Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Jha, U.C.; Nayyar, H.; Wettberg, E.J.B.V.; Naik, Y.D.; Thudi, M.; Siddique, K.H.M. Legume Pangenome for Crop Improvement. Encyclopedia. Available online: https://encyclopedia.pub/entry/37091 (accessed on 06 June 2026).
Jha UC, Nayyar H, Wettberg EJBV, Naik YD, Thudi M, Siddique KHM. Legume Pangenome for Crop Improvement. Encyclopedia. Available at: https://encyclopedia.pub/entry/37091. Accessed June 06, 2026.
Jha, Uday Chand, Harsh Nayyar, Eric J. B. Von Wettberg, Yogesh Dashrath Naik, Mahendar Thudi, Kadambot H. M. Siddique. "Legume Pangenome for Crop Improvement" Encyclopedia, https://encyclopedia.pub/entry/37091 (accessed June 06, 2026).
Jha, U.C., Nayyar, H., Wettberg, E.J.B.V., Naik, Y.D., Thudi, M., & Siddique, K.H.M. (2022, November 29). Legume Pangenome for Crop Improvement. In Encyclopedia. https://encyclopedia.pub/entry/37091
Jha, Uday Chand, et al. "Legume Pangenome for Crop Improvement." Encyclopedia. Web. 29 November, 2022.
Copy Citation
Legume crops play a crucial role in ensuring global food security. Legume reference genomes have been constructed for soybean, chickpea, common bean, pigeonpea, pea, lupin, peanut, cowpea, and mungbean. However, pangenomes are needed to obtain insights into the genome dynamics, gene-content variation, genetic basis of agronomic-trait variation, and evolutionary relationship in various legumes.
pangenome
legumes
structural variation
genome sequence
gene
1. Soybean (Glycine max L.)
In soybean, Li et al. [1] performed pangenome analysis by constructing a de novo genome assembly of seven accessions of Glycine soja collected from different geographic regions to shed light on the genetic basis of untapped genetic diversity and evolution-related novel insights. Comparing the genome sequences obtained from this pangenome analysis and a reference genome sequence of G. max W82 revealed a plethora of loss or gain of copy number variations (CNVs) in multiple genes, ranging from biotic-stress tolerance (including R genes) to various transcription factors contributing to local-environmental acclimation against invading pathogens [1]. In addition, comparing the G. max and G. soja genomes provided novel insights into the FLOWERING LOCUS T gene controlling flowering time, PHYA-regulating light receptor, and LEAFY genes via various ‘indels’ and nonsynonymous SNPs in the given loci in the G. soja genome [1]. In Arabidopsis, 1332 genes related to seed protein and oil contents were annotated in the G. max W82 genome based on homology to genes related to lipid-acyl pathways [2]. A comparative genomics analysis revealed that the G. max and G. soja genomes differ for several indels and large-effect single-nucleotide variants (SNPs) and CNVs for these genes attributing to oil-seed content [1]. A de novo assembly of 26 soybean accessions (including 14 cultivated, 3 wild, and 9 landraces) from 2898 deeply sequenced soybean genomes and pangenome analysis provided novel insights into the thousands of structural variations related to various candidate genes of agronomic interest (including iron-deficiency chlorosis, seed luster, and flowering) and involved in domestication and evolution [3] (Figure 1). The assembled sequences contained 20,623 gene families designated as core genes, with 8163 gene families in 25–26 accessions designated soft-core genes, and 28,679 families in 2–24 accessions designated dispensable genes [3] (Table 1).
