Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Władysław Lasoń | -- | 4673 | 2022-11-25 12:24:08 | | | |
| 2 | Rita Xu | -1 word(s) | 4672 | 2022-11-28 02:31:27 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lasoń, W.; Jantas, D.; Leśkiewicz, M.; Regulska, M.; Basta-Kaim, A. Vitamin D3 and Ischemic Stroke. Encyclopedia. Available online: https://encyclopedia.pub/entry/36620 (accessed on 08 June 2026).
Lasoń W, Jantas D, Leśkiewicz M, Regulska M, Basta-Kaim A. Vitamin D3 and Ischemic Stroke. Encyclopedia. Available at: https://encyclopedia.pub/entry/36620. Accessed June 08, 2026.
Lasoń, Władysław, Danuta Jantas, Monika Leśkiewicz, Magdalena Regulska, Agnieszka Basta-Kaim. "Vitamin D3 and Ischemic Stroke" Encyclopedia, https://encyclopedia.pub/entry/36620 (accessed June 08, 2026).
Lasoń, W., Jantas, D., Leśkiewicz, M., Regulska, M., & Basta-Kaim, A. (2022, November 25). Vitamin D3 and Ischemic Stroke. In Encyclopedia. https://encyclopedia.pub/entry/36620
Lasoń, Władysław, et al. "Vitamin D3 and Ischemic Stroke." Encyclopedia. Web. 25 November, 2022.
Copy Citation
Ischemic stroke is one of the major causes of death and permanent disability worldwide. The only efficient treatment to date is anticoagulant therapy and thrombectomy, which enable restitution of blood flow to ischemic tissues. Numerous promising neuroprotectants have failed in clinical trials. Given the complex pathomechanism of stroke, a multitarget pharmacotherapy seems a more rational approach in stroke prevention and treatment than drugs acting on single molecular targets. Vitamin D3 has emerged as a potential treatment adjunct for ischemic stroke, as it interferes with the key prosurvival pathways and shows neuroprotective, anti-inflammatory, regenerative and anti-aging properties in both neuronal and vascular tissue.
vitamin D3
brain ischemia
neuroprotection
post-stroke depression
1. Introduction
The central nervous system (CNS) is extremely sensitive to a shortage of oxygen and glucose, and a sudden loss of blood circulation to an area of the brain due to systemic hypo-perfusion, thrombosis, or embolism which can result in ischemic stroke. In 1970, the World Health Organization defined stroke as “rapidly developing clinical signs of focal (or global) disturbance of cerebral function, lasting more than 24 h or leading to death, with no apparent cause other than that of vascular origin” [1]. This classic definition of stroke has been updated by the American Heart Association/American Stroke Association, which pointed out that ischemic stroke specifically refers to CNS infarction, which can be defined as “brain, spinal cord, or retinal cell death attributable to ischemia, based on neuropathological, neuroimaging, and/or clinical evidence of permanent injury” [2]. Stroke is considered to be an acute cerebrovascular disease and includes ischemic stroke (about 85–90% of strokes) and hemorrhagic stroke, while the latter is further subdivided into intracerebral hemorrhage and subarachnoid hemorrhage. The current treatment of acute ischemic strokes is based on reperfusion therapies including intravenous administration of thrombolytic agents and endovascular therapy [3][4]. Ischemic stroke is one of the major causes of death and long-term severe disabilities worldwide. According to the World Health Organization, 15 million people suffer from strokes worldwide each year [3]. Moreover, there are estimates that there will be 23 million first-case strokes and 7–8 million stroke deaths in 2030 [4]. Even worse, clinical observations showed that the SARS-CoV-2 pandemic engulfing the entire world in recent years brought about an increase in stroke incidences among COVID-19 patients [5][6][7]. However, a causative link between COVID-19 and stroke has not been proven, yet, and the long-term consequences for cerebrovascular complications among COVID-19 survivors remain unknown [8][9][10].
Despite enormous efforts and substantial investments of the pharmaceutical industry, no clinically efficient and well-tolerated neuroprotective drug has been marketed yet. Nevertheless, it is still expected that a better understanding of pathological events involved in ischemic brain injury, as well as interconnection and crosstalk between various ischemia-induced cell death programs will be helpful in discovering new therapeutic strategies. Because of a high complexity of molecular processes involved in the pathomechanism of stroke, agents with pleiotropic activities rather than those aimed at a single molecular target may be more promising as candidates for neuroprotective drugs. This view is in agreement with the statement of the Stroke Treatment Academic Industry Roundtable (STAIR) which recommended to target multiple mechanisms simultaneously instead of treating a solo pathway after stroke injury [11]. It is proposed that vitamin D3 is one of such multitarget drugs, as it stimulates the key pro-survival pathways and shows neuroprotective, antioxidant, anti-inflammatory, regenerative and anti-aging properties not only in neuronal but also in glia and vascular tissue [12]. The latter is of importance for revascularization of the stroke-affected brain region and recovering normal functions of the blood–brain barrier (BBB), which is regulated by the interactions between the brain endothelium, astrocytes and neurons [13]. Of note, an intimate relationship between the brain and its vessels in the concept of the neurovascular unit has been strongly accentuated [14]. As vitamin D3 stimulates some pro-survival pathways essential for neuroplasticity, including up-regulation of neurotrophin gene expression, it has been suggested that supplementation with vitamin D3 might be beneficial, not only in restoration of neurological functions after stroke, but also in alleviating the post-stroke psychiatric disorders, e.g., depression and anxiety, and improving the deteriorated cognitive functions [15][16]. An increasing body of evidence suggests that vitamin D3 has a positive impact on the prevention of cardiovascular diseases and rehabilitation outcomes in stroke patients; however, due to various methodological limitations of the studies conducted to date it is difficult to draw final conclusions [17].
2. Pathomechanisms of Ischemic Stroke
2.1. Pathomorphological Features of Ischemic Stroke
The ischemia-induced pathophysiological changes in the brain tissue are time-dependent and can be roughly divided into three phases: acute, subacute, and late phase [18]. The acute phase refers to the first 24 h after stroke and its characteristic feature is necrotic death of neuronal and glia cells in the core of the stroke lesions. Pathomorphological analysis shows that the acute irreversible ischemic neuronal injury is characterized by the presence of eosinophilic cytoplasm in the affected neurons, which lacks identifiable substructure, and has a pyknotic or collapsed nucleus. Around the core zone, a much more extended area of moderately ischemic tissue, called ischemic penumbra, can be detected. In this area, insufficiently supplied with blood by collateral arteries, the metabolic and functional activity of neurons deteriorates, but morphological integrity of the cells is still maintained [19]. In the subacute phase, which lasts several days following stroke, the activation of glia cells and neuroimmunological processes appear to play the main role in the extent of brain injury. In the late phase, a glia scar is formed and some reparative processes connected with proliferation and differentiation of cells along with angiogenesis in the stroke-affected brain tissue take place, but inflammation is still evident [20].
