Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Salma A Rizo-Tellez | -- | 2792 | 2022-11-28 16:32:34 | | | |
| 2 | Sirius Huang | Meta information modification | 2792 | 2022-11-29 08:43:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rizo-Téllez, S.A.; Sekheri, M.; Filep, J.G. Myeloperoxidase Regulation of Neutrophil Function. Encyclopedia. Available online: https://encyclopedia.pub/entry/36911 (accessed on 08 August 2026).
Rizo-Téllez SA, Sekheri M, Filep JG. Myeloperoxidase Regulation of Neutrophil Function. Encyclopedia. Available at: https://encyclopedia.pub/entry/36911. Accessed August 08, 2026.
Rizo-Téllez, Salma A., Meriem Sekheri, János G. Filep. "Myeloperoxidase Regulation of Neutrophil Function" Encyclopedia, https://encyclopedia.pub/entry/36911 (accessed August 08, 2026).
Rizo-Téllez, S.A., Sekheri, M., & Filep, J.G. (2022, November 28). Myeloperoxidase Regulation of Neutrophil Function. In Encyclopedia. https://encyclopedia.pub/entry/36911
Rizo-Téllez, Salma A., et al. "Myeloperoxidase Regulation of Neutrophil Function." Encyclopedia. Web. 28 November, 2022.
Copy Citation
Neutrophils, the most abundant white blood cells in humans, are critical for host defense against invading pathogens. Equipped with an array of antimicrobial molecules, neutrophils can eradicate bacteria and clear debris. Among the microbicide proteins is the heme protein myeloperoxidase (MPO), stored in the azurophilic granules, and catalyzes the formation of the chlorinating oxidant HOCl and other oxidants (HOSCN and HOBr). Evolving evidence indicates that MPO can directly modulate the function and fate of neutrophils, thereby shaping immunity. These actions include MPO orchestration of neutrophil trafficking, activation, phagocytosis, lifespan, formation of extracellular traps, and MPO-triggered autoimmunity.
neutrophil
myeloperoxidase
degranulation
neutrophil extracellular traps
host defense
autoimmunity
inflammation
1. Introduction
Neutrophil granulocytes are the most abundant immune cells, constituting about 60–70% of all leukocytes in human blood. Neutrophils are typically the first immune cells to be recruited to sites of infection and tissue injury [1][2][3], and provide the first line of defense to the host against invading pathogens [2][4][5]. This role is best exemplified by the susceptibility of neutropenic patients to repeated bacterial infections [6]. Neutrophils can efficiently kill bacteria through different defense mechanisms [2][7][8], including the mobilization and discharge of microbicide proteins and proteases stored in pre-formed cytosolic granules [9][10]. Granules can fuse with pathogen-containing phagosomes following phagocytosis of bacteria or the plasma membrane in response to pathogens that cannot be phagocytosed, leading to the extracellular release of their content [2][8]. Among the microbicide proteins is the heme-containing enzyme myeloperoxidase (MPO), the most abundantly expressed protein by human neutrophils. MPO utilizes chloride as a co-substrate with H2O2 to generate chlorinating oxidants, such as HOCl, which have one of the highest cytotoxic potentials to bacteria and living cells [11]. However, the release of some neutrophil products, including MPO, to the extracellular milieu can be detrimental to the host by inflicting tissue damage and consequently exacerbating inflammation [5][11][12]. Neutrophil-driven inflammation has been recognized as a common mechanism underlying a wide range of pathologies, including atherosclerosis, cardiovascular, respiratory, autoimmune and neurodegenerative diseases, sepsis, and cancer [5][13]. Elevated plasma MPO levels are frequently detected in patients under these conditions and correlate with disease severity [14][15][16][17][18]. By contrast, recent studies in MPO-deficient animals reported exaggeration of the inflammatory response reviewed in [19], implying protective functions for MPO. Given these diverse effects, the balance between MPO’s deleterious and beneficial effects will likely determine its contribution to the outcome of the inflammatory response.
The prevailing and rather simplistic view of MPO as a cytotoxic agent has undergone substantial revision in the past decade, and novel paradigms have emerged. It is now apparent that the roles of MPO extend beyond its antimicrobial function and inflict bystander tissue damage to the host. Accumulating evidence indicates that MPO can directly modulate function of inflammatory and other cells, including neutrophils, macrophages and dendritic cells, endothelial and epithelial cells, and shape immunity [20][21]. Of particular importance, MPO modulates the functions of neutrophils, which interact with other innate and adaptive immune cells to orchestrate immune responses [13].
2. MPO Regulation of Neutrophil Trafficking
In addition to the intracellular killing of invading pathogens, accumulation evidence indicates an important role for MPO in the regulation of neutrophil trafficking. Neutrophil recruitment is a multistep event allowing capture, adhesion, and extravasation of the cell [1][22]. Non-activated neutrophils adhere to MPO-coated surfaces [23], whereas “free” circulating MPO binds to the β2 integrin CD11b (Mac-1) [24] and modulates intracellular Ca2+ levels and cytoskeleton organization [25]. MPO was found to evoke highly directed neutrophil motility in vitro and to attract neutrophils in preclinical models of hepatic ischemia-reperfusion and cremaster muscle inflammation through its cationic surface charge independent of its catalytic properties [26]. Furthermore, catalytically inactive MPO reduced the thickness of the endothelial glycocalyx through ionic interaction with heparan sulfate side chains and induced shedding of the endothelial glycocalyx core protein syndecan-1 [27]. Administration of human MPO was associated with increased neutrophil accumulation in carrageenan-induced acute lung injury in mice [28]. These effects of MPO are consistent with providing adhesive support for neutrophils. Increased MPO expression was detected on the surface of neutrophils from patients with various inflammatory diseases, including sepsis, ischemia-reperfusion injury, or acute coronary syndromes, and correlated with plasma MPO levels [24]. By contrast, reduced neutrophil MPO expression has been suggested as being a good predictor of a higher risk of mortality in patients with sepsis [29].