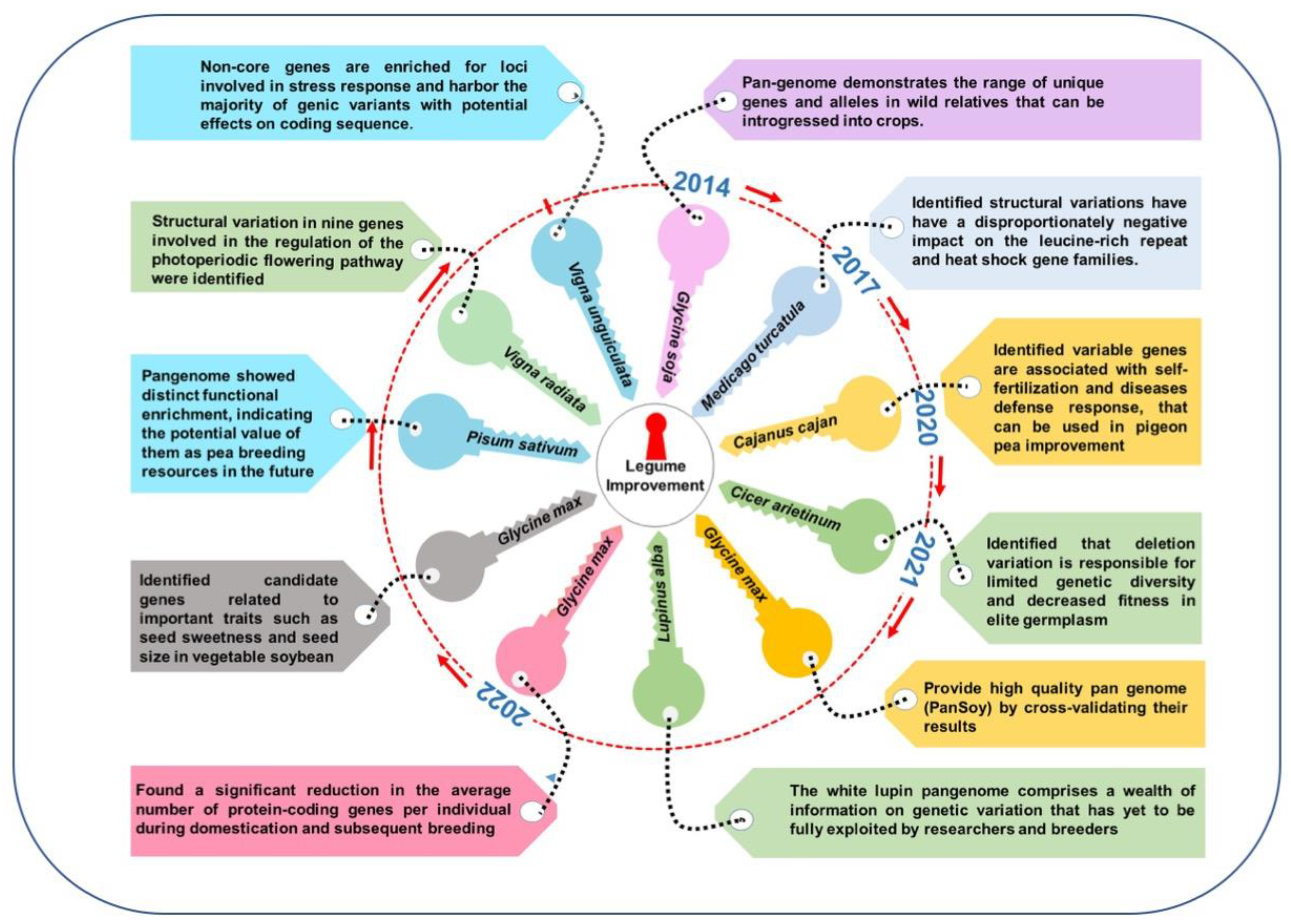
Figure 1. Legume pangenomes and key learnings for crop improvement.
A comparative analysis of this pangenome with the previously assembled reference genomes for Zhonghuang [4][5], W05 [6], and Wm82 [7] indicated the presence of myriad presence–absence variations (PAVs) and CNVs in various accessions of current soybean accessions, demanding a thorough pangenome analysis of diverse and geographically distinct soybean accessions [3]. Torkamaneh et al. [8] developed the soybean pangenome known as ‘Pansoy’ by constructing a de novo assembly of 204 geographically diverse soybean accessions following the ‘map to pan’ approach using the EUPAN pipeline [9]. This pangenome contained 108 Mbp novel genome sequences—with 1659 novel genes not present in the reference Wm82 genome—revealing 49,431 hard-core genes, 1401 soft-core genes, 3402 shell genes, and 297 cloud genes [8]. Thus, the Pansoy resource provided novel insights into intraspecific genetic variation in G. max and offered a platform for genomic diversity and evolutionary and domestication studies for soybeans.