2.2. Biochemical Basis of Ischemic Stroke and Neuroprotective Strategies of Its Treatment
A number of biochemical processes have been implicated in the mechanism of ischemia-induced brain damage, including excitotoxicity, oxidative stress, inflammation, acidotoxicity, and apoptosis: Figure 1 [21]. Biochemical changes in ischemic brain tissue are time-dependent and take the form of complex cascade processes [22]. Early neuronal death in the core of the infarct that occurs within a short time after brain damage is most likely due to necrosis, while delayed neuronal death occurring over days and months shows features of an apoptotic process [23]. In the acute phase of the stroke, deprivation of oxygen and glucose inhibits mitochondrial ATP production resulting in neuronal energy deficit and impairment of Na+/K+-ATPase pumps. These changes cause a disturbance of ion gradients, gradually decrease neuronal membrane potential, and enhance the release of neurotransmitters, mainly represented by glutamate (Glu). Moreover, by reversing Glu, transport ischemia further increases extracellular concentrations of this excitatory neurotransmitter [24][25]. Depolarization of neuronal membrane leads to the activation of voltage-dependent calcium channels and, by removing the magnesium block, it enables the activation of glutamatergic NMDA receptors. Excitotoxicity caused by excessive Glu release, overactivation of NMDA receptors, and increase in intracellular Ca2+ concentration, has long been established as a pivotal mechanism of hypoxia-induced neuronal injury [26]. Indeed, the enhanced Ca2+ influx into cells is considered to be the main factor activating a cascade of enzymatic processes such as those catalyzed by proteases, lipases and DNases which ultimately destroy intracellular proteins, lipids and nucleic acids, leading to neuronal death. Although NMDA receptors play a key role, ischemia-induced neuronal damage, AMPA receptors and metabotropic mGluR1 receptors are also engaged in this process [27]. However, clinical trials with classical NMDA receptor antagonists conducted to date have been terminated because of undesired side effects and insignificant neuroprotective action of NMDA receptor antagonists, as well as in many cases with a limited therapeutic window [28]. More recently, it has been recognized that synaptic NMDA receptors fulfill physiological functions in synaptic plasticity and cognitive functions, whereas extrasynaptic receptors mediate excitotoxicity [29][30]. Recently, it has been found that extrasynaptic NMDAR subunits GluN2A and GluN2B form a complex with transient receptor potential cation channel subfamily M member 4 (TRPM4) and that this complex is accountable for ischemia-related excitotoxicity, since disruption of the complex provides neuroprotection without disturbing physiological NMDAR-induced calcium signals [31]. These findings, which may open new avenues in the pharmacology of NMDA receptors, await further studies. The oversupply of intracellular calcium ions also induces mitochondrial membrane depolarization, resulting in the release of free radicals which contribute to the complex mechanism of ischemic neuronal damage [32]. It is commonly agreed that in the treatment of acute ischemic stroke, blood supply restoration is critical for salvaging the penumbra-marked brain tissue. However, with restitution of blood flow to ischemic tissues, there is a paradoxical increase in the production of superoxide, nitric oxide and peroxynitrate radicals, which, via activation of matrix metalloproteases, have a damaging effect on BBB integrity, enabling infiltration of neutrophils and leukocytes into the brain parenchyma and promoting neuroinflammation [33]. Overall, an ischemia/reperfusion (I/R) induces complex pathological processes involving intracellular and extracellular pathways that result in metabolic, thrombotic, and inflammatory changes in the affected tissues [34]. While prolonged I/R injury leads to further neuronal cell death which could be executed by apoptotic or non-apoptotic (necroptosis, ferroptosis, parthanatos, and pyroptosis) cell death programs, the moderate injury may favor autophagy and activate recovery systems for survival [35][36][37].
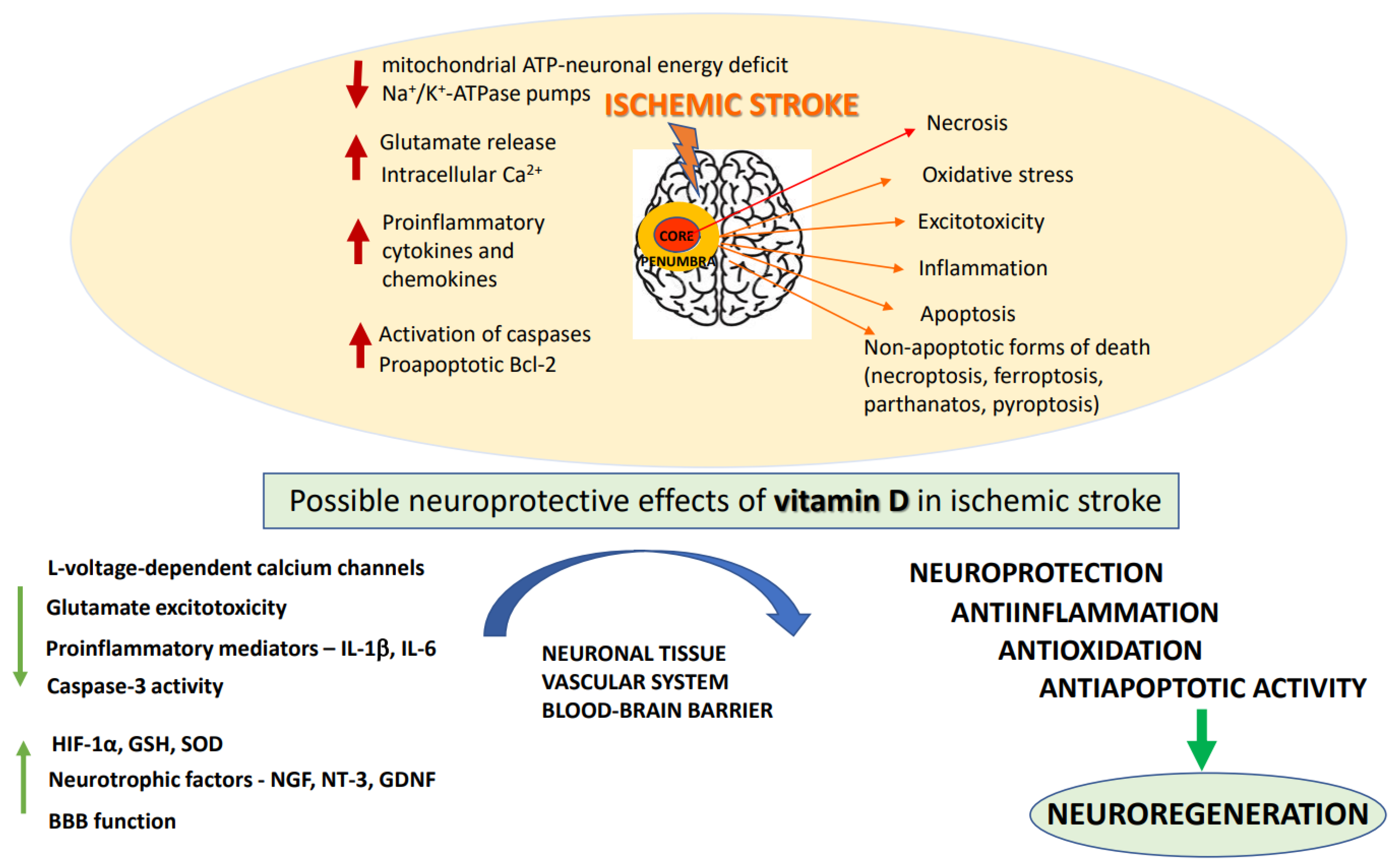
Figure 1. The pathogenic mechanisms of ischemic stroke and neuroprotective effects of vitamin D. BBB: blood–brain–barrier; GDNF: glial-derived neurotrophic factor; GSH: glutathione; HIF: 1a-hypoxia-inducible factor; IL: interleukins; NGF: nerve growth factor; NT: 3-neutrophin-3; SOD: superoxide dismutase.
