Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ali Mussa | -- | 2281 | 2022-11-18 14:25:09 | | | |
| 2 | Catherine Yang | Meta information modification | 2281 | 2022-11-21 03:44:43 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ismail, N.H.; Mussa, A.; Zakaria, N.A.; Al-Khreisat, M.J.; Zahidin, M.A.; Ramli, N.N.; Mohammad, S.N.N.A.; Hassan, R.; Noor, N.H.M.; Iberahim, S.; et al. Targeting Epigenetic Mechanisms in Multiple Myeloma. Encyclopedia. Available online: https://encyclopedia.pub/entry/35310 (accessed on 29 June 2026).
Ismail NH, Mussa A, Zakaria NA, Al-Khreisat MJ, Zahidin MA, Ramli NN, et al. Targeting Epigenetic Mechanisms in Multiple Myeloma. Encyclopedia. Available at: https://encyclopedia.pub/entry/35310. Accessed June 29, 2026.
Ismail, Nor Hayati, Ali Mussa, Nur Atikah Zakaria, Mutaz Jamal Al-Khreisat, Muhamad Aidil Zahidin, Noor Nabila Ramli, Siti Nur Nabeela A’ifah Mohammad, Rosline Hassan, Noor Haslina Mohd Noor, Salfarina Iberahim, et al. "Targeting Epigenetic Mechanisms in Multiple Myeloma" Encyclopedia, https://encyclopedia.pub/entry/35310 (accessed June 29, 2026).
Ismail, N.H., Mussa, A., Zakaria, N.A., Al-Khreisat, M.J., Zahidin, M.A., Ramli, N.N., Mohammad, S.N.N.A., Hassan, R., Noor, N.H.M., Iberahim, S., Zulkafli, Z., Yusoff, S.M., Husin, A., & Johan, M.F. (2022, November 18). Targeting Epigenetic Mechanisms in Multiple Myeloma. In Encyclopedia. https://encyclopedia.pub/entry/35310
Ismail, Nor Hayati, et al. "Targeting Epigenetic Mechanisms in Multiple Myeloma." Encyclopedia. Web. 18 November, 2022.
Copy Citation
Multiple myeloma (MM) is an exceptionally complicated and heterogeneous disease that is caused by the abnormal proliferation of malignant monoclonal plasma cells initiated in the bone marrow. In disease progression, a multistep process including differentiation, proliferation, and invasion is involved.
multiple myeloma
DNA methylation
histone modification
1. Targeting DNA Methylation
Between the years 2004 and 2015, the United States Food and Drug Administration (FDA) granted authorisation for the use of six different epigenetic agents in clinical trials. These epigenetic agents include azacytidine, 5-aza-2′-deoxycytidine, suberoylanilide hydroxamic acid (SAHA), romidepsin, belinostat, panobinostat and chidamide [1]. In addition, the FDA has granted approval for the therapeutic use of 5-aza-2′deoxycytidine (5-aza-AZA; decitabine) and 5-aza-acytidine (5-aza-AZA) for the treatment of patients who suffer from MDS and chronic myelomonocytic leukaemia. These two epigenetic drugs did not have a clinical license to treat myelodysplastic syndromes [2], but they did display anti-myeloma efficacy in vitro and in vivo [3][4]. AZA [5] and DAC [6] accelerate clonal cell cycle arrest by boosting the activation of negative cell cycle regulators, which ultimately leads to apoptosis and senescence pathways (p16 and p15). G0/G1 and G2/M cell cycle arrests involving p21 and p38 were detected following DAC therapy [6]. Curiously, AZA in combination with doxorubicin and bortezomib had synergistic anti-MM efficacy [7] and restored sensitivity to dexamethasone [8]. Both AZA and 5-aza-2′-deoxycytidine (DAC) are capable of exerting detrimental effects by integrating into DNA and blocking covalently bound DNMT enzymes, which in turn causes DNMT damage and passive DNA demethylation [9]. In a model of murine myeloma, treatment with CM-272, an inhibitor combination that blocks both DNMTs and the histone methyltransferase G9a, decreases bone loss associated with the tumour and reduces the overall volume of the tumour. Utilising inhibitors allows for the induction of osteoblast formation in myeloma MSCs and the restoration of the expression of hypermethylated osteogenic regulators [10].