Subsequently, a pangenome measuring 1213 Mbp was constructed from >1000 accessions collected from the USDA Soybean Germplasm collection using the chromosome level ‘Lee’ soybean genome assembly as the reference sequence [10]. The results identified 3765 genes missing in the ‘Lee’ genome sequence assembly, indicating a loss of genetic diversity during domestication and breeding activity [10]. This pangenome also contained an additional 198.34 Mbp sequence assembly compared to the ‘Lee’ reference genome. The PAV analysis revealed 86.8% core genes and 13.2% dispensable genes [10]. The authors also uncovered 110 genomic regions containing 1266 protein-coding genes in a ‘domestication-selective sweep’ and 86 genomic regions containing 1434 genes in a ‘breeding-selective sweep’ [10]. Based on this pangenome analysis, the authors concluded that wild soybean had higher nucleotide diversity than landraces, old cultivars, and modern cultivars due to the loss of diversity during domestication and breeding efforts.
2. Chickpea (Cicer arietinum L.) Pangenome
Chickpea is a major pulse crop, ranking third among pulses for global production and serving as a plant source of protein and many essential micronutrients [11]. In the last decade, unprecedented advancements in bioinformatics and NGS technologies allowed for the assembly of a chickpea genome sequence [12][13], which has served as a tool for chickpea improvement. To further build chickpea-genomic resources and to study speciation, phylogenetic analysis, genomic diversity of cultivated species and its wild progenitor, and migration of cultivated chickpea species, Varshney et al. [11] constructed the first chickpea pangenome (Figure 1) (measuring 592.58 Mb with 29,870 genes) by sequencing 3171 cultivated and 195 wild chickpea accessions using iterative mapping and an assembly approach. From this, 124,833 SNPs were selected to form linkage-disequilibrium blocks; these SNPs and the phenotyping data for 100 seed weight (100 SW) and yield per plant (YPP) from 2980 genotypes were used for genomic prediction to improve these traits [11]. Likewise, 3.94 million SNPs and the phenotyping data for 16 phenological and yield-related traits in 2980 genotypes revealed associations between 205 SNPs and 11 traits, with 152 SNPs associated with 79 unique genes governing seed size and development [11].
3. Cowpea (Vigna ungiculata L.) Pangenome
Cowpea is an important warm season, climate-resilient, multipurpose legume crop in sub-Saharan Africa [14]. Significant progress has been achieved in developing genomics resources for cowpeas. Lonardi et al. [15] developed the genome assembly of cowpea genotype IT97K-499-35. Liang et al. [16] constructed the first cowpea pangenome by producing de novo assemblies of six cowpea accessions with a mean size of 449.91 Mb, identifying 21,330 core and 23,531 non-core genes, with the large number of frameshift variations in the non-core genes attributing large diversity in domesticated cowpeas. Comparative and genomic synteny analyses of the IT97K-499-35 reference genome and the genome assemblies of the six accessions indicated the presence of numerous structural rearrangements, including PAVs, inversions, indels, and CNVs [16]. The authors confirmed that the existing PAVs are responsible for the black seed-coat color in cowpeas.
