 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rajib Sengupta | -- | 4863 | 2022-11-25 13:01:49 | | | |
| 2 | Lindsay Dong | -5 word(s) | 4858 | 2022-11-28 02:49:38 | | |
Video Upload Options
S-nitrosylation of proteins occurs as a consequence of the derivatization of cysteine thiols with nitric oxide (NO) and is often associated with diseases and protein malfunction. Aberrant S-nitrosylation, in addition to other genetic and epigenetic factors, has gained rapid importance as a prime cause of various metabolic, respiratory, and cardiac disorders, with a major emphasis on cancer and neurodegeneration. The S-nitrosoproteome, a term used to collectively refer to the diverse and dynamic repertoire of S-nitrosylated proteins, is relatively less explored in the field of redox biochemistry, in contrast to other covalently modified versions of the same set of proteins. Advancing research is gradually unveiling the enormous clinical importance of S-nitrosylation in the etiology of diseases and is opening up new avenues of prompt diagnosis that harness this phenomenon. Ever since the discovery of the two robust and highly conserved S-nitrosoglutathione reductase and thioredoxin systems as candidate denitrosylases, years of rampant speculation centered around the identification of specific substrates and other candidate denitrosylases, subcellular localization of both substrates and denitrosylases, the position of susceptible thiols, mechanisms of S-denitrosylation under basal and stimulus-dependent conditions, impact on protein conformation and function, and extrapolating these findings towards the understanding of diseases, aging and the development of novel therapeutic strategies.
1. Introduction
Nitric oxide (NO), a tiny, lipophilic, redox-active molecule, acts as a catalyst for the activation of the effector Guanylyl cyclase, which in turn mediates the creation of cyclic-GMP (cGMP) from GTP, triggering an intracellular chain of signaling events when present at the right concentration (low nanomolar levels). The relevance of the molecule in redox signaling and homeostasis is exemplified by intracellular amplification of the message conveyed by NO to the cell and its subsequent translation into large-scale alterations seen in both the extracellular and intracellular milieu.Nitric Oxide Synthases (NOSs) are enzymes that produce NO.Disruptions in the regulatory systems that keep NO levels in check could result in an excess of NO and subsequently other free radicals, which could cause a variety of illnesses such as carcinomas, ulcerative colitis, multiple sclerosis, and juvenile diabetes, in addition to direct tissue toxicity and vascular collapse [1][2][3][4].
Free radicals are produced inside the cell as intermediates of several redox reactions and are crucial for redox signaling in the body. Free radicals can be generically categorized into reactive oxygen species (ROS) and reactive nitrogen species (RNS). ROS comprises toxic intermediates of oxygen metabolism, such as superoxide (O2−), hydroxyl (HO.), alkoxyl, peroxyl, and hydroperoxyl radicals, along with hydrogen peroxide (H2O2). RNS comprises reactive nitrogen intermediates, such as NO, nitrogen dioxide (NO2.), and peroxynitrite (ONOO-) [5][6]. Another important function of NO is post-translational control of protein function through covalent modification of amino acids such ascysteine thiol(ates)s, which results in the formation of S-nitrosylated proteins/S-nitrosoproteins (PSNOs), which may either have positive physiological effects (such as the role of S-nitrosoglutathione (GSNO) as an endogenous bronchodilator and NO-mediated activation of platelet specific integrin αIIbβ3) or negative pathological outcomes (as is the case in neurodegenerative diseases, and various respiratory, metabolic, and cardiac ailments) [7][8][9][10][11].
One can gauge the clinical impact of free radical overload and improper oxidative modifications of proteins by examining the myriad of molecular malfunctions and signal misfiring caused by excessive ROS/RNS, which ultimately result in cell death or neoplasia due to the inactivation or hyperactivation of various kinases/phosphatases such as the phosphatase and tensin homolog (PTEN). A daily supply of antioxidants, either from food or other nutraceuticals, is required to counteract exposure to exogenous and endogenous prooxidants or chemicals that induce a burst of ROS/RNS within the body, particularly because the levels of beneficial antioxidants decline with age. Antioxidants are chemicals that control the levels of ROS and RNS within cells and, in some situations, undo abnormal oxidative alterations to biomolecules. Hence, in addition to controlling the number of free radicals in the cell, antioxidants also make sure that other antioxidants and de-nitrosylases, such as reduced glutathione (GSH), thioredoxin (Trx), and glutaredoxin (Grx), do not suffer from a deficiency [12][13][14][15]. De-nitrosylases are a collection of small molecules and enzymatic systems that are deployed by the cell to reverse the harmful, aberrant modifications of cysteine thiols of key intracellular proteins. The existence of an ornately designed network of multiple denitrosylases indicates the presence of a combinatorial approach towards denitrosylation, facilitated by the synergistic cum complementary, and additive actions of the above-mentioned (and other) denitrosylating systems, ascribable to their ‘redundant’ and ‘substrate specific’ nature, respectively.
2. GSH Synthesis and Denitrosylation
2.1. GSH Synthesis
This tripeptide is a pivotal Non-Protein Thiol (NPSH) present in 5–10 mM concentrations physiologically and is synthesized in the cytosol from glutamate through a feedback mechanism [16]. Several pathways have come to light related to the biosynthesis of ϒ-glutamylcysteine from the conjugation of glutamate and cysteine; the most common being the de novo GSH synthesis through the ϒ-glutamyl cycle, involving ϒ-glutamyl transpeptidase, peptidase, and ϒ-glutamylcysteine synthetase to catalyze the biosynthesis. The same pathway can be simplified with the help of glutathione synthetases (Gsh1, Gsh2, GshF) and prolinases [17][18]. This pathway is the most crucial due to the involvement of the ϒ-glutamyl transpeptidase, the only enzyme localized on the outer plasma membrane, and is directly involved in stress-regulation and cellular GSH homeostasis, besides its role in the extracellular hydrolysis of GSH into cysteine and the intercellular resynthesis of GSH in oxidants such as human beings [19]. Another way is to conjugate cysteine with glutamate, forming ϒ-glutamylcysteine through a two-step ATP-acquiring enzymatic process involving glutamate-cysteine ligase as the catalyst for the first step [18]. The later steps of the pathway are similar for all the studied ways. ϒ-glutamylcysteine, with the help of glutathione synthetases, is reduced to form reduced glutathione (GSH), which is further oxidized (GSSG) depending on the amount of positive or negative feedback generated in the ϒ-glutamyl cycle in the presence of a particular GSH/GSSG ratio in the mammalian system [17][20].