2. Targeting Histone Acetylation
HDACi displays anti-MM activity in cells by activating the apoptotic pathway, inhibiting the proteasome, and decreasing tumorigenesis and treatment resistance [11][12]. WT161, an HDAC6 inhibitor, induces cell death by boosting tubulin acetylation and inhibiting aggresome-dependent protein degradation in MM cells both in vitro and in vivo [13]. Panobinostat (LBH589) is a pan-HDACi that interacts with bortezomib and has been licensed for patients with relapsed or refractory MM [14] due to its ability to inhibit class I, II, and IV HDACs at a low nanomolar concentration [15][16]. This agent inhibits aggresome and proteasome networks and enhances the acetylation of proteins implicated in many carcinogenic pathways in MM cells [17]. Thus, progression-free survival (PFS) and complete and near-complete responses were significantly enhanced. Nevertheless, some individuals had adverse symptoms, including thrombocytopenia, diarrhoea, asthenia, and weariness. The combined treatment of panobinostat, bortezomib, and dexamethasone is predicted to benefit patients with MM cancer who developed bortezomib resistance [18]. Additionally, the first oral selective HDAC6 inhibitor, Ricolinostat (ACY-1215), showed reduced class I HDAC activity when coupled with carfilzomib [19], lenalidomide, and dexamethasone [20], showing anti-MM effects following therapy. HDACi concentrates on bromodomain and the extra-terminal domain (BET) because the BET proteins physically link the enhancer and promoter regions to stimulate the initiation and extension of gene transcription. By blocking the MYC oncogene and its gene expression network, BET inhibitors have shown an anti-MM effect in vitro and/or in vivo, either alone or in combination, highlighting that BET inhibitors might be considered a feasible therapeutic intervention in MM [21].
3. Targeting Histone Methylation
Inhibitors of histone methyltransferase for enhancer of zeste homolog 2 (EZH2) are emerging as an epigenetic therapy strategy for MM disease whether used alone or in combination with other targeted drugs. EZH2, which contains the enzyme component of the polycomb repressive complex 2 (PRC2), is essential for both normal cell development and the progression of disease (PRC2). PRC2 in EZH2 catalyses the methylation of histone H3 lysine tail residue 27 (H3K27me3), which induces the reprogramming of cells associated with stem cell self-renewal, cell cycle, cell differentiation, and cellular transformation. Thus, the discovery of highly selective inhibitors of EZH2’s histone methyltransferase activity has shed light on the function of EZH2 and PRC2 in carcinogenesis and their potential as cancer therapy targets [22].
Since both target combinations allow for the control of gene expression, histone H3 lysine 27 (H3K27) methyltransferase and G9, an H3K9 methyltransferase, have been identified as another promising therapeutic target in MM. To be more specific, the combination of these two inhibitors induces cell cycle arrest and triggers the pathway that leads to apoptosis, which in turn lowers the rate of MM cell growth. In addition, an examination in animals demonstrated an anticancer effect, as shown by a decrease in the formation of MM cell xenografts. There is also a correlation between greater levels of EZH2 and EHMT2 expression (both of which encode G9a) and worse outcomes for patients with MM. In contrast, the inhibition of EZH2/G9a resulted in an increase in the expression of genes that are activated by IFN and a reduction in the expression of genes that are involved in the IRF4-MYC axis in MM cells. This is supported by the observation that the degree of ERV gene expression in MM cells has dramatically risen and that the H3K27/H3K9 methylation levels have decreased, both of which are indicators of an IFN response [23].