4. Pigeonpea (Cajanus cajan L.) Pangenome
Pigeonpea is an important grain legume, mostly grown in India and Africa, with an essential role in ensuring protein nutrition security across the semi-arid tropics [17]. The pigeonpea reference genome sequence [17] has been used to improve various traits in pigeonpea; however, the pangenome assembly of pigeonpea provides further opportunities to harness untapped genetic variation not present in the reference genome sequence. Zhao et al. [18] built the pigeonpea pangenome (measuring 622 Mbp) using iterative mapping and an assembly approach [19], sequencing 89 pigeonpea accessions collected across the globe and the reference pigeonpea with >9.5× coverage. The pangenome contained 55,512 more genes than the previously assembled reference genome (53,612), with 48,067 core genes and 7445 accessory genes [18]. A functional analysis of the ‘variable genes’ indicated their possible role in proteolysis, pollen tube reception, and the signal transduction process [18]. Of 909 identified R genes, 836 were core genes and 73 were variable genes. To confirm the role of PAVs attributing phenotypic diversity, the authors detected several additional SNPs for various phenotypic traits (seed weight, days to 50% flowering, and plant height) compared to Varshney et al. [20], who found a significant association of 1 SNP for pods/plant, 1 SNP for primary branches/plant, and 3 SNP for secondary branches/plant from the study of whole genome resequencing of 292 pigeonpea accessions.
5. Mungbean (Vigna radiata L.) Pangenome
Mungbean is an important grain legume crop with high nutritional benefits, mostly grown in south and southeast Asian countries [21]. Draft genome assemblies are available for mungbean genotypes VC1973A [22] and JL7 [23]. Liu et al. [23] assembled the first mungbean pangenome (measuring 762.92 Mb), with 43,462 annotated genes, by deep-sequencing JL7 and 217 mungbean accessions, including cultivars and landraces. The predicted genes comprised 33,258 hard-core genes, 2872 soft-core genes, 7154 shell genes, and 178 cloud genes [23]. Of the nine PAVs controlling flowering regulation, three genes (jg13350, jg13746, and Pang80812) were present in Chinese landraces and breeding lines [23], while six genes (jg1521, jg5273, jg5274, jg5281, jg5284, and Pang68295 were absent but present in non-Chinese lines [23].
6. White Lupin (Lupinus alba L.) Pangenome
White lupin (Lupinus albus L.) is a protein-rich grain legume crop domesticated in the Mediterranean region [24][25]. However, the presence of quinolizidine alkaloids in lupin seed causes a bitter taste and toxicity; thus, lowering seed alkaloids is a prime objective of lupin-breeding programs. Hufnagel et al. [26] and Xu et al. [27] constructed the white-lupin genome assembly. Hufnagel et al. [28] established the first white-lupin pangenome by sequencing a diverse set of 39 white-lupin accessions using a ‘map-to-pan’ approach [9]. The pangenome comprised 32,068 (78.5%) core genes, 6046 soft-core genes, and 8776 shell genes [28]. Several selection sweeps related to breeding for low-alkaloid accessions were identified on chromosomes 18, 15, 1, and 3, overlapping the low-alkaloid-content QTLs previously reported by Ksiazkiewicz et al. [29] and Lin et al. [30].
7. Barrel Medic (Medicago truncatula) Pangenome
Barrel medic (Medicago truncatula) is an important model legume crop used for investigating symbiotic relationships with rhizobia and mycorrhizae and root development [31][32]. Zhou et al. [33] developed the first M. truncatula pangenome (measuring 388–428 Mbp, with an additional 63 Mbp novel sequence) by de novo assembly of 15 M. truncatula accessions, adding 16% genome space to the reference genome sequence [34] of this model legume. This pangenome sheds light on causative structural variants, including transposable elements in different gene families, such as the nucleotide-binding site leucine-rich repeat rapid evolvement.
8. Lentil (Lens culinaris) Pangenome
Lentil is an important source of dietary protein, carbohydrates, and iron for humans [35]. Due to the large size of the genome (4 Gb) and the presence of a high number repetitive elements, the completion of a lentil genome assembly has taken some time to complete. However, an incomplete-draft genome sequence of Lens culinaris cv. CDC Redberry v2.0 [36][37] has facilitated new directions in lentil genomics research. Thus, in the absence of a complete reference genome sequence, pan-transcriptomes [38] could provide a platform to establish the lentil pangenome. Guerra-Garcia et al. [39] have used an exome capture array built on the draft lentil genome and transcriptomic data [40] to examine copy number variation across cultivated lentil germplasm. Herein, it was uncovered a previously hidden amount of variation across lentils [39]. Furthermore, Gutierrez-Gonzalez et al. [41] assembled and annotated the transcriptomes of eight lentil accessions, including cultivated and wild species (L. orientalis, L. tomentosus, L. ervoides, L. lamottei, L. nigricans, and two L. odemensis), identifying 15,910 core genes and 24,226 accessory genes. A comparative analysis of these assembled transcriptomes with the incomplete CDC Redberry reference genome assembly indicated that transcripts of L. culinaris had the greatest similarity with CDC Redberry transcripts, while transcripts of L. lamottei and L. nigricans had the least [41]. Further long-read-based sequencing efforts could assist in decoding the complete genome and pangenome assembly of lentils.