2.2. S-Nitrosylation of Glutathione
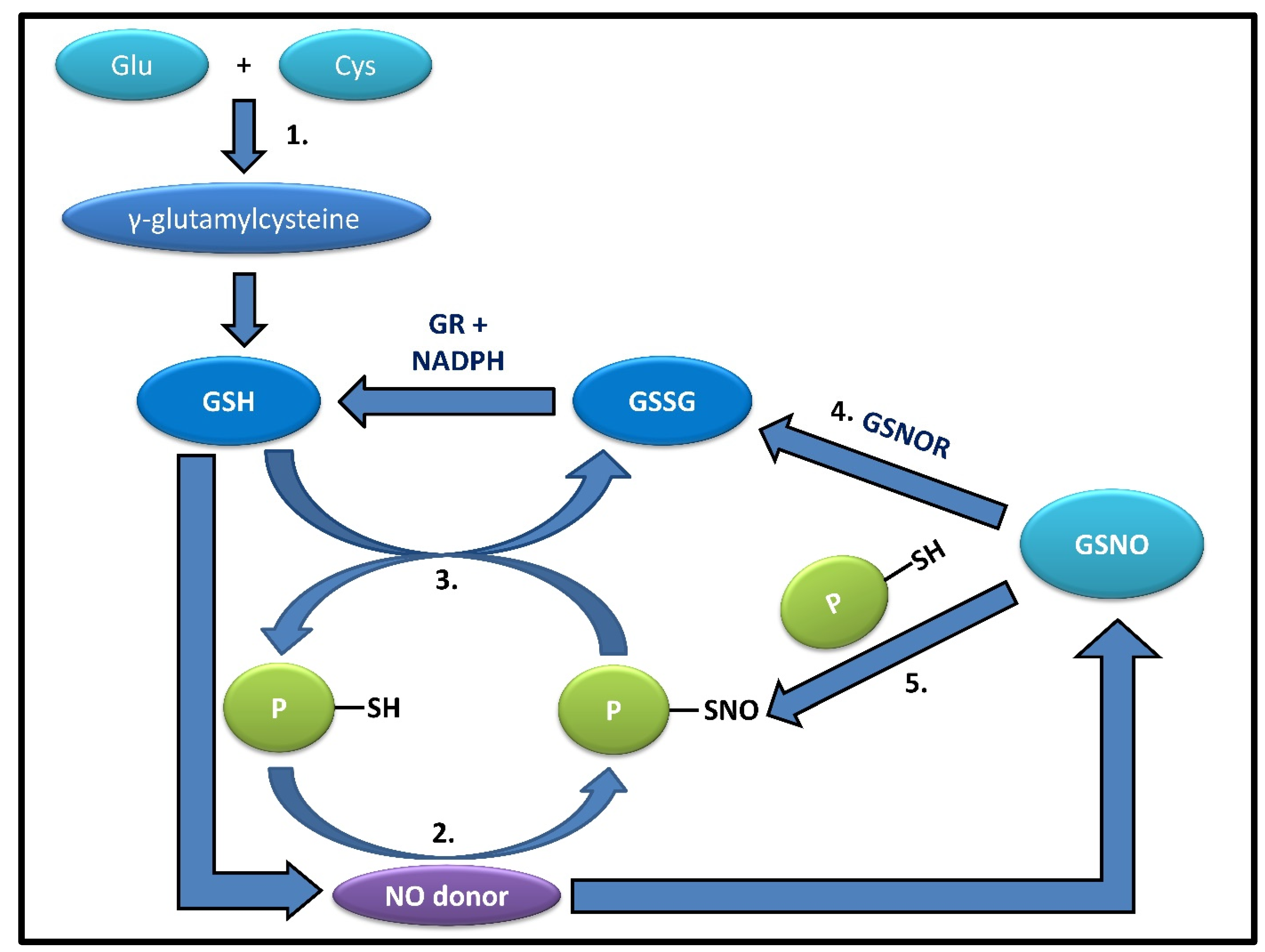
2.3. Role of GSH in S-Denitrosylation
It has been well established that in active multiple sclerosis patients, there is a significant increase in the concentration of NO metabolites and low molecular mass thiols in cerebrospinal fluid. with a larger addition of GSH-EE, the rate of protein denitrosylation increases accordingly [26]. PSNO levels fall at a quicker rate in the presence of endogenous cellular GSH since the addition of GSH-EE causes an increase in NPSH concentration, which mostly consists of GSH, facilitating the increased presence of GSH, suggesting a faster rate of denitrosylation by GSH. This inverse relationship between NPSH and PSNO levels remaining after the chase keeps up with the notion that the cellular concentration of GSH is a primary determinant of nitrosothiols’ stability. Depletion of intracellular GSH prevents protein denitrosylation [27]. When GSH was depleted with GSH depletion DEM, the steady-state level of PSNO remained stable and was higher than that in presence of GSH. GSH depletion facilitates S-nitrosylation in a way where GSH will accept NO from PSNOs generating GSNO, which will, in turn, be metabolized by GSNOR [28][29][30] (Figure 1). This signifies that the depletion of GSH prevents PSNO denitrosylation and that low amounts of GSH are enough to keep protein thiols in a reduced state. Intracellular GSH was also restored by adding GSH-EE and expectedly found an increase in protein denitrosylation. Previously when GSH-EE was added, the PSNOs did not disappear completely, showing resistance to GSH, and during its replenishment, the original PSH level could not be retrieved, signifying that either the oxidized thiols are metabolically stable or inaccessible to GSH because of their location inside hydrophobic protein pockets [27]. GSH carries out the protein denitrosylation in two ways, either via transnitrosylation with PSNOs or by reaction of GSH with PSNO to form S-glutathionylated protein or PSSG (Figure 2). All these findings suggest that decreased GSH levels lead to a decrease in the rate of S-denitrosylation by GSH, resulting in the accumulation of PSNOs in significant amounts in the white matter of MS patients helping in the aggravation of pathogenesis in active MS patients via increased nitrosative stress and protein S-nitrosylation [31]. The most known mechanism is the formation of GSNOR and then the denitrosylation of cysteine thiols by the same. This GSNOR is active as a homodimer with the greatest activity in the liver and found in maximum content in the cytoplasm, which can also be found in the nucleus [24]. It is GSH-dependent formaldehyde dehydrogenase that can reduce GSNO [29]. It is a major regulator that can control both GSNO and PSNO levels to maintain their cellular equilibrium [3]. There was an increase in cellular levels of GSNO and PSNO when the GSNOR gene, i.e., human gene ADH5, was deleted. There is evidence that the genetic Knock Out of GSNOR in mice results in high levels of PSNO and displays multi-organ dysfunction, mortality in models of sepsis, low systemic vascular resistance, and high susceptibility to hypertension, showing that abnormal GSNOR-dependent denitrosylation is relevant to human disease [32]. As stated earlier, GSNOR does not denitrosylate proteins directly; rather, it lowers PSNO levels by pushing the equilibrium from PSNO to GSNO [33]. It is done in a way where GSH will accept NO from PSNOs to generate GSNO, which can then be metabolized by GSNOR [28]. The denitrosylation mechanism involves the reduction of PSNO to PSH and the generation of GSNO by binding one molecule of GSH [34]. This GSNO is reduced by GSNOR using NADH forming GSSG, which is also reduced by GR to replenish GSH back into the cell. The actual function of GSNOR is to metabolize or reduce the endogenously generated GSNO, whose level fluctuates markedly upon the GSH concentration depending on the nitrosative and oxidative stress inside the cell [34]. This prompt disposal of GSNO helps the equilibrium shift towards the denitrosylated state. Hence, it is evident that GSH alone cannot fully terminate SNO-signaling or protect the protein from S-nitrosylation in the absence of GSNOR [30].
