 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rudy J. Castellani | + 2230 word(s) | 2230 | 2020-12-15 03:26:22 | | | |
| 2 | Dean Liu | -45 word(s) | 2185 | 2020-12-22 03:48:14 | | |
Video Upload Options
P-tau accumulates with age in a roughly hierarchical manner, but avoids abundance in the neocortex unless co-occurring with amyloid-β. Neurodegenerative tauopathies tend to have p-tau morphologies that differ from aging and Alzheimer’s disease. Tau isoforms (3R vs. 4R) have a tendency to vary with tauopathy phenotype for unknown reasons. Selective vulnerability to p-tau and spatial-temporal disconnect from amyloid-β are evident in aging. P-tau assessment at autopsy involves tissue decomposition, which may skew microanatomical observations toward limited biological meaning. Two major consensus guidelines for interpreting p-tau at autopsy emphasize the challenges of clinicopathologic correlation, and reinforce the observation that regional neurodegeneration is a better correlate of clinical signs than is proteinopathy.
1. Introduction
Neurofibrillary degeneration was first identified by Alzheimer in his seminal case report linking microscopic lesions to progressive neurological decline[1]. In doing so, he established Alzheimer’s disease (AD) as an entity and neurodegeneration as a category of disease. Much has been learned about AD since then, although the lesions in question remain the same. Indeed, the silver technique used by Alzheimer is still used today to identify the same hallmark lesions[2][3].
Researchers in the early and mid-20th century described neurofibrillary degeneration in copious detail, but were circumspect about its significance with respect to the disease process. Bielschowsky commented that alterations in nerve cells indicate only that “pathological processes have taken place”[4]. Malamud in 1929 concluded from his case series that “etiologically, this clinicopathologic syndrome may be caused by a variety of factors,” and that “the whole process has been so thoroughly completed that a possible pathogenetic theory of it could only be guessed at” [5]. McMenemey in 1940 suggested the changes “as a permanent tombstone to mark the site of the deceased cell” [6]. King noted in 1942 that “the nature and origin of this pathological material remain unsettled”[7]. Early reports on electron microscopy of neurofibrillary change by Kidd[8] and Terry[9][10] were purely descriptive in nature, with Kidd commenting that “it is difficult to speculate on the nature of these structures.” Hirano did not attach special significance to neurofibrillary tangles but instead listed populations of neurons that appeared vulnerable[11].
The paradigm shifted in the mid-1980s as researchers identified individual proteins within microscopic lesions. Brion et al.[12] first reported tau protein (named for its role in polymerization of tubulin[13]), as a major protein component of neurofibrillary tangles. Grundke-Iqbal et al.[14] reported similar findings shortly thereafter and commented that post-translational modifications such as phosphorylation may be driving the histogenesis of neurofibrillary change. Kinase-phosphatase biology would soon follow[15]. Both phosphatase modifiers and kinase inhibitors are in clinical trials today for Alzheimer’s disease (AD) therapeutics[16]. In essence, the hallmark lesion itself has been conceptualized in recent years as an epicenter of pathogenesis and a potential target for therapy.
2. Tau in Aging and Alzheimer’s Disease
P-tau accumulation tends to occur in a stereotyped fashion[17][18], conceptualized as so-called Braak stages. P-tau appears first in the locus ceruleus, followed by the transentorhinal region (e.g., stage I and II), limbic pathways (e.g., stage III and IV), and isocortex (Braak stages V and VI). Primary motor and sensory neocortical areas are relatively spared in the Braak scheme, even in advanced AD[17][18]. P-tau in subcortical areas other than the locus ceruleus occurs with age, but is not otherwise adopted for staging purposes.
Involvement of the locus ceruleus is interesting in that (i) immunolabeling for p-tau appears as early as childhood [19]and (ii) the locus ceruleus has more diverse neuroanatomical connections than any other brain region[20]. These basic observations tend to contradict prion-like p-tau templating and neuroanatomical spread as a meaningful neurodegenerative process in vivo, at least as regards human aging.