Studies on MPO-deficient mice yielded apparently contradictory results. In some studies, genetic deletion of MPO was found to attenuate neutrophil accumulation and distant organ damage after renal ischemia-reperfusion [30], to reduce E. coli septicemia-induced pulmonary bacterial colonization, lung injury, and mortality [31], and to increase cellular protection in ischemic stroke [32]. Furthermore, MPO deficiency was associated with increased glomerular accumulation of neutrophils and induction of CD4+ T cell autoimmunity in a model of lupus nephritis [33]. Of note, MPO deficiency is associated with upregulated baseline expression of inducible NO synthase and increased NO production in the lung, which may partially compensate for the lack of HOCl-mediated bacterial killing [31]. The contribution of enhanced NO production to the protection afforded by MPO deficiency remains to be investigated. By contrast, other studies reported a role for MPO to limit excessive neutrophil accumulation. Thus, MPO-deficient mouse neutrophils display increased surface expression of CD11b and a pro-migratory phenotype in ischemia-reperfusion-induced liver injury [34]. Blockade or genetic deletion of MPO was found to significantly increase endotoxemia-associated mortality, whereas adoptive transfer of wild-type neutrophils protected against mortality [35]. Whether these actions were mediated by CD11b or ionic interactions remains to be investigated. MPO was shown to limit local tissue damage by converting diffusible H2O2 into highly reactive, but locally confined HOCl [36]. MPO may also modulate the inflammatory response through catalyzing the oxidation of lipid mediators [37], as exemplified by elevated plasma levels of cysteinyl leukotrienes and reduced oxidative metabolites of linoleic acid [38]. Another study has suggested that enhanced adhesive interactions were responsible for increased neutrophil migration into the peritoneal cavity in response to zymosan and the inflamed cremaster muscle in MPO-KO mice [39]. Consistently, the administration of recombinant MPO to wild-type mice diminished neutrophil migration and accumulation in the same models [39]. Genetic MPO deletion may have dual consequences; for instance, MPO deficiency prevented neutrophil-mediated renal injury, but aggravated T-cell immunity inducing crescentic glomerulonephritis [40].
Differences in the composition of primary neutrophil granules, in particular lack of defensins and low MPO content (about 10–20% of that of human neutrophils) in mouse neutrophils [41] should be considered for translating the data from mouse studies to the clinical setting. Furthermore, cell contact-dependent CD11b-mediated MPO transfer from neutrophils to endothelial cells can disrupt normal endothelial function [42], leading to endothelial damage and consequently enhanced leukocyte adherence. MPO delivered in extracellular vesicles might also evoke endothelial injury similar to that reported for epithelial cells [43].
3. MPO, Neutrophil Activation and Phagocytosis
MPO can regulate neutrophil function independent of enzymatic properties. MPO binding to CD11b on human neutrophils has been shown to evoke release of primary/azurophilic granule contents, including MPO and elastase, and to upregulation of CD11b expression through yet unidentified molecular mechanisms [24][28]. MPO ligation of CD11b leads to phosphorylation of p38 MAPK, ERK ½, and PI3K [24][28], and activation of NF-κB [24]. p38 MAPK induces phosphorylation of p47phox, the cytoplasmic regulatory subunit of NADPH oxidase [44], leading to superoxide formation [24], and transcription of NF-κB-regulated genes involved in the acute inflammatory response [45]. These findings are consistent with the function of CD11b as a bidirectional allosteric “signaling machine” [46], and CD11b-mediated outside-in signaling in degranulation [47]. These data also imply an MPO-centered feed-forward autocrine/paracrine mechanism for aggravation and prolongation of the inflammatory response, as exemplified in a mouse model of acute lung injury [28][48]. CD11b also functions as a receptor for complement C3b (CR3) that mediates phagocytosis of complement C3b-opsonized microbes and damaged cells [49]. MPO-deficient neutrophils exhibit higher levels of surface expression of CD11b and engulf more zymosan than do wild-type neutrophils [50]. Neutrophil-produced reactive oxygen and nitrogen species, such as H2O2 and ONOO−,have long been recognized as intracellular signaling molecules to regulate the activation of transcription factors, including NF-κB, and cytokine production [51][52]. Thus, it is plausible that MPO or MPO-derived oxidants HOCl (or its derivatives) function as signaling molecules to modulate CD11b-mediated phagocytosis. MPO deficiency may result in the accumulation of intracellular H2O2 [53], which together with reduced HOCl generation might lead to neutrophil activation via the NF-κB and ERK 1/2 pathways. Similar to H2O2, MPO-generated HOSCN targets cysteine residues present in protein tyrosine phosphatases, resulting in increased phosphorylation of p38 MAPK and ERK1/2, impaired antioxidant defenses and induction of pro-inflammatory cytokine gene expression and cytokine release [54][55]. MPO modulation of CD11b expression and/or function may vary on the stimulus and context. Indeed, studies comparing cytokine production of MPO-deficient and wild-type neutrophils revealed substantial stimulus-dependent variations. For instance, MPO-deficient neutrophils were found to produce less KC and MIP-1α, and higher amounts of IL-6, IL-10, and TNF-α than wild-type neutrophils challenged with LPS in vitro [56]. Stimulation of MPO-KO neutrophils with zymosan generated more IL-1α, IL-1β, MIP-1α, MIP-1β, MIP-2, and TNF-α as compared to wild-type neutrophils [57].