9. Pea (Pisum sativum L.) Pangenome
Pea is the fourth-most globally important grain legume, enriched with protein, starch, and minerals [42][43], and is considered a ‘founder crop’ member; it is also one of the oldest domesticated crops [44]. Despite having an assembled pea reference genome [45], genetic diversity in cultivated and wild pea and the domestication history of pea remain elusive. Yang et al. [42] constructed a second pea de novo genome assembly of ZW6 and a pangenome assembly of 116 accessions containing cultivated and wild pea genotypes, identifying 112,117 pangenes, including 15,470 core genes, 6170 soft-core genes, 41,028 shell genes, and 50,108 cloud genes. The study confirmed that P. abyssinicum is separate from P. fulvum and P. sativum. The pangenes from P. abyssinicum were unique in stimulus and chemical response, while those of P. fulvum were involved in growth, tropism, and development; thus, the novel genes harboring in these species could be used to improve disease resistance and yield in modern pea cultivars [42].
10. Pangenome-Derived PAVs for GWAS Analysis to Explore Novel Genes
Pangenome analysis allows the discovery of a plethora of novel structural variations (SVs), including gene PAVs that may be missing in the reference genome assembly [8][23][46]. Thus, these pangenome-derived PAVs can be used in a GWAS to uncover a novel agronomic trait gene allele(s). Liu et al. [23] uncovered five novel genes missing in the mungbean reference genome assembly that contributed to bruchid resistance (Pang34265, Pang44622, Pang57772, Pang58608, and Pang64254). Zhao et al. [18] identified 225 significant SNPs associated with various phenological and yield-related traits (e.g., days to 50% flowering, days to maturity, pods/plant, and yield/plant) from SNPs and PAVs derived from a pigeonpea pangenome analysis, which overlapped those previously identified by Varshney et al. [20]. However, several new SNPs were associated with these studied traits [18]. Likewise, Varshney et al. [11] identified 205 significantly associated SNPs for agronomic traits in a GWAS study on 16 traits in a panel of 2980 chickpea accessions using 3.94 million SNPs, of which 152 SNPs were in 79 unique genes related to seed size and seed development [11]. GWAS analysis using SVs derived from 2898 soybean accessions elucidated the genetic basis of soybean seed luster, uncovering the presence of 10 kb PAVs in the hydrophobic protein-encoding gene causing seed luster [3].
References
- Li, Y.H.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.G.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052.
- Li-Beisson, Y.; Shorrosh, B.; Beisson, F.; Andersson, M.X.; Arondel, V.; Bates, P.D.; Baud, S.; Bird, D.; DeBono, A.; Durrett, T.P.; et al. Acyl-lipid metabolism. Arab. Book/Am. Soc. Plant Biol. 2013, 11, e0161.
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.A.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-genome of wild and cultivated soybeans. Cell 2020, 182, 162–176.
- Shen, Y.; Liu, J.; Geng, H.; Zhang, J.; Liu, Y.; Zhang, H.; Xing, S.; Du, J.; Ma, S.; Tian, Z. De novo assembly of a Chinese soybean genome. Sci. China Life Sci. 2018, 61, 871–884.
- Shen, Y.; Du, H.; Liu, Y.; Ni, L.; Wang, Z.; Liang, C.; Tian, Z. Update soybean Zhonghuang 13 genome to a golden reference. Sci. China Life Sci. 2019, 62, 1257–1260.