While the Braak scheme tends to suggest that p-tau progresses in all people from the brainstem to medial temporal lobe to neocortical areas given enough time, in reality, Braak stages are a composite of healthy aging and AD. Braak stages I through IV in the absence of Aβ are both common and strictly age-related[21][22]. Braak stage III and IV without Aβ (Figure 1), may be encountered in cognitively intact centenarians, for example[23]. Braak stage V/VI is for practical purposes de facto evidence of co-existing Aβ and some neurocognitive deficit[24][25]. There are no data as yet to suggest that people with medial temporal p-tau aggregates without Aβ will invariably progress to full-blown dementia, although once initiated, AD encompasses medial temporal p-tau aggregates as well as Aβ pathology[2][3]. In this sense, p-tau progression through Braak stage IV in the absence of Aβ seems to distinguish “senility” from AD.
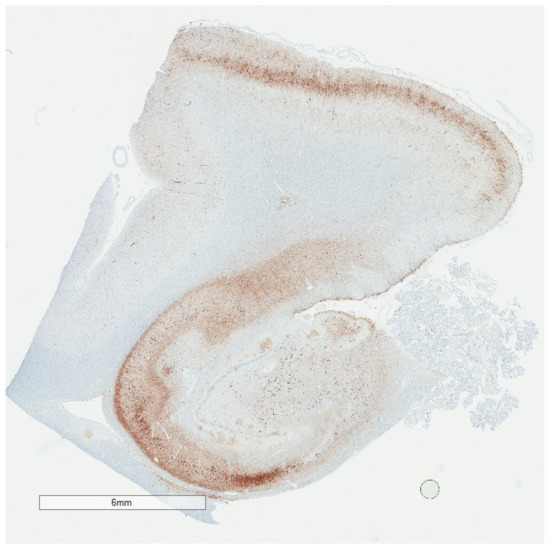
Figure 1. Low magnification p-tau (AT8) immunohistochemistry of the hippocampus showing changes consistent with Braak stage IV. Despite the extensive p-tau labeling, individuals with Braak stage IV may have been cognitively normal during life.
P-tau is generally viewed as more closely aligned with clinical signs than is Aβ. Precise clinicopathologic data in support of this view are lacking, although general observations may be cited[26]. One observation is that Braak stages I-IV selectively involve memory circuitry (entorhinal/transentorhinal region and Ammon’s horn)[17][18]. Since memory dysfunction is among the early clinical manifestations of AD, p-tau in these locations may be viewed as evidence of clinical correlation. A problem with this concept is that p-tau in memory circuitry is common in control brains, in some cases abundant, with no precise association with clinical signs during life[21].
A second observation is that Aβ plaques are occasionally abundant in cognitively intact elderly[24][27], while an abundance of neocortical p-tau is reasonably predictive of cognitive dysfunction [26]. Aβ thus appears to be a more sensitive indicator of the process of AD, whereas p-tau appears to be more specific, but only when present in the neocortex and in considerable abundance. Qualitatively, neither p-tau nor Aβ is specific for clinical signs. This does not preclude the possibility that certain p-tau species may show more or less viability as a disease biomarker once developed. For example, p-tau217 in a recent study appeared to correlate better with other measures of Alzheimer’s disease burden than p-tau181[28]. In terms of pathogenic significance, such correlations are only possible in advanced disease, when neuronal loss by whatever mechanism may be just as meaningful[27].
3. Consensus Guidelines for p-Tau Research: Alzheimer’s Disease versus Chronic Traumatic Encephalopathy
The most recent AD criteria (National Institute on Aging—Alzheimer Association, or NIA-AA criteria) employ Braak staging for p-tau, and Aβ assessment using Thal amyloid phases and CERAD neuritic plaque scores[2][3]. The output of the criteria is an “ABC” score—Amyloid phase, Braak stage, and CERAD plaque score—of “Alzheimer’s disease neuropathologic change,” each component semi-quantitated as 0 to 3. An interpretation of “Alzheimer’s disease neuropathologic change A1, B1, C1” would be an example of mild pathology, while an interpretation of “Alzheimer’s disease neuropathologic change A3, B3, C3” would be an example of advanced pathology.