4. MPO Regulation of Neutrophil Lifespan
During migration from the blood to the inflamed tissue and within the inflammatory locus, neutrophils receive pro-survival cues that extend their lifespan by delaying intrinsic apoptosis [21][58]. CD11b-mediated binding of neutrophils to the endothelial counter-ligand ICAM-1 or fibrinogen induces activation of the PI3k/Akt, MAPK/ERK, and NF-κB signaling pathways [59][60][61], prevention of proteasomal degradation of the anti-apoptotic protein Mcl-1, a key regulator of neutrophil survival [62], and consequently suppression of caspase-3 activity. Adhesion per seis not a prerequisite for prolonged neutrophil survival [63] as ligation of CD11b with MPO or other soluble ligands also activates these signaling pathways [24][28] and generates survival signals for human neutrophils in vitro [28]. The survival signal does not require catalytic activity, for MPO inactivated with the suicide substrate 4-aminobenzoic acid hydrazide (4-ABAH) or the mechanism-based inhibitor 3-amino-1,2,4-triazol plus H2O2 exerted actions similar to that of native MPO [24][28]. Consistently, the administration of human MPO, resulting in levels similar to those detected in patients with inflammatory vascular diseases [14][15], prolongs the lifespan of circulating neutrophils through suppressing apoptosis in rats [28]. Furthermore, MPO suppresses neutrophil apoptosis and delays the spontaneous resolution of inflammation, resulting in the perpetuation of carrageenan-induced acute lung injury in mice [28]. These MPO actions closely resemble those of the pan-caspase inhibitor zVAD-fmk, which aggravates and prolongs carrageenan-elicited acute pleurisy [64] and lung injury [28]. Dissociation of the dimeric MPO into monomeric or hemi-MPO following reductive alkylation attenuates Ca2+ mobilization and degranulation, and results in a partial loss in prolonging neutrophil lifespan and attenuates degranulation [25]. These would imply a regulatory mechanism by which MPO may control the activation and fate of neutrophils. Elevated hemi-MPO levels were detected in the serum of patients with acute inflammation [25], suggesting that this regulatory mechanism may be operational in vivo. However, whether increased hemi-MPO levels were due to increased secretion or decomposition of MPO as well as the mechanism of MPO decomposition in the blood requires further studies.
In contrast to generating survival cues in human neutrophils, MPO was reported to promote apoptosis in HL60 leukemia cells, and this was partially inhibited by the inactivation of MPO with 4-ABAH or methimazole [65][66]. While these studies did not address the involvement of CD11b in the pro-apoptotic action of MPO, it is plausible that ligation of CD11b exerts opposing actions in primary and leukemia cells likely by shifting the balance of pro-survival and pro-apoptosis cues. Interestingly, the pro-apoptotic action partially depends on the catalytic activity of MPO [65][66], whereas pro-survival signaling does not require catalytically active MPO [28]. Nevertheless, it is intriguing that MPO can prolong the lifespan of human neutrophils, the predominant source of this enzyme in spite of its potent cytotoxic properties [28].
5. MPO, NET Formation and Autoimmunity
Neutrophils can release extracellular traps (NET) to trap and kill pathogens extracellularly when phagocytosis is not feasible [8][67][68]. NETs are a meshwork of chromatin decorated with granule proteins, such as MPO, neutrophil elastase, and proteinase-3, and absorb pentraxin 3 [69] and complement [70]. In the suicidal pathway of NET formation (commonly referred to as NETosis), reactive oxygen species produced by NADPH oxidase via activation of the Raf-MEK-ERK and p38 MAPK pathways initiate NET extrusion. Neutrophil elastase translocates to the nucleus, where it partially degrades specific histones and synergizes with MPO in driving chromatin decondensation [71]. The action of MPO is independent of its enzymatic activity. Protein-arginine deiminase 4 (PAD4)-mediated chromatin decondensation ultimately leads to the extrusion of a DNA scaffold studded with citrullinated histones and cytotoxic granular proteins, including MPO [72][73]. Neutrophils from patients with complete MPO deficiency are unable to form NETs, which is associated with several microbial infections, especially with Candida albicans in a subgroup of these patients [74]. Partial MPO deficiency or pharmacological inhibition of MPO only delays and reduces NET formation [74]. The addition of exogenous MPO to MPO-deficient neutrophils does not rescue NET formation, suggesting that this process requires MPO translocation to the appropriate subcellular compartment. Vital NET release is independent of NADPH oxidase and occurs more rapidly than suicidal NET extrusion in response to S. aureus, C. albicans, A. fumigatus, Leishmania promastigotes [75][76], or anaplastic thyroid cancer cells [77] without compromising neutrophil viability. Some studies suggested a role for mitochondrial oxidants and selective extrusion of mitochondrial DNA in forming extracellular traps [78][79]. Thus, the molecular mechanisms governing the release of nuclear or mitochondrial DNA appear to differ [80]. The pathological relevance of suicidal versus vital NET extrusion to innate immunity and the role of MPO in this latter process require further studies. Previous studies showed that phagocytosis of bulky phosphatidylserine-decorated particles, such as apoptotic cells or activated platelets, rendered neutrophils unable to form NET [81][82]. Hence, modulation of phagocytosis by MPO (and other granule constituents) may represent another mechanism to regulate NET extrusion.
Apart from immune defense and infectious diseases, increasing evidence indicates that aberrant NET formation is a central event under a number of pathological conditions, including atherosclerosis, acute respiratory distress syndrome, rheumatoid arthritis, lupus erythematosus, metabolic syndrome, neurological disorders, and cancer [83][84]. NET biology has been the subject of several recent comprehensive reviews [83][84][85][86]. It is important to emphasize that NETs can exert both pro- and anti-inflammatory actions, even under the same pathological conditions, as shown, for example, in a model of rheumatoid arthritis [87]. NETs can limit inflammation by trapping and degrading cytokines and chemokines [88] and orchestrate induction and resolution of sterile crystal-mediated inflammation [89]. The role of MPO in these events is largely unknown.
MPO has long been recognized as a target antigen in different forms of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitides, microscopic polyangiitis, eosinophilic granulomatosis with polyangiitis and to a lesser frequency in granulomatosis with polyangiitis [90][91][92]. Externalization of MPO, together with other well-known antigens, such as double-stranded DNA and histones, through aberrant NET formation or impaired NET degradation, has been implicated in triggering autoimmunity in susceptible individuals [91][93]. Pathogenic ANCA bind to MPO expressed on the surface of cytokine-primed neutrophils, leading to their excessive activation [94]. The resulting necrotizing inflammation of small and medium blood vessels may affect various organs, including the airways, kidneys, skin, and the nervous system [92]. Furthermore, sera from patients with MPO-ANCA-associated microscopic polyangiitis [95] or systemic lupus erythematosus [93] show impaired capacity for NET degradation, partly due to lower serum DNase1 levels [95]. Sera from patients with MPO-ANCA-associated microscopic polyangiitis were found to have a high ability to induce NET formation [95]. These would likely form a vicious cycle that may contribute to disease progression. MPO has also been suggested to trigger autoimmunity during uncontrolled inflammation in mice [96], though it is unclear whether this process involves MPO binding to CD11b and/or NET formation.