The subacute phase, which occurs within few days after stroke, is mainly characterized by the development of inflammation and activation of apoptosis-inducing signaling pathways. The pathological processes initiated during the acute phase led to the release of some endogenous molecules DAMP (damage-associated molecular patterns) from injured cells, which activate Toll-like receptors, stimulating the release of proinflammatory cytokines and chemokines [38]. Of them, Interleukin-1 beta (IL-1β) and tumor necrosis factor alpha (TNF-α) appear to play a particularly important role in stroke pathology, since they have a damaging effect on neuronal and glia cells and enhance the release of proinflammatory prostaglandins. DAMP stimulate PRR receptors (pattern recognition receptors) resulting in the formation of proinflammatory protein complexes (inflammasomes) [39]. During the subacute phase of stroke, the programmed cell death, apoptosis, prevails in the metabolically impaired penumbra region. Apoptosis, contrary to necrosis, is a slow process, requiring energy supply, switching on the gene transcription, and protein synthesis. Delayed neuronal death in focal cerebral ischemia is characterized by cell shrinkage, chromatin condensation, upregulation of proapoptotic Bcl-2 family members (Bax, Bad, Bak) and activation of caspases [40]. Although many preclinical studies showed neuroprotective efficacy of various anti-apoptotic strategies in brain ischemia models, they have not been specifically tested in the clinic due to insufficient knowledge on the role of apoptosis in stroke and its interplay with other pathological events occurring during ischemic stroke [23][41][42]. The occurrence of delayed cell death during brain ischemia creates favorable conditions for a therapeutic window, giving a chance for timely pharmacological intervention in order to salvage neurons in the area of moderate hypoxia [43][44].
During the long-lasting late phase of ischemic stroke, neuroinflammatory and neuroplastic processes, including neurogenesis, angiogenesis and remyelination are highly active [45]. In this phase, microglia of M2 phenotype are engaged in repair of the brain region damaged by ischemia via production of anti-inflammatory cytokines (IL-4, IL-10), enhancement of phagocytosis of cellular debris and promoting neuroplasticity. The involvement of epigenetic processes, including DNA methylation, post-translational modifications of histone proteins and microRNAs in post-ischemic brain repair and neuroplasticity has also been postulated [46][47].
3. The Basics of Vitamin D
3.1. Sources, Biosynthesis and Metabolism
Vitamin D is a fat-soluble vitamin with a pivotal function in the maintenance of calcium and phosphate homeostasis in vertebrates [48][49]. Based on chemical structure, vitamin D belongs to secosteroids and currently its five forms, called D1, D2, D3, D4 and D5, have been identified, which differ in their ultrastructural conformation and origin. The main source of vitamin D in humans (~80–90%) originates from its endogenous non-enzymatic production in the skin epidermis where under sun exposure (UV-B rays, wavelength 290–320 nm) 7-dehydrocholesterol is transformed into an unstable intermediate pre-vitamin D3 which further, in a thermo-sensitive process, isomerizes to vitamin D3 (cholecalciferol) [49][50][51]. About 10–20% of vitamin D could come from the diet (second source) after consumption of plant-derived food enriched in vitamin D2 (ergocalciferol) or animal-based food (e.g., fish oils, fatty fish, beef, liver, eggs or milk) which contain mainly cholecalciferol. Since nowadays a lot of people work indoors and often use sunscreen during sun exposure, their endogenous vitamin D3 production (especially in north latitudes) could be insufficient to maintain health. Thus, various vitamin D supplements (third source) are commercially available, among which the cholecalciferol form predominates as the compound thought to be of a better bioavailability and biological activity [48][49][52][53]. Vitamins D3 and D2 are inactive substances and to evoke their biological effects they need to be activated in a two-step hydroxylation process. The first step takes place in the liver where 25-hydroxylation (mainly via CYP2R1) of cholecalciferol or ergocalciferol leads to the formation of circulating metabolites: 25-hydroxyvitamin D (25(OH)D3 or 25(OH)D2, calcifediol, a clinically used biomarker of serum vitamin D level. In the second step, these metabolites are 1 α-hydroxylated (via CYP27B1) in the kidneys to active forms of vitamin D, 1,25(OH)2D3 (calcitriol) or 1,25(OH)2D2 which possess broad spectrum biological activities [49][50][54]. Since 1,25(OH)2D3 functions as a steroid hormone with a key role in maintaining calcium bone homeostasis, its endogenous production is tightly regulated in the ways typical of the endocrine system, i.e., by feedback inhibition (negative regulation), parathyroid hormone (PTH, stimulant of the renal production of 1,25(OH)2D3), fetal growth factor 23 (FGF-23; inhibits calcitriol production) and by serum concentrations of calcium and phosphate [49][51]. The excessive amount of active (1,25(OH)2D) and storage (25(OH)D) forms of vitamin D are metabolized by 24-hydroxylation (via CYP24A1 to 1,24,25(OH)3D, calcitroic acid or 24,25(OH)2D) to prevent potential toxicity of vitamin D (especially supplemented vitamin D), but some of these metabolites could still possess some biological activities (e.g., 1,24,25(OH)3D in the regulation of bone health). The alternative pathway of vitamin D metabolism involves 20-hydroxylation (via CYP11A1) forming the product 20(OH)D which, together with its metabolite 20,23(OH)2D, are devoid of calcemic activity but possess other biological properties, e.g., anticancer activity [55][56][57]. The amount of free vitamin D metabolites available for biological activity (approx. 0.4% of total 1,25(OH)2D and 0.03% of total 25(OH)D) is also regulated in plasma at the level of their association with DBPs (vitamin D binding proteins). These proteins are characterized by high polymorphism in humans and bind about 58% of the circulating vitamin D metabolites, thus, they are claimed to be responsible for interpersonal differences in vitamin D bioavailability [58]. Other factors which could affect vitamin D metabolism and functions include physical activity, lifestyle, certain medications, environmental pollutants or epigenetics [59].