The NIA-AA criteria are not used to predict neurological signs in the absence of clinical data. In fact, the consensus group commented specifically that the updated criteria are intended to “disentangle” Alzheimer’s disease neuropathologic change from the neurological assessment, given the long known challenges in clinicopathological correlation[2]. The consensus article does provide guidelines for establishing AD as an entity, but only for cases with known dementia during life. Thus, given the presence of dementia, an intermediate degree or more of both p-tau pathology and Aβ pathology is sufficient to conclude that a decedent’s clinical dementia can be assigned to the clinicopathologic entity of Alzheimer’s disease. Embedded in these criteria is that p-tau pathology is by itself insufficient to explain AD dementia without at least intermediate Aβ pathology, which reinforces the concept that p-tau alone, even in abundance, may be devoid of a predictable clinical correlation.
The NINDS/NIBIB consensus effort for chronic traumatic encephalopathy is the only such effort to date that is centered on p-tau predominantly. The criteria were based on a study of ten presumed CTE cases (all contact sport athletes) and fifteen non-CTE tauopathy cases[29], interpreted by consensus invitees with expertise in neurodegenerative diseases. The group was given a priori criteria for CTE which were modified over the course of the study. The group was aware of the finite list of presumptive diagnoses, but was otherwise blinded to clinical information. The study reported a good agreement (Cohen’s kappa 0.78) on the diagnosis of CTE, although there were cases in which some experts diagnosed CTE in non-CTE cases and vice versa. The study did not address whether specific neuropathology correlated with specific neurological signs, and did not adopt CTE stages.
There is broad overlap in tissue sampling between the NINDS/NIBIB consensus and the NIA-AA guidelines, but there are otherwise a number of fundamental differences between the two sets of guidelines: (i) The NINDS/NIBIB paper did not define an upper limit to the extent of sampling and p-tau immunostaining. A minimum of 11 brain regions was recommended, and three additional regions if high suspicion, with the caveat that 20% of cases might be missed by this approach, without, for example, sledge microtome-obtained immunohistochemical stains. In theory one could subject the entire brain to p-tau immunostains looking for the lesion of interest; (ii) The NINDS/NIBIB criteria propose a “pathognomonic lesion” (p-tau aggregates in neurons, astrocytes, and cell processes in an irregular distribution around small blood vessels at the depths of cortical sulci (Figure 2)), i.e., a diagnosis beyond doubt, that infers mechanism, clinical disease, and a neurodegenerative process. The NIA-AA effort, in contrast, specifically avoided clinical inferences and frankly admitted that the disease mechanism is unknown[2]; (iii) Selective vulnerability to p-tau is proposed as a function of the contour of the cortical ribbon (sulcal depth) and vicinity to small blood vessels (“around small blood vessels”), rather than hierarchical involvement of neuronal populations; (iv) the required criterion lacks a lower threshold. One microscopic lesion is sufficient, irrespective of the clinical context. The NIA-AA guidelines require an abundance of two lesions in a specific context (dementia), as noted; (v) The NINDS/NIBIB criteria conceptualize neurodegenerative disease neuropathology and aging-related p-tau as co-morbidities. This appears to be of necessity because of the ubiquity of p-tau with age and the high frequency of neurodegenerative diseases among donors to brain banks. In contrast, NIA-AA guidelines incorporate only Lewy body diseases as co-morbidities. Other explanations for dementia, such as frontal temporal lobar degeneration, would indicate a different disease process and a different diagnosis.