Together with the changing perception of the role of neutrophils in homeostasis and pathogenesis [1][2][20][21], it has become apparent that MPO should no longer be considered exclusively as an enzyme-producing cytotoxic oxidant. Thus, in addition to inflicting tissue damage through enzymatic and non-enzymatic actions, MPO may also exert protective actions and contribute to the dampening of inflammation (Figure 1).
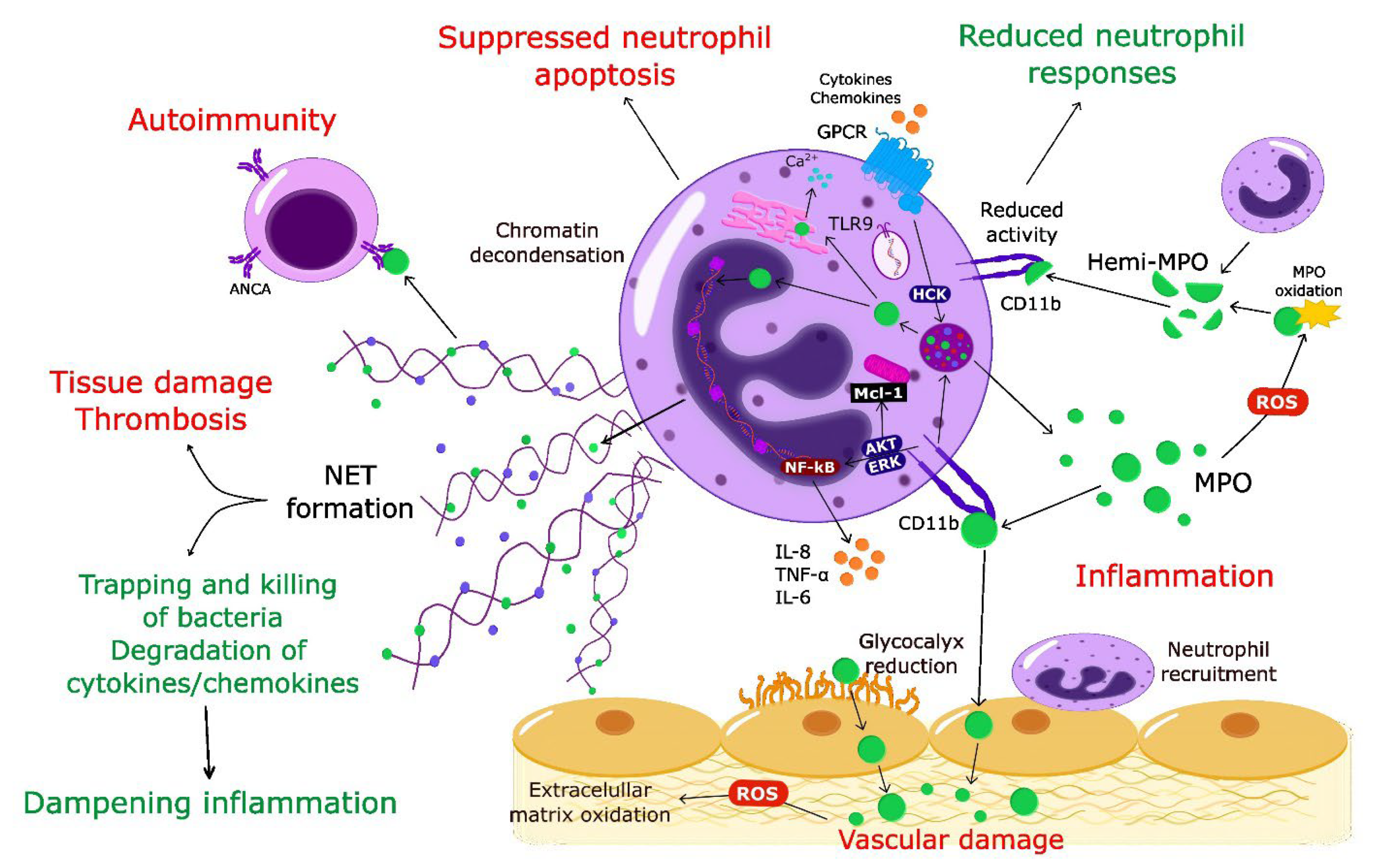
Figure 1. Multifaceted pro-inflammatory and protective actions of MPO. Activation of neutrophils through chemokine/cytokine receptors or TLR9 leads to release of MPO from primary granules MPO catalyzes local formation of highly toxic oxidants that contribute to tissue damage. MPO facilitates neutrophil trafficking into the inflamed site, evokes neutrophil activation, degranulation, and generates survival cues by suppressing constitutive neutrophil apoptosis through non-enzymatic actions on CD11b. MPO-CD11b form a feed-forward mechanism for perpetuating neutrophil-mediated inflammation. Dissociation of MPO into hemi-MPO results in reduced activity and blunted neutrophil responses. Intracellularly released MPO synergizes with neutrophil elastase to drive chromatin decondensation, leading to extrusion of neutrophil extracellular traps. NET-bound MPO contributes to bacterial killing and degrades cytokines/chemokines, thereby dampening inflammation. By contrast, NETs have been implicated in tissue damage and initiation of thrombosis. Aberrant NET formation (or impaired NET degradation) leads to prolonged presentation of MPO antigens, triggering autoimmunity. Red color indicates pro-inflammatory activities of MPO; green color refers to protective functions. ANCA, anti-neutrophil cytoplasmic antibody; GPCR, G protein-coupled receptor; NET, neutrophil extracellular trap; ROS, reactive oxygen species; TLR9, toll-like receptor 9.
References
- Kolaczkowska, E.; Kubes, P. Neutrophil Recruitment and Function in Health and Inflammation. Nat. Rev. Immunol. 2013, 13, 159–175.
- Nauseef, W.M.; Borregaard, N. Neutrophils at Work. Nat. Immunol. 2014, 15, 602–611.
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The Multifaceted Functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218.