3.2. Genomic and Non-Genomic Mechanisms of Vitamin D Action
Biological action of calcitriol could be executed by genomic and non-genomic mechanisms. The former mechanism is structurally and mechanistically well understood as being the main player in the maintenance of vitamin D homeostasis [12][51][60]. The hydrophobic substance 1,25(OH)2D3 passively penetrates the cell membrane or could be bound to DBPs to be actively transported into cells via endocytosis [61]. When reaching cytosol or nucleus, calcitriol binds to the high affinity vitamin D receptor (VDR). VDR has a molecular weight of 55 kDa and, together with receptors for glucocorticosteroids, sex steroids, thyroxine, retinoids, fatty acids and eicosanoids, belongs to the steroid hormone receptor superfamily. The VDR contains two overlapping ligand binding sites, a genomic pocket (VDR-GP) and an alternative pocket (VDR-AP), that respectively bind a bowl-like ligand configuration (gene transcription) or a planar-like ligand shape (rapid responses) [62]. After vitamin D attaches to the ligand-binding domain, the VDR receptor undergoes heterodimerization, with the retinoic acid X receptor (RXR) initiating a change in its spatial conformation. It has been found that the biologically active form of vitamin D3 promotes the generation of non-genomic responses in the 6-s-cis configuration, while secosteroid binding in the 6-s-trans form may be responsible for genomic responses [63]. After vitamin D binding and translocation to the nucleus, VDR-RXR heterodimer interacts with specific DNA-binding domains, VDREs (vitamin D responsive elements). VDREs are located in the promoter region of vitamin D target genes and are composed of a highly conserved N-terminal DNA-binding domain and alpha-helical C-terminal ligand-binding domain [64]. The presence of several VDREs in the gene promoter suggests that they may act synergistically [65]. Such complexes modulate the activity of RNA polymerase II in the regulatory regions of target genes in various cell types. To promote gene expression, first the co-repressors (e.g., NcoR2/SMART) should be released from VDR/RXR/VDRE complex and then the relevant co-activators (e.g., SRC1) are recruited [12]. The inhibition of gene expression by vitamin D3 could be achieved by direct repression of the transcription of a target gene or indirectly by VDR-mediated enhancement of the transcription of negative regulators of target genes [66]. For genomic mechanisms, attaching the nuclear co-regulator proteins activates or inhibits gene transcription. It should be noted that VDR-binding sites are highly dynamic and could be affected by various factors, such as cell differentiation or maturation state, aging or disease activation and all of these could be reflected by gene expression. The VDR-genomic mechanisms regulate crucial enzymes involved in the synthesis and metabolism of vitamin D3, genes involved in the maintenance of bone calcium homeostasis (for example, genes encoding osteocalcin, osteopontin, 24-hydroxylase enzyme (Cyp24a), 1-hydroxylase enzyme (Cyp27b) or calbindin), as well as genes regulating cell proliferation, differentiation and apoptosis. Such genes include, for example, the genes for p21 protein (cell cycle inhibitor), Bcl-2 protein (regulator of apoptosis), p53 protein (suppressor of oncogenes that control cell growth) and the gene for amphiregulin (epithelial growth factor that stimulates the growth of head, neck and breast cancer cells). Moreover, calcitriol not only has a modulatory effect on growth factor/cytokine synthesis but also regulates growth factor signaling [67]. Additionally, the vitamin D system could be also involved in the regulation of various epigenetic events (e.g., posttranscriptional modifications of histone H3 and H4) and by this way could also affect the transcription of various genes. Reciprocally, various epigenetic modifications influence transcription of the VDR gene and in this way could influence the efficiency and interpersonal differences in vitamin D action [59][68]. Interestingly, in the situation of a low level of 1,25(OH)2D3, the VDR can still operate by binding other molecules, including curcumin, polyunsaturated fatty acids and anthocyanidins, which are thought to be low-affinity nutritional ligands for VDR. Other factors such as resveratrol and sirtuin 1 could potentiate nuclear VDR signaling [51][59].
Apart from the genomic mechanisms, the vitamin D via VDR could mediate its faster biological action when it is distributed outside of the nucleus; however, this area is still not well recognized. Similar to other steroid hormones, this non-genomic action could be connected with VDR localization within the membrane and its interaction, for example, with caveolin 1 (CAV1) and SRC (SRC proto-oncogene) in caveolae to down-regulate WNT, sonic hedgehog (Shh) and NOTCH signaling. The rapid VDR-mediated action may also be a result of its direct interaction with other membrane receptors (eg., calcium channels, mitochondrial permeability transition pore) or intracellular pathways [69]. Recently, a novel mitochondrial localization of VDR has been described as the hub linking the control of cell metabolism [59]. Another non-genomic player for vitamin D3, the enzyme PDIA3 (protein disulphide isomerase family A member 3, also known as ERp57 or 1,25D3-MARRS), is found at various subcellular locations (plasma membrane, endoplasmic reticulum, mitochondria) and up until now is the best described membrane-associated protein that binds vitamin D [12][51][61]. For its action, PDIA3 requires also the interaction with CAV1 and is essential for the activation of protein kinases, such as CaMKII (calcium/calmodulin-dependent protein kinase II), PKC (protein kinase C) or phospolipases (PLA2, PLC) facilitating extracellular Ca2+ influx through L-type Ca2+ channels (L-VGCC). Other hypothetical non-genomic targets for the vitamin D fast action associated with PDIA3 could include: PKA (protein kinase A), PI3-K, MAPKs (mitogen-activated protein kinases) or Wnt5a (Wnt family member 5A) signaling pathways [12][61][69]. It is also suggested that PDIA3 may not directly bind vitamin D but may serve as a molecular chaperone for VDR or DBPs or other proteins. It should be noted that calcitriol or active vitamin D metabolites (e.g., 20(OH)D3 and 20,23(OH)2D3), when interacting with membrane VDR or PDIA3, could interact with some transcription factors (e.g., STAT3, NF-κB, Nrf2) and in this way they may influence in the long term the expression of various genes. It is not excluded that these active metabolites of vitamin D could also affect other transcription factors, such as RORα (retinoid-related orphan nuclear receptor alpha) and RORγ (retinoid-related orphan nuclear receptor gamma) or AhR (aryl hydrocarbon receptor) which will broaden the complexity of vitamin D action. Although the non-genomic effects of calcitriol and its metabolites, found mostly in in vitro studies, are still not well understood and seem to be dependent on the development stage or are tissue-specific, it is believed that they take place in vivo mainly to fine-tune the VDR-driven genomic response [61]. Further in vivo studies should confirm physiological and clinical significance of the postulated membrane receptors for vitamin D and their non-genomic actions.
3.3. Vitamin D Analogues
Over three thousand analogs or mimics of vitamin D were synthetized to overcome calcemic side effects (hypercalcemia and hypercalciuria) of supraphysiological concentrations of vitamin D3 which are needed to evoke pro-differentiating, anti-proliferative or anti-inflammatory effects [70][71][72]. The main modifications of 1,25(OH)2D3 structure are applied to its side-chain, A-ring (often together with side-chain changes), triene system, and C-ring and as a result, the increased VDR binding affinity and higher metabolic stability of the molecules were obtained [72]. However, only a few of the proposed and preclinically tested vitamin D analogs are clinically used to date for the treatment of secondary hyperparathyroidism (alfacalcidol, paricalcitol, doxercalciferol, falecalcitriol, maxacalcitol, oxacalcitriol), psoriasis (tacalcitol, calcipotriol, maxacalcitol) or osteoporosis (alfacalcidol, eldecalcitol) [71][72]. Regarding a potential usage of vitamin D analogs in cancer treatment, despite many promising in vitro results, some agents whichreached clinical trials in acute myeloid leukemia or pancreatic cancer (inecalcitol and seocalcitol, respectively), failed in phase II [72]. This dampened the interest of the pharma industry in further development of vitamin D compounds; however, some research is still in progress in academia in order to fully understand the actions of VDR agonists and antagonists, which hopefully could be further developed for treatment of various human diseases [70]. Apart from potential anticancer properties, some low calcemic vitamin D3 analogues showed protective effects against apoptotic- and oxidative stress-induced cell damage in neuronal cultures [73]. Interestingly, in a model of hydrogen peroxide-induced SH-SY5Y injury, a differential involvement of MAPK/ERK1/2 and PI3-K/Akt signaling in neuroprotective effects of 1,25(OH)2D3 and its low-calcemic analogue—PRI-2191 was found [74]. In differentiated SH-SY5Y cells, both 1,25(OH)2D3 and its structural analogue ZK191784 prevented amyloid-β peptide 1-42-induced toxicity via a sphingosine-1-phosphate/ceramide/p38MAPK/ATF4 signaling pathway [75]. In another study, vitamin D analogues, maxacalcitol, calcipotriol, alfacalcidol, paricalcitol, and doxercalciferol decreased amyloid-β formation and increased amyloid-β degradation. Calcipotriol was also shown to suppress calcium-dependent aggregation of α-synuclein (the key aggregating protein in Parkinson’s disease) by stimulating calbindin-D28k expression in SH-SY5Y neuroblastoma cells [76].