The methylation process in histone H3 lysine-4 (H3K4), -36 (H3K36), and -79 (H3K79) caused transcriptional pathway upregulation in MM cells. Gene silencing events, on the other hand, were shown by methylation involving histone H4 lysine 20 (H4K20) [24]. For instance, GSK126, the EZH2 inhibitor, has been administrated to patients with MM with relapsed or refractory phases in phase I clinical trials [25]. Furthermore, MMSET histone methyltransferase was discovered as a promising target for epigenetic treatment in MM due to the anti-tumour activity shown following the shRNA-mediated suppression of MM cells in vitro and in vivo [26]. Consequently, LEM-06 has been introduced as an MMSET inhibitor to serve as an alternative model for assessing the therapeutic potential of MMSET in MM [27].
Table 1 outlines the types of epigenetic inhibitors administered to patients with MM, along with their mode of action.
Table 1. The types of epigenetic inhibitors administered to patients with MM.
| Epigenetic Inhibitors | Mechanisms | References |
|---|---|---|
| DNMT inhibitors | ||
| Azacytidine | Destructs proteosome DNMT and decondenses chromatin Enhances necrosis via oxidative stress |
[28] [29] |
| 5-aza-2′deoxycytidine/decitabine | Damages DNA via gamma-H2AX foci formation G0/G1- or G2/M-phase arrest and caspase-mediated apoptosis Activates DDR |
[30] |
| HDAC inhibitor | ||
| Entinostat | HDACi Class I, effectively inhibits HDAC1 and HDAC3 Induces apoptosis via the downregulation of erbB3 expression Loosens the chromatin conformation and exposes DNA structure to destructive agents |
[31] [32] [3] |
| Panobinostat (LBH-589) |
HDACi Class I, II, and IV Dysregulates canonical Wnt signalling and key player β-catenin Reactivates cancer-suppressed genes and promotes cell death Significant toxicity across all HDAC classes |
[33] [34] [35] |
| Vorinostat | HDACi class I and II Enhances cancer cell-cycle arrest and apoptosis Upregulates the E-cadherin gene and is less toxic than monotherapy or combination therapy |
[4] [36] |
| Romidepsin | Cyclic tetrapeptide HDAC inhibitor HDACi Class 1, Class 2, and Class 6 Enhances cancer suppressor genes p21 and p53 Suppresses antiapoptotic molecules, e.g., Bcl-2, Bcl-XL, BAX, and MCL-1 Initiates apoptosis in a dose-dependent fashion Enhances p53, agitates the function of HSP90, tubulin, and the endoplasmic reticulum, and forms aggresomes Induces cell-cycle arrest (via the p21 and AKT pathways) |
[37] [38] [39][40] [41] |
| ACY-241 (citarinostat) | Second-generation selective HDAC6 inhibitor More selective (13 to 18-fold) HDAC6 in comparison to HDAC1-3 Promotes higher serum concentrations Higher rating of HDAC inhibition including HDAC1/2 Alternative for potent and well-tolerated oral HDAC inhibitor |
[42] [43] |
| ACY-1215 (ricolinostat) | Enhances α-tubulin acetylation and accumulation of ubiquitinated proteins Reduces the inhibition of class I HDACs Lower toxicity than nonselective HDAC inhibitors |
[43][44] |
| Trichostatin A (TSA) | HDACi class I and II Effectively inhibits cell proliferation and initiates cell cycle arrest and apoptosis Sensitises TNF-related apoptosis-inducing factor (TRAIL)-resistant cells by suppressing the antiapoptotic BCL2 proteins Decreases expression of MM proliferation-associated factors |
[45] [46] [47] |
DNMT: DNA methyltransferase, H2AX: H2A.X variant histone, DDR: DNA damage response and repair, HDAC: histone deacetylase, HDACi: histone deacetylase inhibitors, erbB3: erb-b2 receptor tyrosine kinase 3, Bcl-2: B-cell lymphoma-2, Bcl-XL: B-cell lymphoma-extra-large, BAX: BCL2 associated X, MCL-1: myeloid leukaemia 1, HSP90: heat shock protein 90, p21: inhibitor of a cyclin-dependent kinase, AKT: protein kinase B, TRAIL: TNF-related apoptosis-inducing factor, and BCL2: B-cell lymphoma-2.