- Reeves, E.P.; Lu, H.; Jacobs, H.L.; Messina, C.G.; Bolsover, S.; Gabella, G.; Potma, E.O.; Warley, A.; Roes, J.; Segal, A.W. Killing Activity of Neutrophils is Mediated through Activation of Proteases by K+ flux. Nature 2002, 416, 291–297.
- Liew, P.X.; Kubes, P. The Neutrophil’s Role during Health and Disease. Physiol. Rev. 2019, 99, 1223–1248.
- Malech, H.L.; Gallin, J.I. Current Concepts: Immunology. Neutrophils in Human Diseases. N. Engl. J. Med. 1987, 317, 687–694.
- Gordon, S. Phagocytosis: An Immunobiologic Process. Immunity 2016, 44, 463–475.
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535.
- Rørvig, S.; Østergaard, O.; Heegaard, N.H.H.; Borregaard, N. Proteome Profiling of Human Neutrophil Granule Subsets, Secretory Vesicles, and Cell Membrane: Correlation with Transcriptome Profiling of Neutrophil Precursors. J. Leukoc. Biol. 2013, 94, 711–721.
- Cassatella, M.; Östberg, N.K.; Tamassia, N.; Soehnlein, O. Biological Roles of Neutrophil-derived Granule Proteins and Cytokines. Trends Immunol. 2019, 40, 648–664.
- Klebanoff, S.J. Myeloperoxidase: Friend and Foe. J. Leukoc. Biol. 2005, 77, 598–625.
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882.
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the Activation and Regulation of Innate and Adaptive Immunity. Nat. Rev. Immunol. 2011, 11, 519–531.
- Baldus, S.; Heeschen, C.; Meinertz, T.; Zeiher, A.M.; Eiserich, J.P.; Münzel, T.; Simoons, M.L.; Hamm, C.W.; CAPTURE Investigators. Myeloperoxidase Serum Levels Predict Risk in Patients with Acute Coronary Syndromes. Circulation 2003, 108, 1440–1445.
- Brennan, M.L.; Penn, M.S.; Van Lente, F.; Nambi, V.; Shishehbor, M.H.; Aviles, R.J.; Goormastic, M.; Pepoy, M.L.; McErlean, E.S.; Topol, E.J.; et al. Prognostic Value of Myeloperoxidase in Patients with Chest Pain. N. Engl. J. Med. 2003, 349, 1595–1604.
- Nicholls, S.J.; Hazen, S.L. Myeloperoxidase and Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1102–1111.
- Kothari, N.; Keshari, R.S.; Bogra, J.; Kohli, M.; Abbas, H.; Malik, A.; Dikshit, M.; Barthwal, M.K. Increased Myeloperoxidase Enzyme Activity in Plasma is an Indicator of Inflammation and Onset of Sepsis. J. Crit. Care 2011, 26, e1–e7.
- Zhu, A.; Ge, D.; Zhang, J.; Teng, Y.; Yuan, C.; Huang, M.; Adcock, I.M.; Barnes, P.J.; Yao, X. Sputum Myeloperoxidase in Chronic Obstructive Pulmonary Disease. Eur. J. Med. Res. 2014, 19, 12.
- Aratani, Y. Myeloperoxidase: Its Role for Host Defense, Inflammation, and Neutrophil Function. Arch. Biochem. Biophys. 2018, 640, 47–52.
- Silvestre-Roig, C.; Hidalgo, A.; Soehnlein, O. Neutrophil Heterogeneity: Implications for Homeostasis and Pathogenesis. Blood 2016, 127, 2173–2181.
- Filep, J.G.; Ariel, A. Inflammation: From Cellular Mechanisms to Immune Cell Education: Neutrophil Heterogeneity and Fate in Inflamed Tissues: Implications for the Resolution of Inflammation. Am. J. Physiol. Cell Physiol. 2020, 319, C510–C532.
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat. Rev. Immunol. 2007, 7, 678–689.
- Johansson, M.W.; Patarroyo, M.; Oberg, F.; Siegbahn, A.; Nilsson, K. Myeloperoxidase Mediates Cell Adhesion via the Alpha M Beta 2 Integrin (Mac-1, CD11b/CD18). J. Cell. Sci. 1997, 110, 1133–1139.
- Lau, D.; Mollnau, H.; Eiserich, J.P.; Freeman, B.A.; Daiber, A.; Gehling, U.M.; Brümmer, J.; Rudolph, V.; Münzel, T.; Heitzer, T.; et al. Myeloperoxidase Mediates Neutrophil Activation by Association with CD11b/CD18 Integrins. Proc. Natl. Acad. Sci. USA 2005, 102, 431–436.
- Gorudko, I.V.; Grigorieva, D.V.; Sokolov, A.V.; Shamova, E.V.; Kostevich, V.A.; Kudryavtsev, I.V.; Syromiatnikova, E.D.; Vasilyev, V.B.; Cherenkevich, S.N.; Panasenko, O.M. Neutrophil Activation in Response to Monomeric Myeloperoxidase. Biochem. Cell. Biol. 2018, 96, 592–601.
- Klinke, A.; Nussbaum, C.; Kubala, L.; Friedrichs, K.; Rudolph, T.K.; Rudolph, V.; Paust, H.J.; Schröder, C.; Benten, D.; Lau, D.; et al. Myeloperoxidase Attracts Neutrophils by Physical Forces. Blood 2011, 117, 1350–1358.
- Manchanda, K.; Kolarova, H.; Kerkenpaß, C.; Mollenhauer, M.; Vitecek, J.; Rudolph, V.; Kubala, L.; Baldus, S.; Adam, M.; Klinke, A. MPO (Myeloperoxidase) Reduces Endothelial Glycocalyx Thickness Dependent on Its Cationic Charge. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1859–1867.
- El Kebir, D.; József, L.; Pan, W.; Filep, J.G. Myeloperoxidase Delays Neutrophil Apoptosis through CD11b/CD18 Integrins and Prolongs Inflammation. Circ. Res. 2008, 103, 352–359.