4. The Effects of Vitamin D3 in the CNS
Neurosteroids is the group 1,25(OH)2D3 belongs to, since it could be locally synthetized due to the presence of the key rate-limiting enzyme involved in the production of active form of vitamin D3, i.e., 1α-hydroxylase (CYP27B1) in various regions of the rodent and human brain [60][77]. In the brain, vitamin D3 could modulate multiple brain functions by itself or by cross-talking with other steroids signaling molecules, such as estrogens, progesterone or glucocorticoids [53]. In fact, a vast body of knowledge about the importance of sufficient amounts of vitamin D for proper brain development and its well-being during adulthood and during aging has been gathered in the last two decades, coming from experimental studies with vitamin D deficient animals as well as from clinical observations [60][78][79]. Although there are still many gaps in this area regarding, for example, the timing and duration of the critical window throughout life when low vitamin D may have detrimental impact on the brain, a causative link between low neonatal 25-OHD concentrations and an increased risk of schizophrenia [78][79] and hypoxic–ischemic encephalopathy (HIE) [80] has been evidenced.
Regarding the adult brain, it was postulated in the “two-hit hypothesis” that low vitamin D status may rather exacerbate brain lesions-induced by other detrimental events than being harmful by itself [78]. Various experimental studies showed beneficial effects of vitamin D supplementation in various in vitro and in vivo models of neurological or neuropsychiatric diseases at the level of modulation of neurotransmission, neuroprotection, neuroinflammation or neuroregeneration, and the latter was achieved by up-regulation of a wide variety of neurotrophins mainly in astrocytes (nerve growth factor (NGF), neurotrophin 3 (NT-3) or glial-derived neurotrophic factor (GDNF)) [12][60][79][81]. Since VDRs are abundantly expressed in neuronal and glial cells in various brain regions (prefrontal cortex, hippocampus, cingulate gyrus, thalamus, hypothalamus, and substantia nigra), the potential role of vitamin D in the treatment of the central nervous system (CNS) disorders under consideration, e.g., multiple sclerosis, dementia, Parkinson’s disease, depression, schizophrenia, and autism, has been suggested [78][79]. Of note, the distribution of nVDR receptors in the human and rodent brain is very similar [77]. Neurons and glial cells have been shown to contain 1α-hydroxylase, the enzyme that limits synthesis of the active form of vitamin D3, indicating that vitamin D can be synthesized and metabolized locally in the CNS. The highest concentrations of both 1α-hydroxylase and nVDRs are found in the hypothalamus and substantia nigra [77][78].
Alternatively, 1,25(OH)2D3 can also induce rapid non-genomic actions in the CNS via PDIA3 since this transcript is abundantly expressed in neurons, astrocytes, oligodendrocytes, microglia and endothelial cells [82]. However, the precise molecular mechanisms by which vitamin D affects the brain are still unclear and could engage both genomic and non-genomic mechanisms. Some researchers even suggest a predominant role of non-genomic effects of vitamin D in the brain, which may explain its rapid effect on calcium brain homeostasis, neurotransmission, oxidative status or intracellular signaling [12][82]. The non-genomic actions of vitamin D involve rapid response processes that are not dependent on transcriptional gene regulation. It is observed that 1,25(OH)2D3, by affecting the synthesis of neurotransmitters, growth factors and cytokines, modulates many functions of the CNS both during development and in adults [60][83][84]. In vitro and in vivo studies indicate that the synthesis of nerve growth factor (NGF), glial-derived neurotrophic factor (GDNF) and neurotrophin 3 (NT-3) was stimulated by vitamin D3. It also regulates gene expression of the low-affinity NGF receptor, p75NRT [85]. Moreover, 1,25(OH)2D3 was shown to be neuroprotective in an in vitro model of Alzheimer’s Disease through the restoration of Aβ-induced decrease in GDNF level and activation of the phosphatidylinositol 3 kinase (PI3K)/protein kinase B (Akt)/glycogen synthase kinase-3β (GSK-3β) pathway [86]. Both epidemiological data and results from animal experiments suggest that vitamin D3 deficiency may be a significant factor in increasing the risk of multiple sclerosis, diabetes, schizophrenia, and certain cancers, as well as SARS-CoV-2 virus infections [87][88]. Vitamin D deficiency also affects the expression of genes encoding mitochondrial, cytoskeletal and synaptic proteins in the adult rat brain [89] and causes permanent changes in the developing rat brain, disrupting the balance between neural stem cell proliferation and programmed cell death in the offspring [90]. Currently, as opposed to classical aminergic theories, the importance of neuroplastic and neuroinflammatory processes in the pathomechanism of depression, anxiety and cognitive deficits is underlined. Accordingly, regardless of their primary molecular targets, therapeutic efficacy of antidepressant drugs seems to depend on their ability to reverse brain-derived neurotrophic factor (BDNF) deficit and restore normally functioning neuronal networks in the brain structures relevant to these disorders [91][92]. Vitamin D3 shows antidepressant properties in experimental animal models and its mechanism of action is likely to involve the enhanced neurotrophin synthesis, neuromodulatory activity, as well as antioxidant and anti-inflammatory effects [93]. The hypothesis on the therapeutic potential of vitamin D3 in post-stroke depression is supported by experimental data which showed that vitamin D3 improved motor function and attenuated depression-like behaviors in a post-stroke depression model in mice by up-regulation of hippocampal VDR and BDNF expression [15]. Vitamin D’s ability to ameliorate neuroinflammatory processes supports the notion that this compound may prevent development of some psychiatric disorders in post-stroke patients [94].
References
- Aho, K.; Harmsen, P.; Hatano, S.; Marquardsen, J.; Smirnov, V.E.; Strasser, T. Cerebrovascular Disease in the Community: Results of a WHO Collaborative Study. Bull. World Health Organ. 1980, 58, 113–130.
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.; Culebras, A.; Elkind, M.S.V.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An Updated Definition of Stroke for the 21st Century: A Statement for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089.
- Wang, H.; Naghavi, M.; Allen, C.; Barber, R.M.; Bhutta, Z.A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; Coates, M.M.; et al. Global, Regional, and National Life Expectancy, All-Cause Mortality, and Cause-Specific Mortality for 249 Causes of Death, 1980–2015: A Systematic Analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544.
- Swanepoel, A.C.; Pretorius, E. Prevention and Follow-up in Thromboembolic Ischemic Stroke: Do We Need to Think out of the Box? Thromb. Res. 2015, 136, 1067–1073.
- Berger, J.R. COVID-19 and the Nervous System. J. Neurovirol. 2020, 26, 143–148.