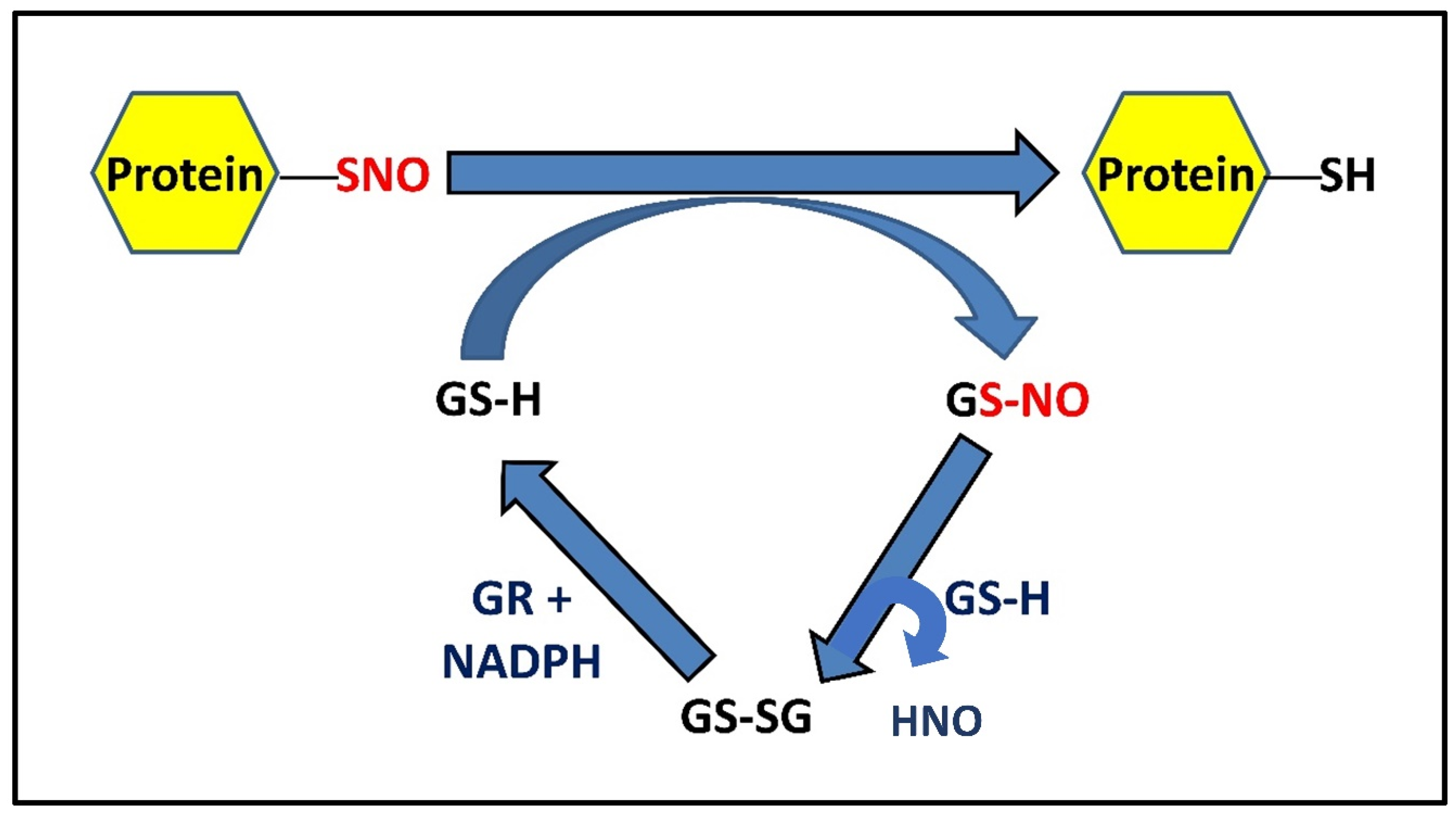
2.4. PSNO Denitrosylation and Exceptions Amongst PSNOs
The cellular redox state is often altered by palmitoylation, henceforth affecting the functionality of cysteine thiols to maintain proper cytosolic levels of GSH required for protein denitrosylation [36][37][38]. Protein nitrosylation and denitrosylation have exerted an effect on the majority of cellular proteins. Denitrosylation of these proteins is catalyzed by certain enzymes such as GSH, glutathione peroxidase (GPx), Grx, and Trx, while the perfect ratio of GSH/GSSG is ensured by glutathione reductase (GR) and GPx [17][39].
The occurrence of S-glutathionylation of proteins is largely dependent on the GSH/GSSG ratio [40][41]. Since denitrosylation is catalyzed by GSH, and S-glutathionylation releases GSH, both are interdependent; results establish the direct proportionality of GSNO reduction to that of protein-SNO reduction while also suggesting the much larger redox pool of nM levels of GSNO and protein-SNO against mM level of GSH [42][43]. NaK-ATPase, etc.,have been explored in the context of post-translational regulatory mechanisms that can be crucial in modulating and fine-tuning the protein/enzymatic activity, thereby restoring cellular redox balance under basal homeostatic as well as in pathophysiological conditions [44]. Similarly, about a hundred proteins are capable of S-(de)nitrosylation, another highly stringent post-translational redox modification within the intracellular milieu, where both the S-nitrosylated as well as denitrosylated proteins are responsible for cellular NO-signaling. Very few protein-SNOs are stable or are not denitrosylated in physiological GSH concentrations; these proteins include caspase-3, α-tubulin, β-tubulin, collapsin response mediator protein-2, Creatine kinase (B chain), extracellular signal-regulated kinase-2, glutathione-S-transferase (GST) pi, glyceraldehyde-3-phosphate dehydrogenase (GADPH), hemoglobin (β chain), pyruvate kinase, and Prx6 [27][45][46][47]. Amongst all these protein-SNOs, the most prominent is caspase-3, which cannot be denitrosylated by GSH or GSNOR, but exclusively by Trx; this proves the S-nitrosylation of this apoptotic protein by NO donors, but the failure of the activity regeneration in the absence of Trx, which again establishes a link between caspases and cancer and eliminates the direct role of GSH in the same, while indirectly GSH as well as reduced human Trxs have the potential to denitrosylated S-nitroso thioredoxins, inactivating caspase-3, linking GSH to the apoptotic pathways [12][48]. All these co-relations between the proteins and the antioxidant entrench a bigger picture, linking the well-established roles of GSH to tumor progression and regression while also suggesting the importance of the maintenance of the GSH/GSSG ratio.