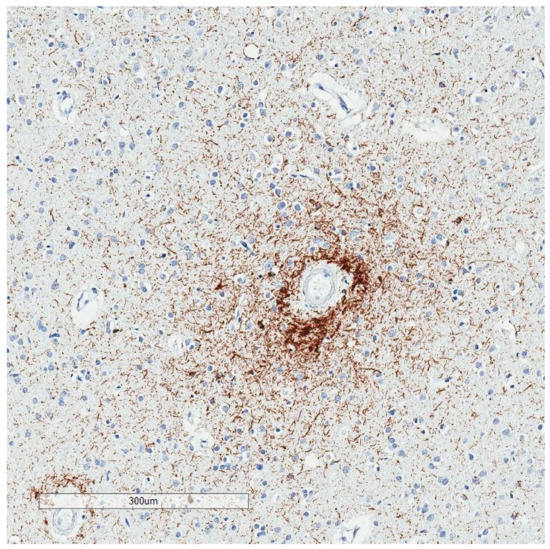
Figure 2. Irregular p-tau aggregates with “vasculocentric neurites” at a sulcal depth, consistent with the required criterion for CTE. The decedent was a 78 year old man with Parkinson’s disease and Alzheimer’s disease pathology. His traumatic brain injury and contact sport history are unknown.
Table 4. Contrasts between NIA-AA 2012 (AD) and NINDS/NIBIB 2016 (CTE) guidelines.
| Consensus Guidelines for p-Tau Assessment at Autopsy | NIA-AA 2012 AD Consensus Guidelines | NINDS/NIBIB 2016 Consensus Criteria for CTE |
|---|---|---|
| Lower threshold of p-tau for clinical correlation? | Yes | No |
| Upper limit for sampling? | Yes | No |
| Clinical context required? | Yes | No |
| Other disease processes exclusionary? | Yes | No |
| Diagnosis implies mechanism? | No | Yes |
To permit AD as a clinicopathologic entity, the AD guidelines require an intermediate degree or more of both p-tau and Aβ as a lower threshold. Five samples for p-tau immunostaining is a suggested upper limit for tissue sampling, with some articles suggesting that fewer samples may be acceptable. The AD guidelines specify an extent of “neuropathologic change” rather than a clinical diagnosis. AD is only suggested if the decedent had dementia dura life (and abundant proteinopathy). The guidelines are not used to predict a clinical state. If another pathological entity (e.g., frontotemporal lobar degeneration, prion disease) is present, AD is excluded. The AD guidelines specifically state that the disease mechanism is unknown, despite the many advances in AD research. In contrast, CTE criteria have no lower threshold. A single microscopic lesion is sufficient for diagnosis. No upper limit of sampling is specified, with sampling and immunostaining beyond standard methods being necessary in about 20% of cases. There is no requirement for dementia or other clinical problems when applying the NINDS/NIBIB criteria. Any clinical context is acceptable. No disease process, neurodegenerative or otherwise, is exclusionary. The CTE diagnosis also implies a specific mechanism (neurotrauma), and is a diagnosis beyond doubt, i.e., pathognomonic, per the consensus criteria.
The NINDS/NIBIB methodology and criteria appear useful as a screen for the lesion of interest, which will benefit the understanding of the pattern or patterns of immunoreactivity. Some caution might be warranted for clinical diagnostic interpretation, however. Each major neurodegenerative disease category includes subtypes with pathogenic mutations, as well as genetic polymorphisms that confer disease susceptibility. Interpreting neurodegenerative diseases as co-morbid to a CTE diagnosis runs the risk of assigning a genetic disorder to an environmental cause.
The appropriateness of either set of criteria as stand-alone guidelines for patient care is questionable. This was recognized in the 1991 CERAD article, which explicitly stated that the “protocol is not intended to characterize each case definitively”[30]. Likewise, the NINDS/NIBIB article points out that the proposed criteria are “preliminary” and a “first step” in the validation process[29]. The primary purpose of these consensus articles is more to facilitate research across institutions than to establish standards of practice for clinical diagnosis.
References
- Alzheimer, A. Über eine eigenartige Erkrankung der Hirnrinde. Allg. Zeitschr. Psychiatr. 1907, 64, 146–148.
- Hyman, B.T.; Phelps, C.H.; Beach, T.G. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012, 8, 1–13.
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11.
- Bielschowsky, M. Histopathology of nerve cells. In Cytology and Cellular Pathology of the Nervous System; W. Penfield, McGill University. Paul B. Hoeber, Inc.: New York, NY, USA, 1932; p. 132.