- Xie, M.; Chung, C.Y.; Li, M.W.; Wong, F.L.; Wang, X.; Liu, A.; Wang, Z.; Leung, A.K.; Wong, T.H.; Tong, S.W.; et al. A reference-grade wild soybean genome. Nat. Commun. 2019, 10, 1216.
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183.
- Torkamaneh, D.; Lemay, M.A.; Belzile, F. The pan-genome of the cultivated soybean (PanSoy) reveals an extraordinarily conserved gene content. Plant Biotechnol. J. 2021, 19, 1852–1862.
- Hu, Z.; Sun, C.; Lu, K.C.; Chu, X.; Zhao, Y.; Lu, J.; Shi, J.; Wei, C. EUPAN enables pan-genome studies of a large number of eukaryotic genomes. Bioinformatics 2017, 33, 2408–2409.
- Bayer, P.E.; Valliyodan, B.; Hu, H.; Marsh, J.I.; Yuan, Y.; Vuong, T.D.; Patil, G.; Song, Q.; Batley, J.; Varshney, R.K.; et al. Sequencing the USDA core soybean collection reveals gene loss during domestication and breeding. Plant Genome 2022, 15, e20109.
- Varshney, R.K.; Roorkiwal, M.; Sun, S.; Bajaj, P.; Chitikineni, A.; Thudi, M.; Singh, N.P.; Du, X.; Upadhyaya, H.D.; Khan, A.W.; et al. A chickpea genetic variation map based on the sequencing of 3,366 genomes. Nature 2021, 599, 622–627.
- Varshney, R.K.; Song, C.; Saxena, R.K.; Azam, S.; Yu, S.; Sharpe, A.G.; Cannon, S.; Baek, J.; Rosen, B.D.; Tar’an, B.; et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 2013, 31, 240–246.
- Jain, M.; Misra, G.; Patel, R.K.; Priya, P.; Jhanwar, S.; Khan, A.W.; Shah, N.; Singh, V.K.; Garg, R.; Jeena, G.; et al. A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J. 2013, 74, 715–729.
- Timko, M.P.; Singh, B.B. Cowpea, a multifunctional legume. In Genomics of Tropical Crop Plants; Moore, P.H., Ming, R., Eds.; Springer: New York, NY, USA, 2008; pp. 227–258.
- Lonardi, S.; Muñoz-Amatriaín, M.; Liang, Q.; Shu, S.; Wanamaker, S.I.; Lo, S.; Tanskanen, J.; Schulman, A.H.; Zhu, T.; Luo, M.C.; et al. The genome of cowpea (Vigna unguiculata Walp.). Plant J. 2019, 98, 767–782.
- Liang, Q.; Munoz-Amatriain, M.; Shu, S.; Lo, S.; Wu, X.; Carlson, J.W.; Davidson, P.; Goodstein, D.M.; Phillips, J.; Janis, N.M.; et al. A view of the pan-genome of domesticated cowpea (Vigna unguiculata Walp.). bioRxiv 2022.
- Varshney, R.K.; Chen, W.; Li, Y.; Bharti, A.K.; Saxena, R.K.; Schlueter, J.A.; Donoghue, M.T.; Azam, S.; Fan, G.; Whaley, A.M.; et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2012, 30, 83.
- Zhao, J.; Bayer, P.E.; Ruperao, P.; Saxena, R.K.; Khan, A.W.; Golicz, A.A.; Nguyen, H.T.; Batley, J.; Edwards, D.; Varshney, R.K. Trait associations in the pangenome of pigeon pea (Cajanus cajan). Plant Biotechnol. J. 2020, 18, 1946–1954.
- Golicz, A.A.; Bayer, P.E.; Barker, G.C.; Edger, P.P.; Kim, H.; Martinez, P.A.; Chan, C.K.; Severn-Ellis, A.; McCombie, W.R.; Parkin, I.A.; et al. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 2016, 7, 13390.
- Varshney, R.; Saxena, R.; Upadhyaya, H.; Khan, A.; Yu, Y.; Kim, C.; Rathore, A.; Kim, D.; Kim, J.; An, S.; et al. Whole-genome resequencing of 292 pigeonpea accessions identifies genomic regions associated with domestication and agronomic traits. Nat. Genet. 2017, 49, 1082–1088.