3. Grx-Mediated denitrosylation and Deglutathionylation
Evidence from numerous research demonstrates the synergistic as well as the additive nature of the interaction of GSH with these enzymatic systems, such as the Trx and Grx systems. While Grx3 and Grx5 are mitochondrial and have only one cysteine in their active site, Grx1 and Grx2 are largely cytosolic (although Grx2a is found in the mitochondrial matrix) and marked by the presence of a dyad of redox-active thiols, instead of one. In the dithiol mechanism, the two cysteines are present in a CXXC motif and are distinguished by a difference in their pKa values. This difference causes one of the cysteines to ionize at the physiological pH and causes the other to remain in the protonated form. The NO attached to substrate nitrosothiols is quickly exchanged by the ionized cysteine, thus reducing them. Subsequent formation of a redox-active disulfide bridge causes Grx to quickly transform from its intermediate nitrosylated form to its completely oxidized form (accompanied by the concomitant release of nitroxyl [HNO] radical). Two molecules of GSH are necessary for the subsequent restoration of Grx activity through the reduction of oxidized Grx. In the monothiol process, the solitary cysteine in the active site directly reduces the substrate RSNOs, which is followed by the production of Grx-SNO, which is then subsequently acted upon and reduced by a molecule of GSH, releasing GSNO in the process (Figure 3). GSNO can then either serve as an NO donor, S-nitrosylating protein thiols, or it can combine with another GSH molecule to produce GSSG.
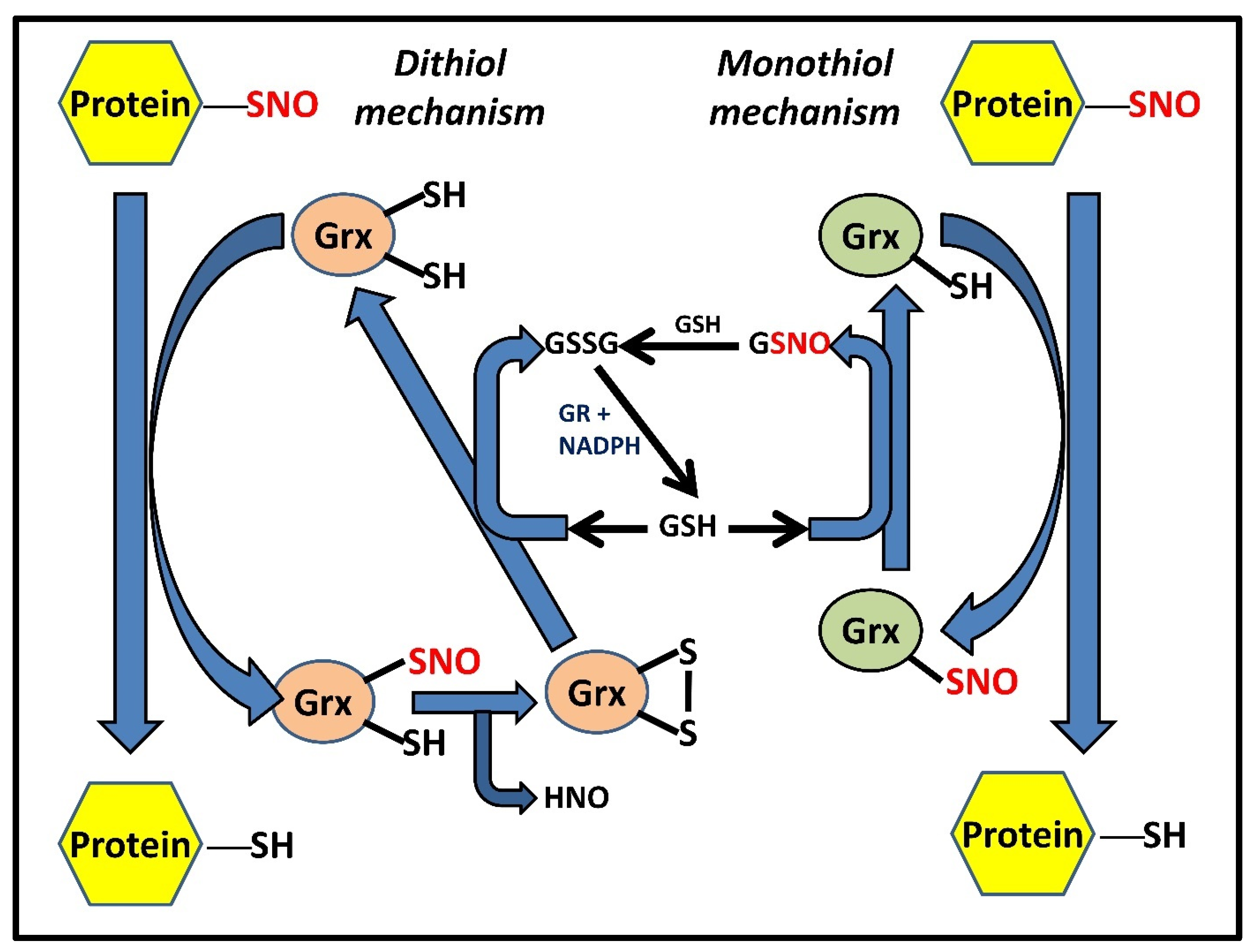
4. Trx Isoforms and Trx-Mediated Denitrosylation
The NADPH-dependent conversion of Trx from its oxidized form into its corresponding dithiol is essentially catalyzed by a homodimeric flavoprotein TrxR . Mammalian TrxRs (114 kDa or more) consist of two active sites, the first one being Cys59-Val-Asn-Val-Gly-Cys64, located in the FAD domain, and the second one being a conserved active site Gly-Cys497-SeCys498-Gly, located at its flexible C-terminal tail, which along with the redox-active SeCys residue, explains their broad substrate specificity compared to their bacterial, fungal, and plant counterparts. The reaction mechanism for reducing Trx and other substrates begins with NADPH binding to the oxidized enzyme. Being a flavoprotein, TrxR utilizes its FAD domain to transfer electrons from NADPH to the first active site (CVNVGC), present as a disulfide. Reduction of the first active site results in an intermediate Cys59 with intact thiol and FAD bound Cys64, further reduced to a dithiol pair. The second half-reaction involves the transfer of electrons to the C-terminal selenylsulfide (Cys497–Cys498) in another redox-active site (GCUG) in the neighboring subunit, which in turn, is reduced to a selenol thiol pair. At physiological pH, the selenol is converted into a corresponding selenolate anion that carries out a nucleophilic attack on the disulfide of the oxidized Trx, forming an enzyme–substrate intermediate. Further, Cys497 attacks this bond and forms an eight-membered ring of the C-terminal by participating in a selenylsulfide bond with SeCys. In order to keep the SeCys in its anionic form, Cys59 will donate its electrons, thereby maintaining the enzyme in its reduced form. Thus, the dual nature of the selenocysteine moiety to act as a nucleophile and an electrophile confers a reductase activity to the enzyme towards a vast plethora of protein as well as non-protein TrxR substrates such as oxidized Trx, Grx2, PDI, and certain small molecules including selenites, dehydroascorbate, DTNB, alloxan, hydrogen peroxide, lipoic acid, cytochrome c, ubiquinone, and motexafin gadolinium [49][50][51]. In mammals, TrxR exists in three isoforms as well, the cytosolic TrxR1 (TXNRD1), mitochondrial TrxR2 (TXNRD2), and testes-specific TrxR3 or TGR (TXNRD3), a unique enzyme with an additional N-terminal Grx domain, mainly found in maturing sperm [51]. TrxR1 has been shown to be particularly dependent upon the selenocysteine residue and follows a fixed pathway for reducing substrates due to the restrictive movement of the C-terminal, which is controlled by the ‘guiding bar’ made up of side chains of Asn418, Asn419, and Trp407 [52][53].