- Malamud, W.L.K. Alzheimer’s Disease. Archiv. Neurol. Psychiatry 1929, 21, 805–827.
- McMenemey, W. Alzheimer’s disease: A report of six cases. J. Neurol. Pscyhiatry 1940, 3, 211–240.
- King, L. Pathology of senile brains I. Silver-reducing structures in the hippocampus. Archiv. Neurol. Psychiatry 1942, 48, 241–256.
- Kidd, M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature 1963, 197, 192–193.
- Terry, R.D. The Fine Structure of Neurofibrillary Tangles in Alzheimer’s Disease. J. Neuropathol. Exp. Neurol. 1963, 22, 629–642.
- Terry, R.D.; Gonatas, N.K.; Weiss, M. Ultrastructural Studies in Alzheimer’s Presenile Dementia. Am. J. Pathol. 1964, 44, 269–297.
- Hirano, A.; Zimmerman, H.M. Alzheimer’s neurofibrillary changes. A topographic study. Arch. Neurol. 1962, 7, 227–242.
- Brion, J.P.; Nunez, J.H.; Flament-Durand, J. Mise en ’evidence immunologique de la prot´eine tau au niveau des l´esions de d´eg´en´erescence neurofibrillaire de la maladie d’Alzheimer. Arch. Biol. 1985, 95, 229–352.
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862.
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089.
- Hagestedt, T.; Lichtenberg, B.; Wille, H.; Mandelkow, E.M. Tau protein becomes long and stiff upon phosphorylation: Correlation between paracrystalline structure and degree of phosphorylation. J. Cell Biol. 1989, 109, 1643–1651.
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415.
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404.
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259.
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969.
- Jones, B.E. Noradrenergic locus coeruleus neurons: Their distant connections and their relationship to neighboring (including cholinergic and GABAergic) neurons of the central gray and reticular formation. Prog. Brain Res. 1991, 88, 15–30.
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766.
- Haroutunian, V.; Purohit, D.P.; Perl, D.P.; Marin, D.; Khan, K.; Lantz, M.; Davis, K.L.; Mohs, R.C. Neurofibrillary tangles in nondemented elderly subjects and mild Alzheimer disease. Arch. Neurol. 1999, 56, 713–718.
- Ganz, A.B.; Beker, N.; Hulsman, M.; Sikkes, S.; Bank, N.B.; Scheltens, P.; Smit, A.B.; Rozemuller, A.J.M.; Hoozemans, J.J.M.; Holstege, H. Neuropathology and cognitive performance in self-reported cognitively healthy centenarians. Acta Neuropathol. Commun. 2018, 6, 1–13.
- Knopman, D.S.; Parisi, J.E.; Salviati, A.; Floriach-Robert, M.; Boeve, B.F.; Ivnik, R.J.; Smith, G.E.; Dickson, D.W.; Johnson, K.A.; Petersen, L.E.; et al. Neuropathology of Cognitively Normal Elderly. J. Neuropathol. Exp. Neurol. 2003, 62, 1087–1095.
- Santacruz, K.S.; Sonnen, J.A.; Pezhouh, M.K.; Desrosiers, M.F.; Nelson, P.T.; Tyas, S.L. Alzheimer Disease Pathology in Subjects Without Dementia in 2 Studies of Aging: The Nun Study and the Adult Changes in Thought Study. J. Neuropathol. Exp. Neurol. 2011, 70, 832–840.
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381.
- Giannakopoulos, P.; Herrmann, F.R.; Bussiere, T. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003, 60, 495–500.
- Janelidze, S.; Stomrud, E.; Smith, R.; Palmqvist, S.; Mattsson-Carlgren, N.; Airey, D.C.; Proctor, N.K.; Chai, X.; Shcherbinin, S.; Sims, J.R.; et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat. Commun. 2020, 11, 1–12.
- McKee, A.C.; Cairns, N.J.; Dickson, D.W.; Folkerth, R.D.; Keene, C.D.; Litvan, I.; Perl, D.P.; Stein, T.D.; Vonsattel, J.G.; The TBI/CTE Group; et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016, 131, 75–86.
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L.; et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479.