- Demaret, J.; Venet, F.; Friggeri, A.; Cazalis, M.A.; Plassais, J.; Jallades, L.; Malcus, C.; Poitevin-Later, F.; Textoris, J.; Lepape, A.; et al. Marked Alterations of Neutrophil Functions during Sepsis-induced Immunosuppression. J. Leukoc. Biol. 2015, 98, 1081–1090.
- Matthijsen, R.A.; Huugen, D.; Hoebers, N.T.; de Vries, B.; Peutz-Kootstra, C.J.; Aratani, Y.; Daha, M.R.; Tervaert, J.W.C.; Buurman, W.A.; Heeringa, P. Myeloperoxidase Is Critically Involved in the Induction of Organ Damage after Renal Ischemia Reperfusion. Am. J. Pathol. 2007, 171, 1743–1752.
- Brovkovych, V.; Gao, X.P.; Ong, E.; Brovkovych, S.; Brennan, M.L.; Su, X.; Hazen, S.L.; Malik, A.B.; Skidgel, R.A. Augmented Inducible Nitric Oxide Synthase Expression and Increased NO Production Reduce Sepsis-Induced Lung Injury and Mortality in Myeloperoxidase-Null Mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L96–L103.
- Kim, H.J.; Wei, Y.; Lee, J.Y.; Wu, Y.; Zheng, Y.; Moskowitz, M.A.; Chen, J.W. Myeloperoxidase Inhibition Increases Neurogenesis after Ischemic Stroke. J. Pharmacol. Exp. Ther. 2016, 359, 262–272.
- Odobasic, D.; Muljadi, R.C.; O’Sullivan, K.M.; Kettle, A.J.; Dickerhof, N.; Summers, S.A.; Kitching, A.R.; Holdsworth, S.R. Suppression of Autoimmunity and Renal Disease in Pristane-Induced Lupus by Myeloperoxidase. Arthritis Rheumatol. 2015, 67, 1868–1880.
- Tseng, A.; Kim, K.; Li, J.; Cho, J. Myeloperoxidase Negatively Regulates Neutrophil-Endothelial Cell Interactions by Impairing αMβ2 Integrin Function in Sterile Inflammation. Front. Med. 2018, 5, 134.
- Reber, L.L.; Gillis, C.M.; Starkl, P.; Jönsson, F.; Sibilano, R.; Marichal, T.; Gaudenzio, N.; Bérard, M.; Rogalla, S.; Contag, C.H.; et al. Neutrophil Myeloperoxidase Diminishes the Toxic Effects and Mortality Induced by Lipopolysaccharide. J. Exp. Med. 2017, 214, 1249–1258.
- Schürmann, N.; Forrer, P.; Casse, O.; Li, J.; Felmy, B.; Burgener, A.V.; Ehrenfeuchter, N.; Hardt, W.D.; Recher, M.; Hess, C.; et al. Myeloperoxidase Targets Oxidative Host Attacks to Salmonella and Prevents Collateral Tissue Damage. Nat. Microbiol. 2017, 2, 16268.
- Zhang, R.; Brennan, M.-L.; Shen, Z.; MacPherson, J.C.; Schmitt, D.; Molenda, C.E.; Hazen, S.L. Myeloperoxidase Functions as a Major Enzymatic Catalyst for Initiation of Lipid Peroxidation at Sites of Inflammation. J. Biol. Chem. 2002, 277, 46116–46122.
- Kubala, L.; Schmelzer, K.R.; Klinke, A.; Kolarova, H.; Baldus, S.; Hammock, B.D.; Eiserich, J.P. Modulation of Arachidonic and Linoleic Acid Metabolites in Myeloperoxidase-deficient Mice During Acute Inflammation. Free Radic. Biol. Med. 2010, 48, 1311–1320.
- Rehring, J.F.; Bui, T.M.; Galán-Enríquez, C.S.; Urbanczyk, J.M.; Ren, X.; Wiesolek, H.L.; Sullivan, D.P.; Sumagin, R. Released Myeloperoxidase Attenuates Neutrophil Migration and Accumulation in Inflamed Tissue. Front. Immunol. 2021, 12, 654259.
- Odobasic, D.; Kitching, A.R.; Semple, T.J.; Holdsworth, S.R. Endogenous Myeloperoxidase Promotes Neutrophil-mediated Renal Injury, but Attenuates T Cell Immunity Inducing Crescentic Glomerulonephritis. J. Am. Soc. Nephrol. 2007, 18, 760–770.
- Rausch, P.G.; Moore, T.G. Granule Enzymes of Polymorphonuclear Neutrophils: A Phylogenetic Comparison. Blood 1975, 46, 913–919.
- Jerke, U.; Rolle, S.; Purfürst, B.; Luft, F.C.; Nauseef, W.M.; Kettritz, R. β2 Integrin-Mediated Cell-Cell Contact Transfers Active Myeloperoxidase from Neutrophils to Endothelial Cells. J. Biol. Chem. 2013, 288, 12910–12919.
- Slater, T.W.; Finkielsztein, A.; Mascarenhas, L.A.; Mehl, L.C.; Butin-Israeli, V.; Sumagin, R. Neutrophil Microparticles Deliver Active Myeloperoxidase to Injured Mucosa to Inhibit Epithelial Wound Healing. J. Immunol. 2017, 198, 2886–2897.
- Babior, B.M. NADPH Oxidase. Curr. Opin. Immunol. 2004, 16, 42–47.
- Park, K.J.; Gaynor, R.B.; Tae Kwak, Y. Heat Shock Protein 27 Association with the IκB Kinase Complex Regulates Tumor Necrosis Factor α-Induced NF-κB Activation. J. Biol. Chem. 2003, 278, 35272–35278.
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687.
- Harris, E.S.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A. The Leukocyte Integrins. J. Biol. Chem. 2000, 275, 23409–23412.
- El Kebir, D.; József, L.; Pan, W.; Wang, L.; Petasis, N.A.; Serhan, C.N.; Filep, J.G. 15-Epi-Lipoxin A4 Inhibits Myeloperoxidase Signaling and Enhances Resolution of Acute Lung Injury. Am. J. Respir. Crit. Care Med. 2009, 180, 311–319.