- Higgins, V.; Sohaei, D.; Diamandis, E.P.; Prassas, I. COVID-19: From an Acute to Chronic Disease? Potential Long-Term Health Consequences. Crit. Rev. Clin. Lab. Sci. 2021, 58, 297–310.
- Montalvan, V.; Lee, J.; Bueso, T.; De Toledo, J.; Rivas, K. Neurological Manifestations of COVID-19 and Other Coronavirus Infections: A Systematic Review. Clin. Neurol. Neurosurg. 2020, 194, 105921.
- Nannoni, S.; de Groot, R.; Bell, S.; Markus, H.S. Stroke in COVID-19: A Systematic Review and Meta-Analysis. Int. J. Stroke 2021, 16, 137–149.
- Sagris, D.; Papanikolaou, A.; Kvernland, A.; Korompoki, E.; Frontera, J.A.; Troxel, A.B.; Gavriatopoulou, M.; Milionis, H.; Lip, G.Y.H.; Michel, P.; et al. COVID-19 and Ischemic Stroke. Eur. J. Neurol. 2021, 28, 3826–3836.
- Silva Andrade, B.; Siqueira, S.; de Assis Soares, W.R.; de Souza Rangel, F.; Santos, N.O.; dos Santos Freitas, A.; Ribeiro da Silveira, P.; Tiwari, S.; Alzahrani, K.J.; Góes-Neto, A.; et al. Long-COVID and Post-COVID Health Complications: An Up-to-Date Review on Clinical Conditions and Their Possible Molecular Mechanisms. Viruses 2021, 13, 700.
- Albers, G.W.; Goldstein, L.B.; Hess, D.C.; Wechsler, L.R.; Furie, K.L.; Gorelick, P.B.; Hurn, P.; Liebeskind, D.S.; Nogueira, R.G.; Saver, J.L. Stroke Treatment Academic Industry Roundtable (STAIR) Recommendations for Maximizing the Use of Intravenous Thrombolytics and Expanding Treatment Options with Intra-Arterial and Neuroprotective Therapies. Stroke 2011, 42, 2645–2650.
- Cui, X.; Gooch, H.; Petty, A.; McGrath, J.J.; Eyles, D. Vitamin D and the Brain: Genomic and Non-Genomic Actions. Mol. Cell. Endocrinol. 2017, 453, 131–143.
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-Endothelial Interactions at the Blood-Brain Barrier. Nat. Rev. Neurosci. 2006, 7, 41–53.
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42.
- Xu, Y.; Liang, L. Vitamin D3/Vitamin D Receptor Signaling Mitigates Symptoms of Post-Stroke Depression in Mice by Upregulating Hippocampal BDNF Expression. Neurosci. Res. 2021, 170, 306–313.
- Torrisi, M.; Bonanno, L.; Formica, C.; Arcadi, F.A.; Cardile, D.; Cimino, V.; Bramanti, P.; Morini, E. The Role of Rehabilitation and Vitamin D Supplementation on Motor and Psychological Outcomes in Poststroke Patients. Medicine 2021, 100, e27747.
- Marek, K.; Cicho, N. The Role of Vitamin D in Stroke Prevention and the Effects of Its Supplementation for Post-Stroke Rehabilitation. Nutrients 2022, 14, 2761.
- Detante, O.; Jaillard, A.; Moisan, A.; Barbieux, M.; Favre, I.M.; Garambois, K.; Hommel, M.; Remy, C. Biotherapies in Stroke. Rev. Neurol. 2014, 170, 779–798.
- Hossmann, K.-A. Viability Thresholds and the Penumbra of Focal Ischemia. Ann. Neurol. 1994, 36, 557–565.
- Sakai, S.; Shichita, T. Inflammation and Neural Repair after Ischemic Brain Injury. Neurochem. Int. 2019, 130, 104316.
- Doyle, K.; Simon, R.; Stenzel-poore, M. Mechanisms of Ischemic Brain Damage—Review Article. Neuropharmacology 2008, 55, 310–318.
- Sekerdag, E.; Solaroglu, I.; Gursoy-Ozdemir, Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr. Neuropharmacol. 2018, 16, 1396–1415.
- Radak, D.; Katsiki, N.; Resanovic, I.; Jovanovic, A.; Sudar-Milovanovic, E.; Zafirovic, S.; Mousad, S.A.; Isenovic, E.R. Apoptosis and Acute Brain Ischemia in Ischemic Stroke. Curr. Vasc. Pharmacol. 2016, 15, 115–122.
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate Release in Severe Brain Ischaemia Is Mainly by Reversed Uptake. Nature 2000, 403, 316–321.
- Krzyzanowska, W.; Pomierny, B.; Filip, M.; Pera, J. Glutamate Transporters in Brain Ischemia: To Modulate or Not? Acta Pharmacol. Sin. 2014, 35, 444–462.
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953.
- Bruno, V.; Battaglia, G.; Copani, A.; D’Onofrio, M.; Di Iorio, P.; De Blasi, A.; Melchiorri, D.; Flor, P.J.; Nicoletti, F. Metabotropic Glutamate Receptor Subtypes as Targets for Neuroprotective Drugs. J. Cereb. Blood Flow Metab. 2001, 21, 1013–1033.
- Ogden, K.K.; Traynelis, S.F. New Advances in NMDA Receptor Pharmacology. Trends Pharmacol. Sci. 2011, 32, 726–733.
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs Oppose Synaptic NMDARs by Triggering CREB Shut-off and Cell Death Pathways. Nat. Neurosci. 2002, 5, 405–414.
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA Receptor Involvement in Central Nervous System Disorders. Neuron 2014, 82, 279–293.
- Yan, J.; Peter Bengtson, C.; Buchthal, B.; Hagenston, A.M.; Bading, H. Coupling of NMDA Receptors and TRPM4 Guides Discovery of Unconventional Neuroprotectants. Science 2020, 370, eaay3302.
- Szydlowska, K.; Tymianski, M. Calcium, Ischemia and Excitotoxicity. Cell Calcium 2010, 47, 122–129.
- Crack, P.J.; Taylor, J.M. Reactive Oxygen Species and the Modulation of Stroke. Free Radic. Biol. Med. 2005, 38, 1433–1444.
- Gürsoy-Özdemir, Y.; Can, A.; Dalkara, T. Reperfusion-Induced Oxidative/Nitrativie Injury to Neurovascular Unit after Focal Cerebral Ischemia. Stroke 2004, 35, 1449–1453.
- Datta, A.; Sarmah, D.; Mounica, L.; Kaur, H.; Kesharwani, R.; Verma, G.; Veeresh, P.; Kotian, V.; Kalia, K.; Borah, A.; et al. Cell Death Pathways in Ischemic Stroke and Targeted Pharmacotherapy. Transl. Stroke Res. 2020, 11, 1185–1202.
- Tuo, Q.Z.; Zhang, S.T.; Lei, P. Mechanisms of Neuronal Cell Death in Ischemic Stroke and Their Therapeutic Implications. Med. Res. Rev. 2022, 42, 259–305.
- Zhou, Y.; Liao, J.; Mei, Z.; Liu, X.; Ge, J. Insight into Crosstalk between Ferroptosis and Necroptosis: Novel Therapeutics in Ischemic Stroke. Oxid. Med. Cell. Longev. 2021, 2021, 9991001.