- Jha, U.C.; Nayyar, H.; Parida, S.K.; Bakır, M.; von Wettberg, E.J.; Siddique, K.H.M. Progress of Genomics-Driven Approaches for Sustaining Underutilized Legume Crops in the Post-Genomic Era. Front. Genet. 2022, 13, 831656.
- Kang, Y.J.; Kim, S.K.; Kim, M.Y.; Lestari, P.; Kim, K.H.; Ha, B.K.; Jun, T.H.; Hwang, W.J.; Lee, T.; Lee, J.; et al. Genome sequence of mungbean and insights into evolution within Vigna species. Nat. Commun. 2014, 5, 5443.
- Liu, C.; Wang, Y.; Peng, J.; Fan, B.; Xu, D.; Wu, J.; Cao, Z.; Gao, Y.; Wang, X.; Li, S.; et al. High-quality genome assembly and pan-genome studies facilitate genetic discovery in mung bean and its improvement. Plant Commun. 2022, 26, 100352.
- Wolko, B.; Clements, J.C.; Naganowska, B.; Nelson, M.; Huaan, Y. Lupinus. In Wild Crop Relatives: Genomic and Breeding Resources; Kole, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 153–206.
- Taylor, J.L.; De Angelis, G.; Nelson, M.N. How have narrow-leafed lupin genomic resources enhanced our understanding of lupin domestication? In The Lupin Genome; Singh, K.B., Kamphuis, L.G., Nelson, M.N., Eds.; Springer: Cham, Switzerland, 2020; pp. 95–108.
- Hufnagel, B.; Marques, A.; Soriano, A.; Marquès, L.; Divol, F.; Doumas, P.; Sallet, E.; Mancinotti, D.; Carrere, S.; Marande, W.; et al. High-quality genome sequence of white lupin provides insight into soil exploration and seed quality. Nat. Commun. 2020, 11, 492.
- Xu, W.; Zhang, Q.; Yuan, W.; Xu, F.; Muhammad Aslam, M.; Miao, R.; Li, Y.; Wang, Q.; Li, X.; Zhang, X.; et al. The genome evolution and low-phosphorus adaptation in white lupin. Nat. Commun. 2020, 11, 1069.
- Hufnagel, B.; Soriano, A.; Taylor, J.; Divol, F.; Kroc, M.; Sanders, H.; Yeheyis, L.; Nelson, M.; Péret, B. Pangenome of white lupin provides insights into the diversity of the species. Plant Biotechnol. J. 2021, 19, 2532–2543.
- Ksiazkiewicz, M.; Nazzicari, N.; Yang, H.; Nelson, M.N.; Renshaw, D.; Rychel, S.; Ferrari, B.; Carelli, M.; Tomaszewska, M.; Stawiński, S.; et al. A high-density consensus linkage map of white lupin highlights synteny with narrow-leafed lupin and provides markers tagging key agronomic traits. Sci. Rep. 2017, 7, 15335.
- Lin, R.; Renshaw, D.; Luckett, D.; Clements, J.; Yan, G.; Adhikari, K.; Buirchell, B.; Sweetingham, M.; Yang, H. Development of a sequence-specific PCR marker linked to the gene “pauper” conferring low-alkaloids in white lupin (Lupinus albus L.) for marker assisted selection. Mol. Breed. 2009, 23, 153–161.
- Young, N.D.; Udvardi, M. Translating Medicago truncatula genomics to crop legumes. Curr. Opin. Plant Biol. 2009, 12, 193–201.
- von Wettberg, E.J.; Porter, S.S.; Moriuchi, K.S.; Mukherjee, J.R. Chapter 2: Medicago as a model forage and ecological model. In The Model Legume Medicago Truncatula; de Bruijn, F.J., Ed.; Wiley Publishers: Hoboken, NJ, USA, 2020.
