie, A.; Corbett, M.A.; Norton, B.Y.; et al. Mutations disrupting neuritogenesis genes confer risk for cerebral palsy. Nat. Genet. 2020, 52, 1046–1056.
- Moreno-De-Luca, A.; Millan, F.; Pesacreta, D.R.; Elloumi, H.Z.; Oetjens, M.T.; Teigen, C.; Wain, K.E.; Scuffins, J.; Myers, S.M.; Torene, R.I.; et al. Molecular Diagnostic Yield of Exome Sequencing in Patients With Cerebral Palsy. JAMA 2021, 325, 467–475.
- Li, N.; Xu, Y.; Li, G.; Yu, T.; Yao, R.E.; Wang, X.; Wang, J. Exome sequencing identifies a de novo mutation of CTNNB1 gene in a patient mainly presented with retinal detachment, lens and vitreous opacities, microcephaly, and developmental delay: Case report and literature review. Medicine 2017, 96, e6914.
- Schwarz-Romond, T.; Metcalfe, C.; Bienz, M. Dynamic recruitment of axin by Dishevelled protein assemblies. J. Cell Sci. 2007, 120, 2402–2412.
- Xing, Y.; Takemaru, K.; Liu, J.; Berndt, J.D.; Zheng, J.J.; Moon, R.T.; Xu, W. Crystal structure of a full-length beta-catenin. Structure 2008, 16, 478–487.
- Huber, A.H.; Weis, W.I. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell 2001, 105, 391–402.
- Huber, A.H.; Nelson, W.J.; Weis, W.I. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell 1997, 90, 871–882.
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.; Zheng, G. E-cadherin/beta-catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011, 567305.
- Mo, R.; Chew, T.L.; Maher, M.T.; Bellipanni, G.; Weinberg, E.S.; Gottardi, C.J. The terminal region of beta-catenin promotes stability by shielding the Armadillo repeats from the axin-scaffold destruction complex. J. Biol. Chem. 2009, 284, 28222–28231.
- Neu-Yilik, G.; Amthor, B.; Gehring, N.H.; Bahri, S.; Paidassi, H.; Hentze, M.W.; Kulozik, A.E. Mechanism of escape from nonsense-mediated mRNA decay of human beta-globin transcripts with nonsense mutations in the first exon. RNA 2011, 17, 843–854.
- Wright, C.F.; Fitzgerald, T.W.; Jones, W.D.; Clayton, S.; McRae, J.F.; van Kogelenberg, M.; King, D.A.; Ambridge, K.; Barrett, D.M.; Bayzetinova, T.; et al. Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genome-wide research data. Lancet 2015, 385, 1305–1314.
- Segel, R.; Ben-Pazi, H.; Zeligson, S.; Fatal-Valevski, A.; Aran, A.; Gross-Tsur, V.; Schneebaum-Sender, N.; Shmueli, D.; Lev, D.; Perlberg, S.; et al. Copy number variations in cryptogenic cerebral palsy. Neurology 2015, 84, 1660–1668.
- Ho, S.; Tsang, M.H.; Fung, J.L.; Huang, H.; Chow, C.B.; Cheng, S.S.; Luk, H.M.; Chung, B.H.; Lo, I.F. CTNNB1-related neurodevelopmental disorder in a Chinese population: A case series. Am. J. Med. Genet. Part A 2021, 188, 130–137.
- Wang, H.; Zhao, Y.; Yang, L.; Han, S.; Qi, M. Identification of a novel splice mutation in CTNNB1 gene in a Chinese family with both severe intellectual disability and serious visual defects. Neurol. Sci. 2019, 40, 1701–1704.
- Panagiotou, E.S.; Sanjurjo Soriano, C.; Poulter, J.A.; Lord, E.C.; Dzulova, D.; Kondo, H.; Hiyoshi, A.; Chung, B.H.; Chu, Y.W.; Lai, C.H.Y.; et al. Defects in the Cell Signaling Mediator beta-Catenin Cause the Retinal Vascular Condition FEVR. Am. J. Hum, Genet. 2017, 100, 960–968.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
961
Revisions:
3 times
(View History)
Update Date:
05 Dec 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No