- Kono, H.; Rock, K.L. How Dying Cells Alert the Immune System to Danger. Nat. Rev. Immunol. 2008, 8, 279–289.
- Bohacek, I.; Cordeau, P.; Lalancette-Hébert, M.; Gorup, D.; Weng, Y.C.; Gajovic, S.; Kriz, J. Toll-like Receptor 2 Deficiency Leads to Delayed Exacerbation of Ischemic Injury. J. Neuroinflamm. 2012, 9, 1–17.
- Linnik, M.D.; Zobrist, R.H.; Hatfield, M.D. Evidence Supporting a Role for Programmed Cell Death in Focal Cerebral Ischemia in Rats. Stroke 1993, 24, 2002–2008.
- Alonso De Leciñana, M.; Díez-Tejedor, E.; Gutierrez, M.; Guerrero, S.; Carceller, F.; Roda, J.M. New Goals in Ischemic Stroke Therapy: The Experimental Approach—Harmonizing Science with Practice. Cerebrovasc. Dis. 2005, 20, 159–168.
- Uzdensky, A.B. Apoptosis Regulation in the Penumbra after Ischemic Stroke: Expression of pro- and Antiapoptotic Proteins. Apoptosis 2019, 24, 687–702.
- Charriaut-Marlangue, C.; Margaill, I.; Represa, A.; Popovici, T.; Plotkine, M.; Ben-Ari, Y. Apoptosis and Necrosis after Reversible Focal Ischemia: An in Situ DNA Fragmentation Analysis. J. Cereb. Blood Flow Metab. 1996, 16, 186–194.
- MacManus, J.P.; Hill, I.E.; Huang, Z.-G.; Rasquinha, I.; Xue, D.; Buchan, A.M. DNA Damage Consistent with Apoptosis in Transient Focal Ischaemic Neocortex. Neuroreport 1994, 5, 493–496.
- Yilmaz, G.; Granger, D.N. Cell Adhesion Molecules and Ischemic Stroke. Neurol. Res. 2008, 30, 783–793.
- Kassis, H.; Shehadah, A.; Chopp, M.; Zhang, Z.G. Epigenetics in Stroke Recovery. Genes 2017, 8, 89.
- Hu, Z.; Zhong, B.; Tan, J.; Chen, C.; Lei, Q.; Zeng, L. The Emerging Role of Epigenetics in Cerebral Ischemia. Mol. Neurobiol. 2017, 54, 1887–1905.
- Bendik, I.; Friedel, A.; Roos, F.F.; Weber, P.; Eggersdorfer, M. Vitamin D: A Critical and Essential Micronutrient for Human Health. Front. Physiol. 2014, 5, 248.
- Norman, A.W. From Vitamin D to Hormone D: Fundamentals of the Vitamin D Endocrine System Essential for Good Health. Am. J. Clin. Nutr. 2008, 88, 491S–499S.
- Bikle, D.D. Vitamin D Metabolism, Mechanism of Action, and Clinical Applications. Chem. Biol. 2014, 21, 319–329.
- Gil, Á.; Plaza-Diaz, J.; Mesa, M.D. Vitamin D: Classic and Novel Actions. Ann. Nutr. Metab. 2018, 72, 87–95.
- Carlberg, C. Nutrigenomics of Vitamin D. Nutrients 2019, 11, 676.
- Norlin, M. Effects of Vitamin D in the Nervous System: Special Focus on Interaction with Steroid Hormone Signalling and a Possible Role in the Treatment of Brain Cancer. J. Neuroendocrinol. 2020, 32, e12799.
- Bivona, G.; Gambino, C.M.; Iacolino, G.; Ciaccio, M. Vitamin D and the Nervous System. Neurol. Res. 2019, 41, 827–835.
- Slominski, A.T.; Janjetovic, Z.; Fuller, B.E.; Zmijewski, M.A.; Tuckey, R.C.; Nguyen, M.N.; Sweatman, T.; Li, W.; Zjawiony, J.; Miller, D.; et al. Products of Vitamin D3 or 7-Dehydrocholesterol Metabolism by Cytochrome P450scc Show Anti-Leukemia Effects, Having Low or Absent Calcemic Activity. PLoS ONE 2010, 5, 7–10.
- Slominski, A.T.; Kim, T.; Hobrath, J.V.; Oak, A.S.W.; Edith, K.Y.; Tieu, E.W.; Li, W.; Tuckey, R.C.; Jetten, A.M. Endogenously Produced Nonclassical Vitamin D Hydroxy-Metabolites Act as “Biased” Agonists on Vdr and Inverse Agonists on Rorα and Rorγ. J. Steroid Biochem. Mol. Biol. 2017, 173, 42–56.
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol. Rev. 2015, 96, 365–408.
- Bikle, D.D.; Schwartz, J. Vitamin D Binding Protein, Total and Free Vitamin D Levels in Different Physiological and Pathophysiological Conditions. Front. Endocrinol. 2019, 10, 317.
- Wimalawansa, S.J. Vitamin D deficiency: Effects on oxidative stress, epigenetics, gene regulation, and aging. Biology 2019, 8, 30.
- Garcion, E.; Wion-Barbot, N.; Montero-Menei, C.N.; Berger, F.; Wion, D. New Clues about Vitamin D Functions in the Nervous System. Trends Endocrinol. Metab. 2002, 13, 100–105.
- Zmijewski, M.A.; Carlberg, C. Vitamin D Receptor(s): In the Nucleus but Also at Membranes? Exp. Dermatol. 2020, 29, 876–884.
- Haussler, M.R.; Jurutka, P.W.; Mizwicki, M.; Norman, A.W. Vitamin D Receptor (VDR)-Mediated Actions of 1α,25(OH) 2 Vitamin D 3: Genomic and Non-Genomic Mechanisms. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 543–559.
- Norman, A.W. Minireview: Vitamin D Receptor: New Assignments for an Already Busy Receptor. Endocrinology 2006, 147, 5542–5548.
- Shaffer, P.L.; Gewirth, D.T. Structural Basis of VDR-DNA Interactions on Direct Repeat Response Elements. EMBO J. 2002, 21, 2242–2252.
- Pertile, R.A.N.; Cui, X.; Eyles, D.W. Vitamin D Signaling and the Differentiation of Developing Dopamine Systems. Neuroscience 2016, 333, 193–203.
- Alroy, I.; Towers, T.L.; Freedman, L.P. Transcriptional Repression of the Interleukin-2 Gene by Vitamin D3: Direct Inhibition of NFATp/AP-1 Complex Formation by a Nuclear Hormone Receptor. Mol. Cell. Biol. 1995, 15, 5789–5799.
- Gurlek, A.; Pittelkow, M.R.; Kumar, R. Modulation of Growth Factor/Cytokine Synthesis and Signaling by 1α,25-Dihydroxyvitamin D3: Implications in Cell Growth and Differentiation. Endocr. Rev. 2002, 23, 763–786.
- Snegarova, V.; Naydenova, D. Vitamin D: A Review of Its Effects on Epigenetics and Gene Regulation. Folia Med. 2020, 62, 662–669.
- Ricca, C.; Aillon, A.; Bergandi, L.; Alotto, D.; Castagnoli, C.; Silvagno, F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int. J. Mol. Sci. 2018, 19, 1672.