- Zhou, P.; Silverstein, K.A.; Ramaraj, T.; Guhlin, J.; Denny, R.; Liu, J.; Farmer, A.D.; Steele, K.P.; Stupar, R.M.; Miller, J.R.; et al. Exploring structural variation and gene family architecture with De Novo assemblies of 15 Medicago genomes. BMC Genom. 2017, 18, 261.
- Young, N.D.; Debellé, F.; Oldroyd, G.E.; Geurts, R.; Cannon, S.B.; Udvardi, M.K.; Benedito, V.A.; Mayer, K.F.; Gouzy, J.; Schoof, H.; et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 520–524.
- de la Vega, M.P.; Torres, A.M.; Cubero, J.I.; Kole, C. (Eds.) Genetics, Genomics and Breeding of Cool Season Grain Legumes; CRC Press: Boca Raton, FL, USA, 2011; pp. 98–150.
- Ramsay, L.; Koh, C.; Konkin, D.; Cook, D.; Penmetsa, V.; Dongying, G.; Coyne, C.; Humann, J.; Kaur, S.; Dolezel, J.; et al. Lens Culinaris CDC Redberry Genome Assembly v2.0. Available online: https://knowpulse.usask.ca/genome-assembly/Lcu.2RBY (accessed on 7 April 2021).
- Ramsay, L.; Koh, C.S.; Kagale, S.; Gao, D.; Kaur, S.; Haile, T.; Gela, T.S.; Chen, L.A.; Cao, Z.; Konkin, D.J.; et al. Genomic rearrangements have consequences for introgression breeding as revealed by genome assemblies of wild and cultivated lentil species. bioRxiv 2021.
- Hirsch, C.N.; Foerster, J.M.; Johnson, J.M.; Sekhon, R.S.; Muttoni, G.; Vaillancourt, B.; Peñagaricano, F.; Lindquist, E.; Pedraza, M.A.; Barry, K.; et al. Insights into the maize pan-genome and pan-transcriptome. Plant Cell 2014, 26, 121–135.
- Guerra-Garcia, A.; Haile, T.; Ogutcen, E.; Bett, K.E.; von Wettberg, E.J. An evolutionary look into the history of lentil reveals unexpected diversity. Evol. Appl. 2022, 15, 1313–1325.
- Ogutcen, E.; Ramsay, L.; von Wettberg, E.B.; Bett, K.E. Capturing variation in Lens (Fabaceae): Development and utility of an exome capture array for lentil. Appl. Plant Sci. 2018, 6, e01165.
- Gutierrez-Gonzalez, J.J.; García, P.; Polanco, C.; González, A.I.; Vaquero, F.; Vences, F.J.; Pérez de la Vega, M.; Sáenz de Miera, L.E. Multi-Species Transcriptome Assemblies of Cultivated and Wild Lentils (Lens sp.) Provide a First Glimpse at the Lentil Pangenome. Agronomy 2022, 12, 1619.
- Yang, T.; Liu, R.; Luo, Y.; Hu, S.; Wang, D. Improved pea reference genome and pan-genome highlight genomic features and evolutionary characteristics. Nat. Genet. 2022, 54, 1553–1563.
- Dahl, W.J.; Foster, L.M.; Tyler, R.T. Review of the health benefits of peas (Pisum sativum L.). Br. J. Nutr. 2012, 108, S3–S10.
- Zohary, D.; Hopf, M. Domestication of pulses in the Old World: Legumes were companions of wheat and barley when agriculture began in the Near East. Science 1973, 182, 887–894.
- Kreplak, J.; Madoui, M.A.; Cápal, P.; Novák, P.; Labadie, K.; Aubert, G.; Bayer, P.E.; Gali, K.K.; Syme, R.A.; Main, D.; et al. A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 2019, 51, 1411–1422.
- Bayer, P.E.; Scheben, A.; Golicz, A.A.; Yuan, Y.; Faure, S.; Lee, H.; Chawla, H.S.; Anderson, R.; Bancroft, I.; Raman, H.; et al. Modelling of gene loss propensity in the pangenomes of three Brassica species suggests different mechanisms between polyploids and diploids. Plant Biotechnol. J. 2021, 19, 2488–2500.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
30 Nov 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No