Trx-related protein 14 (TRP14, also called TXNDC17/TXNL5) is a cytosolic protein with a molecular weight of 14kDa, the oxidized form of which is ideally reduced by TrxR1 in an NADPH-dependent reaction [54]. Being able to be reduced only by TrxR1 but not supporting the classic substrates of Trx1 makes TRP14 a perfect candidate for redox signaling regulation, with unique additional yet uncharacterized functions complementing that of Trx1.
Sharing the similar active site sequence of Cys-Gly-Pro-Cys and functionality with mammalian thioredoxin, another interesting member of the Trx superfamily is the Trx-related protein 32 (TRP32, also called TXL or TXNL), a novel LMW (32 kDa) protein, ubiquitously expressed within the cytoplasm and nucleus, bearing an N-terminal Trx domain that caters to its reducing potential [85]. Trx2 and nucleoredoxin are two recently reported yet sparsely characterized mammalian Trx-related proteins with restricted subcellular localization in mitochondria and nucleus, respectively, which might hint toward their specific compartmentalized roles within the cytoplasmic milieu [55].
Trx can reduce disulfides as well as denitrosylate RSNOs/PSNOs in a likely similar biochemical mechanism, and this capability is attributed to its redox-active dithiol (Cys32 and Cys35) moiety. Moreover, S-nitrosylated Trx can further engage in transnitrosylation by transferring its own S-NO moiety to other target proteins, such as Caspase 3. Consistent with the novelty of further findings, inhibition of the redox-active cysteines of mammalian Trx1, coupled with the nitrosative (PS-NO) modification of only structural Cys residues (Cys62, Cys69, and Cys73), mainly Cys69 and Cys73, can bring its hypothesized transnitrosylase activity into the picture [3][56]. Trx1, when S-nitrosylated at either of its vicinal dithiols (Cys32 and Cys35), is auto-denitrosylated and leads to disulfide bond formation, further compromising Trx1 disulfide reductase and denitrosylase activity [3]. Denitrosylation of multiple mammalian PSNOs by Trx, driven by substrate proteins (both LMW and HMW) and certain intracellular regulators, can exert profound effects by modulating diverse cellular processes such as redox signaling, endocytosis, inflammation, angiogenesis, and apoptosis, etc., thereby conferring cellular protection from nitrosative stress. Regulation of this activity has been seen to be different for the various isoforms, with Trx2 acting as a denitrosylase in response to Fas-signaling or NO suppressing Txnip, allowing Trx1 to act as a denitrosylase [57][58]. S-nitrosylated Caspase-3, the pivotal enzyme in apoptosis, was reportedly denitrosylated by both Trx and TRP14 systems [14][58]. This leads to the activation of the enzyme and elicits an apoptotic response from the cell. Similarly, Trx1 can denitrosylate the S-nitrosylated caspase-8, reactivating it for the extrinsic apoptotic pathway [59]. This characteristic of Trx to denitrosylate PSNOs, though vast, is specific, and this specificity has not yet been explained well in mammalian cells. However, the intracellular redox status, subcellular localization of S-nitroso-Trx1/PSNOs, substrate specificity, vicinity of strong reductants such as GSH or Trx-(SH)2, and stability and/or half-life of Trx1-SNO bond to be able to catalyze the NO transfer, substantiated by more in vitro and in vivo studies would be crucial determinants for delineating the possibilities of both S-denitrosylation and trans-S-nitrosylation by Trxs and further studying their impact on protein activity profiles and expression levels in normal and pathophysiological conditions.
5. Redundancy of Redoxin Systems
Cellular existence is based on redox homeostasis. The main pillars of this balance are the two antioxidant pathways, the Trx-mediated pathway and the glutathioredoxin-mediated pathway. These two pathways exhibit functional redundancy; in essence, the redox system in a cell that is Trx-deficient can reprogram itself to work with the Grx system and survive [60]. Ever since their discovery as an alternative hydrogen donor system for RNR with active NADPH-dependent deoxyribonucleotide synthesis in E. coli mutants lacking Trxs, GSH and Grx have been eventually established as putative worthy players that play synergistic roles in maintaining redox homeostasis, along with the Trx system, under normal physiological conditions. TrxR, previously being the only physiological Trx reductant, underwent aurothioglucose (ATG) induced inhibition but exhibited no change in the cell viability along with an unaltered Trx1 redox state; the findings could be extrapolated to its reduction catalyzed by physiological GSH concentrations (5–10 mM), NADPH, and GR, strongly stimulated by Grx [61]. Cumulative pieces of evidence down the line identified Trx (Trx-S2) as an excellent substrate for GSH and Grx and, alternatively, oxidized GSH (GSSG) as a substrate for the Trx system, thereby establishing the eminent redundancy of either of the redoxins under nitro-oxidative stress being compensated by the other system [61][62][63][64][65][66]. This ensures cell survival and progression and further highlights a crucial perspective of potentiating current conventional or novel therapeutic approaches with careful consideration of inactivating both the GSH-Grx and Trx systems to halt the cellular viability.