- Chen, J.; Tang, Z.; Slominski, A.T.; Li, W.; Żmijewski, M.A.; Liu, Y.; Chen, J. Vitamin D and Its Analogs as Anticancer and Anti-Inflammatory Agents. Eur. J. Med. Chem. 2020, 207, 112738.
- Leyssens, C.; Verlinden, L.; Verstuyf, A. The Future of Vitamin D Analogs. Front. Physiol. 2014, 5, 122.
- Maestro, M.A.; Molnár, F.; Carlberg, C. Vitamin D and Its Synthetic Analogs. J. Med. Chem. 2019, 62, 6854–6875.
- Regulska, M.; Leśkiewicz, M.; Budziszewska, B.; Kutner, A.; Jantas, D.; Basta-Kaim, A.; Kubera, M.; Jaworska-Feil, L.; Lasoń, W. Inhibitory Effects of 1,25-Dihydroxyvitamin D3 and Its Low-Calcemic Analogues on Staurosporine-Induced Apoptosis. Pharmacol. Rep. 2007, 59, 393–401.
- Regulska, M.; Leśkiewicz, M.; Budziszewska, B.; Kutner, A.; Basta-Kaim, A.; Kubera, M.; Jaworska-Feil, L.; Lasoń, W. Involvement of P13-K in Neuroprotective Effects of the 1,25-Dihydroxyvitamin D3 Analogue—PRI-2191. Pharmacol. Rep. 2006, 58, 900–907.
- Pierucci, F.; Garcia-Gil, M.; Frati, A.; Bini, F.; Martinesi, M.; Vannini, E.; Mainardi, M.; Luzzati, F.; Peretto, P.; Caleo, M.; et al. Vitamin D3 Protects against Aβ Peptide Cytotoxicity in Differentiated Human Neuroblastoma SH-SY5Y Cells: A Role for S1P1/P38MAPK/ATF4 Axis. Neuropharmacology 2017, 116, 328–342.
- Grimm, M.O.W.; Thiel, A.; Lauer, A.A.; Winkler, J.; Lehmann, J.; Regner, L.; Nelke, C.; Janitschke, D.; Benoist, C.; Streidenberger, O.; et al. Vitamin D and Its Analogues Decrease Amyloid-β (Aβ) Formation and Increase Aβ-Degradation. Int. J. Mol. Sci. 2017, 18, 2764.
- Eyles, D.W.; Smith, S.; Kinobe, R.; Hewison, M.; McGrath, J.J. Distribution of the Vitamin D Receptor and 1α-Hydroxylase in Human Brain. J. Chem. Neuroanat. 2005, 29, 21–30.
- Cui, X.; Gooch, H.; Groves, N.J.; Sah, P.; Burne, T.H.; Eyles, D.W.; McGrath, J.J. Vitamin D and the Brain: Key Questions for Future Research. J. Steroid Biochem. Mol. Biol. 2015, 148, 305–309.
- Lang, F.; Ma, K.; Leibrock, C.B. 1,25(OH)2D3 in Brain Function and Neuropsychiatric Disease. Neurosignals 2019, 27, 40–49.
- Lowe, D.W.; Hollis, B.W.; Wagner, C.L.; Bass, T.; Kaufman, D.A.; Horgan, M.J.; Givelichian, L.M.; Sankaran, K.; Yager, J.Y.; Katikaneni, L.D.; et al. Vitamin D Insufficiency in Neonatal Hypoxic–ischemic Encephalopathy. Pediatr. Res. 2017, 82, 55–62.
- Chen, K.B.; Lin, A.M.Y.; Chiu, T.H. Systemic Vitamin D3 Attenuated Oxidative Injuries in the Locus Coeruleus of Rat Brain. Ann. NY Acad. Sci. 2003, 993, 313–324.
- Landel, V.; Stephan, D.; Cui, X.; Eyles, D.; Feron, F. Differential Expression of Vitamin D-Associated Enzymes and Receptors in Brain Cell Subtypes. J. Steroid Biochem. Mol. Biol. 2018, 177, 129–134.
- Neveu, I.; Naveilhan, P.; Jehan, F.; Baudet, C.; Wion, D.; De Luca, H.F.; Brachet, P. 1,25-Dihydroxyvitamin D3 Regulates the Synthesis of Nerve Growth Factor in Primary Cultures of Glial Cells. Mol. Brain Res. 1994, 24, 70–76.
- Neveu, I.; Naveilhan, P.; Baudet, C.; Brachet, P.; Metsis, M. 1,25-Dihydroxyvitamin D3 Regulates NT-3, NT-4 but Not BDNF MRNA in Astrocytes. Neuroreport 1994, 6, 124–126.
- Pertile, R.A.N.; Cui, X.; Hammond, L.; Eyles, D.W. Vitamin D Regulation of GDNF/Ret Signaling in Dopaminergic Neurons. FASEB J. 2018, 32, 819–828.
- Lin, C.-I.; Chang, Y.; Kao, N.; Lee, W.; Cross, T.; Lin, S. 1,25(OH)2D3 Alleviates Aβ(25-35)-Induced Tau Hyperphosphorylation, Excessive Reactive Oxygen Species, and Apoptosis Through Interplay with Glial Cell Line-Derived Neurotrophic Factor Signaling in SH-SY5Y Cells. Int. J. Mol. Sci. 2020, 21, 4215.
- McGrath, J.J.; Féron, F.P.; Burne, T.H.J.; MacKay-Sim, A.; Eyles, D.W. Vitamin D3—Implications for Brain Development. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 557–560.
- Ilie, P.C.; Stefanescu, S.; Smith, L. The Role of Vitamin D in the Prevention of Coronavirus Disease 2019 Infection and Mortality. Aging Clin. Exp. Res. 2020, 32, 1195–1198.
- Eyles, D.; Almeras, L.; Benech, P.; Patatian, A.; Mackay-Sim, A.; McGrath, J.; Féron, F. Developmental Vitamin D Deficiency Alters the Expression of Genes Encoding Mitochondrial, Cytoskeletal and Synaptic Proteins in the Adult Rat Brain. J. Steroid Biochem. Mol. Biol. 2007, 103, 538–545.
- Levenson, C.W.; Figueirôa, S.M. Gestational Vitamin D Deficiency: Long-Term Effects on the Brain. Nutr. Rev. 2008, 66, 726–729.
- Monteggia, L.M.; Björkholm, C. BDNF—A Key Transducer of Antidepressant Effects. Neuropharmacology 2016, 102, 72–79.
- Duman, R.S.; Deyama, S.; Fogaça, M.V. Role of BDNF in the Pathophysiology and Treatment of Depression: Activity-dependent Effects Distinguish Rapid-acting Antidepressants. Eur. J. Neurosci. 2021, 53, 126–139.
- Kouba, B.R.; Camargo, A.; Gil-Mohapel, J.; Rodrigues, A.L.S. Molecular Basis Underlying the Therapeutic Potential of Vitamin D for the Treatment of Depression and Anxiety. Int. J. Mol. Sci. 2022, 23, 7077.
- Marazziti, D.; Parra, E.; Palermo, S.; Barberi, F.M.; Buccianelli, B.; Ricciardulli, S.; Cappelli, A.; Mucci, F.; Dell’Osso, L. Vitamin D: A Pleiotropic Hormone with Possible Psychotropic Activities. Curr. Med. Chem. 2021, 28, 3843–3864.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
28 Nov 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No