- Dupuy, A.G.; Caron, E. Integrin-Dependent Phagocytosis–Spreadingfrom Microadhesion to New Concepts. J. Cell. Sci. 2008, 121, 1773–1783.
- Fujimoto, K.; Motowaki, T.; Tamura, N.; Aratani, Y. Myeloperoxidase Deficiency Enhances Zymosan Phagocytosis Associated with Up-Regulation of Surface Expression of CD11b in Mouse Neutrophils. Free Radic. Res. 2016, 50, 1340–1349.
- Müller, J.M.; Rupec, R.A.; Bauerle, P.A. Study of Gene Regulation by NF-κB and AP-1 in Response to Reactive Oxygen Intermediates. Methods 1997, 11, 301–312.
- József, L.; Zouki, C.; Petasis, N.A.; Serhan, C.N.; Filep, J.G. Lipoxin A4 and Aspirin-triggered 15-epi-lipoxin A4 Inhibit Peroxynitrite Formation, NF-κB and AP-1 Activation, and IL-8 Gene Expression in Human Leukocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 13266–13271.
- Winterbourn, C.C.; Hampton, M.B.; Livesey, J.H.; Kettle, A.J. Modeling the Reactions of Superoxide and Myeloperoxidase in the Neutrophil Phagosome: Implications for Microbial Killing. J. Biol. Chem. 2006, 281, 39860–39869.
- Lane, A.E.; Tan, J.T.M.; Hawkins, C.L.; Heather, A.K.; Davies, M.J. The Myeloperoxidase-Derived Oxidant HOSCN Inhibits Protein Tyrosine Phosphatases and Modulates Cell Signalling via the Mitogen-Activated Protein Kinase (MAPK) Pathway in Macrophages. Biochem. J. 2010, 430, 161–169.
- Guo, C.; Davies, M.J.; Hawkins, C.L. Role of Thiocyanate in the Modulation of Myeloperoxidase-Derived Oxidant Induced Damage to Macrophages. Redox. Biol. 2020, 36, 101666.
- Haegens, A.; Heeringa, P.; van Suylen, R.J.; Steele, C.; Aratani, Y.; O’Donoghue, R.J.J.; Mutsaers, S.E.; Mossman, B.T.; Wouters, E.F.M.; Vernooy, J.H.J. Myeloperoxidase Deficiency Attenuates Lipopolysaccharide-Induced Acute Lung Inflammation and Subsequent Cytokine and Chemokine Production. J. Immunol. 2009, 182, 7990–7996.
- Endo, D.; Saito, T.; Umeki, Y.; Suzuki, K.; Aratani, Y. Myeloperoxidase Negatively Regulates the Expression of Proinflammatory Cytokines and Chemokines by Zymosan-Induced Mouse Neutrophils. Inflamm. Res. 2015, 65, 151–159.
- Savill, J.; Dransfield, I.; Gregory, C.; Haslett, C. A Blast from the Past: Clearance of Apoptotic Cells Regulates Immune Responses. Nat. Rev. Immunol. 2002, 2, 965–975.
- Rubel, C.; Gómez, S.; Fernández, G.C.; Isturiz, M.A.; Caamaño, J.; Palermo, M.S. Fibrinogen-CD11b/CD18 Interaction Activates the NF-κB Pathway and Delays Apoptosis in Human Neutrophils. Eur. J. Immunol. 2003, 33, 1429–1438.
- Yan, M.; Mao, J.; Wei, Y.; Zhong, J.; Yang, S.; Xu, R. Study on Intracellular Trafficking of Mac-1 by Direct Visualization. Sci. China C Life Sci. 2004, 47, 521–529.
- Whitlock, B.B.; Gardai, S.; Fadok, V.; Bratton, D.; Henson, P.M. Differential Roles for αMβ2 Integrin Clustering or Activation in the Control of Apoptosis via Regulation of Akt and ERK Survival Mechanisms. J. Cell Biol. 2000, 151, 1305–1320.
- Dzhagalov, I.; St. John, A.; He, Y.W. The Antiapoptotic Protein Mcl-1 Is Essential for the Survival of Neutrophils but Not Macrophages. Blood 2007, 109, 1620–1626.
- Pluskota, E.; Soloviev, D.A.; Szpak, D.; Weber, C.; Plow, E.F. Neutrophil Apoptosis: Selective Regulation by Different Ligands of Integrin αMβ2. J. Immunol. 2008, 181, 3609–3619.
- Rossi, A.G.; Sawatzky, D.A.; Walker, A.; Ward, C.; Sheldrake, T.A.; Riley, N.A.; Caldicott, A.; Martinez-Losa, M.; Walker, T.R.; Duffin, R.; et al. Cyclin-Dependent Kinase Inhibitors Enhance the Resolution of Inflammation by Promoting Inflammatory Cell Apoptosis. Nat. Med. 2006, 12, 1056–1064, Erratum in Nat. Med. 2006, 12, 1434.
- Wagner, B.A.; Buettner, G.R.; Oberley, L.W.; Darby, C.J.; Burns, C.P. Myeloperoxidase Is Involved in H2O2-Induced Apoptosis of HL-60 Human Leukemia Cells. J. Biol. Chem. 2000, 275, 22461–22469.
- Kanayama, A.; Miyamoto, Y. Apoptosis Triggered by Phagocytosis-Related Oxidative Stress through FLIPS down-Regulation and JNK Activation. J. Leukoc. Biol. 2007, 82, 1344–1352.
- Bardoel, B.W.; Kenny, E.F.; Sollberger, G.; Zychlinsky, A. The Balancing Act of Neutrophils. Cell Host Microbe 2014, 15, 526–536.
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil Extracellular Traps Capture and Kill Candida albicans Yeast and Hyphal Forms. Cell Microbiol. 2006, 8, 668–676.
- Erreni, M.; Manfredi, A.A.; Garlanda, C.; Mantovani, A.; Rovere-Querini, P. The Long Pentraxin PTX3: A Prototypical Sensor of Tissue Injury and a Regulator of Homeostasis. Immunol. Rev. 2017, 280, 112–125.