6. Substrate Specificity of GSH and Redoxin Systems
Extensive research in the last two decades has revealed the presence of a wide-ranging network of intracellular denitrosylases and reductases that enable the cell to combat against and survive in the presence of continual bursts of ROS/RNS, generated as a response to various extrinsic and intrinsic stimuli. Reduced GSH efficiently denitrosylates small molecule nitrosothiols and a large percentage of PSNOs; however, a subset of PSNOs continues to be stable, even in the presence of cellular levels of GSH [46][47]. This suggests the presence of preferential selectivity or specificity of one substrate over the other by various denitrosylases. Proteins have complex, three-dimensional, tertiary, and quaternary structures; nitrosylation and other covalent modifications reportedly distort these tertiary structures, which as a consequence, sterically shields and thus, greatly limits the access of the nitrosylated cysteine residues to various denitrosylases. This phenomenon especially becomes relevant in some ‘hyper-nitrosylated’ proteins, having more than one nitrosylated cysteine, which differs in their reactivity and, thus, stability in the presence of GSH. Another context of great importance is the uneven abundance and the tissue-specific distribution of various proteins throughout the body. One such protein of prime importance is caspase-3; the epidermal keratinocytes, myocardiocytes, bronchial epithelium, hepatocytes, thymocytes, and renal tubule epithelium have been documented to have high amounts of caspase-3, although the majority of neurons in the brain and spinal cord have low levels of this protein [67]. Interestingly, S-nitrosocaspase-3 (both cytosolic and mitochondrial), although stable in the presence of GSH, is successfully denitrosylated by both Trx and Grx, which in turn bears great clinical impact, as the nitrosylation of caspase-3 reportedly inhibits its function as a pro-apoptotic agent, thereby engendering a tumorigenic transformation [3][12][13][42]. The existence of such specificity towards selective substrates confers differential weight and importance to certain denitrosylases in certain tissues. Experiments conducted in the rat-spinal cord failed to detect any decline in the efficiency of denitrosylation upon the chemical inhibition of TrxR; however, HepG2 cells (a widely used cellular model for hepatic cancer) devoid of TrxR exhibited a significant reduction in the denitrosylation of caspase-3 [12][27]. This further exemplifies the differential roles played by different denitrosylases under different circumstances.
7. Drugs and Treatments: Current Potential and Challenges
The two members of the thiol-disulfide oxidoreductase superfamily, Trx and Grx, have been unequivocally recognized as critical mediators of redox homeostasis, involved in both intra- and extra-cellular redox signaling. Tinkering with the structure and function of the redoxin domain of proteins and their selective inhibition upon a knock-out, knock-down, or gene silencing have generated many new insights into the molecular structures and biochemistry of each of the components with novel functions. Several drugs, inhibitors, mimetics, and LMW thiols targeting either of the redoxin components have been developed to harness the therapeutic benefits, which further ascertain their increasingly important functional relevance in the growing discipline of redox biology.
Unlike other pathophysiological conditions, the Trx system can act as a double-edged sword in cancer progression, depending upon the stage of cancer. At the onset of cancer, it exhibits direct antioxidant properties by defending against nitro-oxidative stress caused by carcinogens and xenobiotics, along with the support of other antioxidant molecules/enzymes such as GSH, Prx, and Msr, thereby preventing cell malignancy and further metastasis. However, once tumor formation has initiated, elevated levels of Trx/TrxR relative to normal cells help in coping with elevated ROS levels and facilitate tumor progression and metastasis owing to their growth-promoting, apoptosis-resisting, and angiogenesis-supporting functions, implicating themselves as crucial harbingers of chemotherapeutic resistance [68][69]. Despite the growing interest in developing novel drugs for antioxidant-based therapies, potentiating their efficacies in clinical trials remains at a standstill. Higher reactivity but a lower abundance of selenol groups compared to thiols, risks of cross-reactivity with other enzymes or cellular thiols, lack of major binding sites for inhibitors/ complexes, small surface area, and weak interactions between Trx-TrxR complex are some of the major challenges that limit the possibilities of drug designing concerning these areas [70].
8. Conclusions
Within the vast cellular repertoire, the redox homeostasis is maintained by a unique S-nitrosylation/S-denitrosylation dependent redox switch, which, when dysregulated, alleviates nitro-oxidative stress that further orchestrates cellular dysfunction implicated in a wide plethora of diseases and disorders. Skepticism pertaining to the putative targets of S-nitrosylation of various proteins, and the consequent functional implications of such modifications, comprise a significant portion of the largely incomplete jigsaw of S-nitrosoproteome study. Considerable research into gaining a deeper understanding of the complex, inter-twined properties of the intracellular denitrosylating network, using rapid, effective, and accurate proteomic and biochemical techniques, is elementary in order to enable scientists to devise efficient and prompt diagnostic tools and therapeutic molecules. Decrypting the complex overlap between denitrosylases and their multiple substrates would thereby aid in mustering missing pieces of information and provide crucial answers to open-ended concerns, such as the putative reduction of TRP14 by GSH or the possibility of Txnip mediated inhibition of TRP14. Keeping in mind the novel additions to the Trx superfamily, such as TRP14, and its biochemical characterization as a comparably efficient disulfide reductase as Trx, careful consideration of exploring and expanding the substrate-specificities of GSH and Txnip on TRP14 seems like a notion possible to reconcile experimentally, thereby improving its propensity as another attractive target for drug designing.
References
- Foster, M.W.; Hess, D.T.; Stamler, J.S. Protein S-nitrosylation in health and disease: A current perspective. Trends Mol. Med. 2009, 15, 391–404.
- Nakamura, T.; Tu, S.; Akhtar, M.W.; Sunico, C.R.; Okamoto, S.; Lipton, S.A. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 2013, 78, 596–614.
- Sengupta, R.; Holmgren, A. The role of thioredoxin in the regulation of cellular processes by S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 689–700.
- Villanueva, C.; Giulivi, C. Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radic. Biol. Med. 2010, 49, 307–316.
- Ren, X. Thioredoxin and Glutaredoxin Systems under Oxidative and Nitrosative Stress. Ph.D. Thesis, Karolinska Institutet, Department of Medical Biochemistry and Biophysics, Stockholm, Sweden, 2017.
- Csillag, A.; Boldogh, I.; Pazmandi, K.; Magyarics, Z.; Gogolak, P.; Sur, S.; Rajnavolgyi, E.; Bacsi, A. Pollen-induced oxidative stress influences both innate and adaptive immune responses via altering dendritic cell functions. J. Immunol. 2010, 184, 2377–2385.
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta 2010, 1802, 29–44.
- Rizza, S.; Cardaci, S.; Montagna, C.; Giacomo, G.D.; Zio, D.D.; Bordi, M.; Maiani, E.; Campello, S.; Borreca, A.; Puca, A.A.; et al. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. USA 2018, 115, E3388–E3397.
- Walsh, G.M.; Leane, D.; Moran, N.; Keyes, T.E.; Forster, R.J.; Kenny, D.; O’Neill, S. S-nitrosylation of platelet alphaIIbbeta3 as revealed by Raman spectroscopy. Biochemistry 2007, 46, 6429–6436.
- Que, L.G.; Liu, L.; Yan, Y.; Whitehead, G.S.; Gavett, S.H.; Schwartz, D.A.; Stamler, J.S. Protection from experimental asthma by an endogenous bronchodilator. Science 2005, 308, 1618–1621.
- Zima, A.V.; Mazurek, S.R. Functional impact of ryanodine receptor oxidation on intracellular calcium regulation in the heart. Rev. Physiol. Biochem. Pharmacol. 2016, 171, 39–62.
- Sengupta, R.; Ryter, S.W.; Zuckerbraun, B.S.; Tzeng, E.; Billiar, T.R.; Stoyanovsky, D.A. Thioredoxin catalyzes the denitrosation of low-molecular-mass and protein S-nitrosothiols. Biochemistry 2007, 46, 8472–8483.
- Ren, X.; Sengupta, R.; Lu, J.; Lundberg, J.O.; Holmgren, A. Characterization of mammalian glutaredoxin isoforms as S-denitrosylases. FEBS Lett. 2019, 593, 1799–1806.
- Pader, I.; Sengupta, R.; Cebula, M.; Xu, J.; Lundberg, J.O.; Holmgren, A.; Johansson, K.; Arnér, E.S.J. Thioredoxin-related protein of 14 kDa is an efficient L-cystine reductase and S-denitrosylase. Proc. Natl. Acad. Sci. USA 2014, 111, 6964–6969.
- Sircar, E.; Rai, S.R.; Wilson, M.A.; Schlossmacher, M.G.; Sengupta, R. Neurodegeneration: Impact of S-nitrosylated Parkin, DJ-1 and PINK1 on the pathogenesis of Parkinson’s disease. Arch. Biochem. Biophys. 2021, 704, 108869.
- Russell, T.M.; Azad, M.G.; Richardson, D.R. The Relationship of Glutathione-S-Transferase and Multi-Drug Resistance-Related Protein 1 in Nitric Oxide (NO) Transport and Storage. Molecules 2021, 26, 5784.
- Oestreicher, J.; Morgan, B. Glutathione: Subcellular distribution and membrane transport. Biochem. Cell. Biol. 2019, 97, 270–289.
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3143–3153.
- Zhang, H.; Forman, H.J.; Choi, J. Gamma-glutamyl transpeptidase in glutathione biosynthesis. Methods Enzymol. 2005, 401, 468–483.
- Raj Rai, S.; Bhattacharyya, C.; Sarkar, A.; Chakraborty, S.; Sircar, E.; Dutta, S.; Sengupta, R. Glutathione: Role in oxidative/nitrosative stress, antioxidant defense, and treatments. ChemistrySelect 2021, 6, 4566–4590.
- Corpas, F.J.; Alché, J.D.; Barroso, J.B. Current overview of S-nitrosoglutathione (Gsno) in higher plants. Front. Plant Sci. 2013, 4, 126.
- Won, J.S.; Kim, J.; Annamalai, B.; Shunmugavel, A.; Singh, I.; Singh, A.K. Protective role of s-nitrosoglutathione (Gsno) against cognitive impairment in rat model of chronic cerebral hypoperfusion. J. Alzheimer’s Dis. 2013, 34, 621–635.
- Barnett, S.D.; Buxton, I.L.O. The role of S-nitrosoglutathione reductase (Gsnor) in human disease and therapy. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 340–354.
- Kalinina, E.; Novichkova, M. Glutathione in Protein Redox Modulation through S-Glutathionylation and S-Nitrosylation. Molecules 2021, 26, 435.
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837.
- Anderson, M.E.; Powrie, F.; Puri, R.N.; Meister, A. Glutathione monoethyl ester: Preparation, uptake by tissues, and conversion to glutathione. Arch. Biochem. Biophys. 1985, 239, 538–548.
- Romero, J.M.; Bizzozero, O.A. Intracellular glutathione mediates the denitrosylation of protein nitrosothiols in the rat spinal cord. J. Neurosci. Res. 2009, 87, 701–709.
- Forrester, M.T.; Foster, M.W.; Stamler, J.S. Assessment and application of the biotin switch technique for examining protein s-nitrosylation under conditions of pharmacologically induced oxidative stress. J. Biol. Chem. 2007, 282, 13977–13983.
- Liu, L.; Hausladen, A.; Zeng, M.; Que, L.; Heitman, J.; Stamler, J.S. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 2001, 410, 490–494.
- Liu, L.; Yan, Y.; Zeng, M.; Zhang, J.; Hanes, M.A.; Ahearn, G.; McMahon, T.J.; Dickfeld, T.; Marshall, H.E.; Que, L.G.; et al. Essential roles of s-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 2004, 116, 617–628.
- Calabrese, V.; Scapagnini, G.; Ravagna, A.; Bella, R.; Butterfield, D.A.; Calvani, M.; Pennisi, G.; Giuffrida Stella, A.M. Disruption of thiol homeostasis and nitrosative stress in the cerebrospinal fluid of patients with active multiple sclerosis: Evidence for a protective role of acetylcarnitine. Neurochem. Res. 2003, 28, 1321–1328.
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein s-nitrosylation: Determinants of specificity and enzymatic regulation of s-nitrosothiol-based signaling. Antioxid. Redox Signal. 2019, 30, 1331–1351.
- Sengupta, R.; Holmgren, A. Thioredoxin and thioredoxin reductase in relation to reversible s-nitrosylation. Antioxid. Redox Signal. 2013, 18, 259–269.
- Chatterji, A.; Banerjee, D.; Billiar, T.R.; Sengupta, R. Understanding the role of S-nitrosylation/nitrosative stress in inflammation and the role of cellular denitrosylases in inflammation modulation: Implications in health and diseases. Free Radic. Biol. Med. 2021, 172, 604–621.
- Benhar, M.; Forrester, M.T.; Stamler, J.S. Protein denitrosylation: Enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol. 2009, 10, 721–732.
- Cha, S.J.; Kim, H.; Choi, H.J.; Lee, S.; Kim, K. Protein glutathionylation in the pathogenesis of neurodegenerative diseases. Oxid. Med. Cell. Longev. 2017, 2017, 2818565.
- Conibear, E.; Davis, N.G. Palmitoylation and depalmitoylation dynamics at a glance. J. Cell Sci. 2010, 123, 4007–4010.
- Fukata, Y.; Fukata, M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 2010, 11, 161–175.
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155.
- Gilbert, H.F. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 1995, 251, 8–28.
- Gilbert, H.F. Molecular and cellular aspects of thiol-disulfide exchange. Adv. Enzymol. Relat. Areas Mol. Biol. 1990, 63, 69–172.
- Stoyanovsky, D.A.; Scott, M.J.; Billiar, T.R. Glutathione and thioredoxin type 1 cooperatively denitrosate HepG2 cells-derived cytosolic S-nitrosoproteins. Org. Biomol. Chem. 2013, 11, 4433–4437.