- Yuen, J.; Pluthero, F.G.; Douda, D.N.; Riedl, M.; Cherry, A.; Ulanova, M.; Kahr, W.H.; Palaniyar, N.; Licht, C. NETosing Neutrophils Activate Complement both on their own NETs and Bacteria via Alternative and Non-alternative Pathways. Front. Immunol. 2016, 7, 137.
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil Elastase and Myeloperoxidase Regulate the Formation of Neutrophil Extracellular Traps. J. Cell Biol. 2010, 191, 677–691.
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J. Cell. Biol. 2007, 176, 231–241.
- Tessarz, P.; Kouzarides, T. Histone Core Modifications Regulating Nucleosome Structure and Dynamics. Nat. Rev. Mol. Cell. Biol. 2014, 15, 703–708.
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is Required for Neutrophil Extracellular Trap Formation: Implications for Innate Immunity. Blood 2011, 117, 953–959.
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425.
- Byrd, A.S.; O’Brien, X.M.; Johnson, C.M.; Lavigne, L.M.; Reichner, J.S. An Extracellular Matrix-Based Mechanism of Rapid Neutrophil Extracellular Trap Formation in Response to Candida albicans. J. Immunol. 2013, 190, 4136–4148.
- Cristinziano, L.; Modestino, L.; Loffredo, S.; Varricchi, G.; Braile, M.; Ferrara, A.L.; de Paulis, A.; Antonelli, A.; Marone, G.; Galdiero, M.R. Anaplastic Thyroid Cancer Cells Induce the Release of Mitochondrial Extracellular DNA Traps by Viable Neutrophils. J. Immunol. 2020, 204, 1362–1372.
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable Neutrophils Release Mitochondrial DNA to Form Neutrophil Extracellular Traps. Cell Death Differ. 2009, 16, 1438–1444.
- Cristinziano, L.; Modestino, L.; Antonelli, A.; Marone, G.; Simon, H.U.; Varricchi, G.; Galdiero, M.R. Neutrophil Extracellular Traps in Cancer. Semin. Cancer Biol. 2022, 79, 91–104.
- Yousefi, S.; Stojkov, D.; Germic, N.; Simon, D.; Wang, X.; Benarafa, C.; Simon, H.U. Untangling “NETosis” from NETs. Eur. J. Immunol. 2019, 49, 221–227.
- Manfredi, A.A.; Ramirez, G.A.; Rovere-Querini, P.; Maugeri, N. The Neutrophil’s Choice: Phagocytose vs Make Neutrophil Extracellular Traps. Front. Immunol. 2018, 9, 288.
- Maugeri, N.; Rovere-Querini, P.; Evangelista, V.; Covino, C.; Capobianco, A.; Bertilaccio, M.T.S.; Piccoli, A.; Totani, L.; Cianflone, D.; Maseri, A.; et al. Neutrophils Phagocytose Activated Platelets in Vivo: A Phosphatidylserine, P-Selectin, and β2 Integrin-Dependent Cell Clearance Program. Blood 2009, 113, 5254–5265.
- Papayannopoulos, V. Neutrophil Extracellular Traps in Immunity and Disease. Nat. Rev. Immunol. 2018, 18, 134–147.
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2021, 61, 194–211.
- Sørensen, O.E.; Borregaard, N. Neutrophil Extracellular Traps—the Dark Side of Neutrophils. J. Clin. Investig. 2016, 126, 1612–1620.
- Jorch, S.K.; Kubes, P. An Emerging Role for Neutrophil Extracellular Traps in Noninfectious Disease. Nat. Med. 2017, 23, 279–287.
- Ribon, M.; Seninet, S.; Mussard, J.; Sebbag, M.; Clavel, C.; Serre, G.; Boissier, M.C.; Semerano, L.; Decker, P. Neutrophil Extracellular Traps Exert both Pro- and Anti-inflammatory Actions in Rheumatoid Arthritis that are Modulated by C1q and LL-37. J. Autoimmun. 2019, 98, 122–131.
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weidner, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated Neutrophil Extracellular Traps Resolve Inflammation by Proteolysis of Cytokines and Chemokines and Protection from Antiproteases. FASEB J. 2019, 33, 1401–1414.
- Li, Y.; Cao, X.; Liu, Y.; Zhao, Y.; Herrmann, M. Neutrophil Extracellular Traps Formation and Aggregation Orchestrate Induction and Resolution of Sterile Crystal-Mediated Inflammation. Front. Immunol. 2018, 9, 1559.
- Jennette, J.C.; Xiao, H.; Falk, R.J. Pathogenesis of Vascular Inflammation by Anti-Neutrophil Cytoplasmic Antibodies. J. Am. Soc. Nephrol. 2006, 17, 1235–1242.
- Gupta, S.; Kaplan, M.J. The Role of Neutrophils and NETosis in Autoimmune and Renal Diseases. Nat. Rev. Nephrol. 2016, 12, 402–413.
- Austin, K.; Janagan, S.; Wells, M.; Crawshaw, H.; McAdoo, S.; Robson, J.C. ANCA Associated Vasculitis Subtypes: Recent Insights and Future Perspectives. J. Inflamm. Res. 2022, 15, 2567–2582.
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of Neutrophil Extracellular Trap Degradation Is Associated with Lupus Nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818.
- Csernok, E. Anti-Neutrophil Cytoplasmic Antibodies and Pathogenesis of Small Vessel Vasculitides. Autoimmun. Rev. 2003, 2, 158–164.
- Nakazawa, D.; Shida, H.; Tomaru, U.; Yoshida, M.; Nishio, S.; Atsumi, T.; Ishizu, A. Enhanced Formation and Disordered Regulation of NETs in Myeloperoxidase-ANCA-Associated Microscopic Polyangiitis. J. Am. Soc. Nephrol. 2014, 25, 990–997.
- Xiao, H.; Heeringa, P.; Hu, P.; Liu, Z.; Zhao, M.; Aratani, Y.; Maeda, N.; Falk, R.J.; Jennette, J.C. Antineutrophil Cytoplasmic Autoantibodies Specific for Myeloperoxidase Cause Glomerulonephritis and Vasculitis in Mice. J. Clin. Investig. 2002, 110, 955–963.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Revisions:
2 times
(View History)
Update Date:
29 Nov 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No