- Stamler, J.S.; Lamas, S.; Fang, F.C. Nitrosylation. the prototypic redox-based signaling mechanism. Cell 2001, 106, 675–683.
- Dominko, K.; Đikić, D. Glutathionylation: A regulatory role of glutathione in physiological processes. Arh. Hig. Rada Toksikol. 2018, 69, 196018.
- Sen, C.K. Redox signaling and the emerging therapeutic potential of thiol antioxidants. Biochem. Pharmacol. 1998, 55, 1747–1758.
- Paige, J.S.; Xu, G.; Stancevic, B.; Jaffrey, S.R. Nitrosothiol reactivity profiling identifies s-nitrosylated proteins with unexpected stability. Chem. Biol. 2008, 15, 1307–1316.
- Chatterji, A.; Sengupta, R. Stability of S-nitrosothiols and S-nitrosylated proteins: A struggle for cellular existence! J. Cell. Biochem. 2021, 122, 1579–1593.
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Tyurin, V.A.; Anand, D.; Mandavia, D.N.; Gius, D.; Ivanova, J.; Pitt, B.; Billiar, T.R.; Kagan, V.E. Thioredoxin and lipoic acid catalyze the denitrosation of low molecular weight and protein s -nitrosothiols. J. Am. Chem. Soc. 2005, 127, 15815–15823.
- Zhong, L.; Arnér, E.S.; Holmgren, A. Structure and mechanism of mammalian thioredoxin reductase: The active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Natl. Acad. Sci. USA 2000, 97, 5854–5859.
- Lothrop, A.P.; Snider, G.W.; Ruggles, E.L.; Patel, A.S.; Lees, W.J.; Hondal, R.J. Selenium as an electron acceptor during the catalytic mechanism of thioredoxin reductase. Biochemistry 2014, 53, 654–663.
- Arnér, E.S. Focus on mammalian thioredoxin reductases—Important selenoproteins with versatile functions. Biochim. Biophys. Acta 2009, 1790, 495–526.
- Lothrop, A.P.; Snider, G.W.; Ruggles, E.L.; Hondal, R.J. Why is mammalian thioredoxin reductase 1 so dependent upon the use of selenium? Biochemistry 2014, 53, 554–565.
- Fritz-Wolf, K.; Urig, S.; Becker, K. The structure of human thioredoxin reductase 1 provides insights into C-terminal rearrangements during catalysis. J. Mol. Biol. 2007, 370, 116–127.
- Jeong, W.; Yoon, H.W.; Lee, S.R.; Rhee, S.G. Identification and characterization of TRP14, a thioredoxin-related protein of 14 kDa. New insights into the specificity of thioredoxin function. J. Biol. Chem. 2004, 279, 3142–3150.
- Lee, K.K.; Murakawa, M.; Takahashi, S.; Tsubuki, S.; Kawashima, S.; Sakamaki, K.; Yonehara, S. Purification, Molecular Cloning, and Characterization of TRP32, a Novel Thioredoxin-related Mammalian Protein of 32 kDa. J. Biol. Chem. 1998, 273, 19160–19166.
- Wu, C.; Parrott, A.M.; Fu, C.; Liu, T.; Marino, S.M.; Gladyshev, V.N.; Jain, M.R.; Baykal, A.T.; Li, Q.; Oka, S.; et al. Thioredoxin 1-mediated post-translational modifications: Reduction, transnitrosylation, denitrosylation, and related proteomics methodologies. Antioxid. Redox Signal. 2011, 15, 2565–2604.
- Benhar, M.; Forrester, M.T.; Hess, D.T.; Stamler, J.S. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 2008, 320, 1050–1054.
- Forrester, M.T.; Seth, D.; Hausladen, A.; Eyler, C.E.; Foster, M.W.; Matsumoto, A.; Benhar, M.; Marshall, H.E.; Stamler, J.S. Thioredoxin-interacting protein (Txnip) is a feedback regulator of S-nitrosylation. J. Biol. Chem. 2009, 284, 316160–316166.
- Sengupta, R.; Billiar, T.R.; Kagan, V.E.; Stoyanovsky, D.A. Nitric oxide and thioredoxin type 1 modulate the activity of caspase 8 in HepG2 cells. Biochem. Biophys. Res. Commun. 2010, 391, 1127–1130.
- Arnér, E.S.; Holmgren, A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006, 16, 420–426.
- Du, Y.; Zhang, H.; Lu, J.; Holmgren, A. Glutathione and Glutaredoxin Act as a Backup of Human Thioredoxin Reductase 1 to Reduce Thioredoxin 1 Preventing Cell Death by Aurothioglucose. J. Biol. Chem. 2012, 287, 38210–38219.
- Tan, S.X.; Greetham, D.; Raeth, S.; Grant, C.M.; Dawes, I.W.; Perrone, G.G. The thioredoxin-thioredoxin reductase system can function in vivo as an alternative system to reduce oxidized glutathione in Saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 6118–6126.
- Rollins, M.F.; van der Heide, D.M.; Weisend, C.M.; Kundert, J.A.; Comstock, K.M.; Suvorova, E.S.; Capecchi, M.R.; Merrill, G.F.; Schmidt, E.E. Hepatocytes lacking thioredoxin reductase 1 have normal replicative potential during development and regeneration. J. Cell Sci. 2010, 123, 2402–2412.
- Trotter, E.W.; Grant, C.M. Non-reciprocal regulation of the redox state of the glutathione-glutaredoxin and thioredoxin systems. EMBO Rep. 2003, 4, 184–188.
- Han, C.; Kim, M.J.; Ding, D.; Park, H.J.; White, K.; Walker, L.; Gu, T.; Tanokura, M.; Yamasoba, T.; Linser, P.; et al. GSR is not essential for the maintenance of antioxidant defenses in mouse cochlea: Possible role of the thioredoxin system as a functional backup for GSR. PLoS ONE 2017, 12, e0180817.
- Mandal, P.K.; Schneider, M.; Kölle, P.; Kuhlencordt, P.; Förster, H.; Beck, H.; Bornkamm, G.W.; Conrad, M. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res. 2010, 70, 9505–9514.
- Krajewska, M.; Wang, H.G.; Krajewski, S.; Zapata, J.M.; Shabaik, A.; Gascoyne, R.; Reed, J.C. Immunohistochemical analysis of in vivo patterns of expression of CPP32 (Caspase-3), a cell death protease. Cancer Res. 1997, 57, 1605–1613.
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176.
- Jovanović, M.; Podolski-Renić, A.; Krasavin, M.; Pešić, M. The Role of the Thioredoxin Detoxification System in Cancer Progression and Resistance. Front. Mol. Biosci. 2022, 9, 883297.
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808.