4. Targeting MicroRNAs in MM
miRNAs have been suggested as a new class of agents for MM therapeutic intervention since extensive research indicated their deregulation effect on MM cells and the possible targeting of several oncogenes or tumour suppressor genes, hence modifying MM development in vitro and in vivo [48]. Dysregulation of the tumour suppressor miRNA, miR-155, leads to the suppression of MM cell proliferation and treatment resistance. miR-155 overexpression increased drug-resistant MM cells’ sensitivity to bortezomib in a dosage and time-dependent manner. miRNA-155, on the other hand, targets TNF-mediated apoptosis by decreasing caspase-8 activity, inhibiting BID cleavage and caspase-3 activation [49][50]. Furthermore, the potentiality of this inhibitor to reduce CD47 on the cell surface activates the phagocytosis process through macrophage activity, resulting in tumour regression and enhanced bone resorption in animal models [51].
The hsa_circRNA_101237 has been investigated as a potential candidate for a circular RNA (circRNA) diagnostic biomarker for multiple myeloma (MM) malignancy. When hsa_circRNA_101237 was upregulated, it was discovered that a few signalling pathways such as PI3K-Akt signalling and chemokine signalling, that are cell cycle-related, as well as the signalling pathway of cytokines and their receptors were affected. These complex interactions are associated with significantly reduced OS and PFS rates [52]. Patients with MM who received four cycles of bortezomib-containing therapy had a differential response, with a higher M protein decrease correlating with a declined expression of hsa_circRNA_101237 [52]. This is observed in contrast to patients with MM who received treatment that did not contain bortezomib. Patients who received treatment that did not include bortezomib did not have a differential response. In addition, individuals who had a deletion of 13q14, an amplification of 1q21, a deletion of the P53 gene, and mutations in the t(4,14), t(14,16), and t(11,14) genes were observed to have overexpression, but mutations in the t(11,14) gene had the opposite effect. Overexpression has a substantial effect on the prognosis of patients with MM. In addition to this, patients who had multiple myeloma that relapsed or was resistant to treatment exhibited higher expression of the hsa_circRNA_101237 [52].
Additionally, miR145-3p inhibits the growth of MM cells by initiating the apoptotic pathway in vitro and in vivo experiments. Increased rates of the pro-apoptotic protein BCL2L11 and the inactivation of mTORC1 result in the activation of the autophagic flux, which in turn causes increased autophagy and cell death [53]. Furthermore, a significantly lower expression of circ-MYBL2 was indicated in MM bone marrow and serum [54], indicating a poor prognosis due to their advanced clinical stage and other factors. Treatment with exogenous circ-MYBL2 resulted in a robust MM cell death rate and enhanced DNA production and proliferation machinery. In a manner analogous to this, the treatment of circ-MYBL2 has been shown to inhibit the growth of subcutaneous xenograft tumours in experimental animal models [55]. Additionally, miRNA-338-3p was a substrate to circ_000784, and both miRNA-338-3p and circ_0007841 showed anti-MM effects on the development and progression of multiple myeloma. Because of the efforts made by miRNA-338-3p, the PI3K/AKT signalling pathway was made more effective by circular 0007841 [53][56]. Circ_0007841 displayed its oncogene activity through the miRNA-338-3p/BRD4 complex to increase cell proliferation and cell cycle mechanisms, eventually evading the cell apoptosis and senescence route [56][57]. The effects of ncRNAs on the expression of oncogenes and tumour suppressor genes in MM are illustrated in Figure 1.
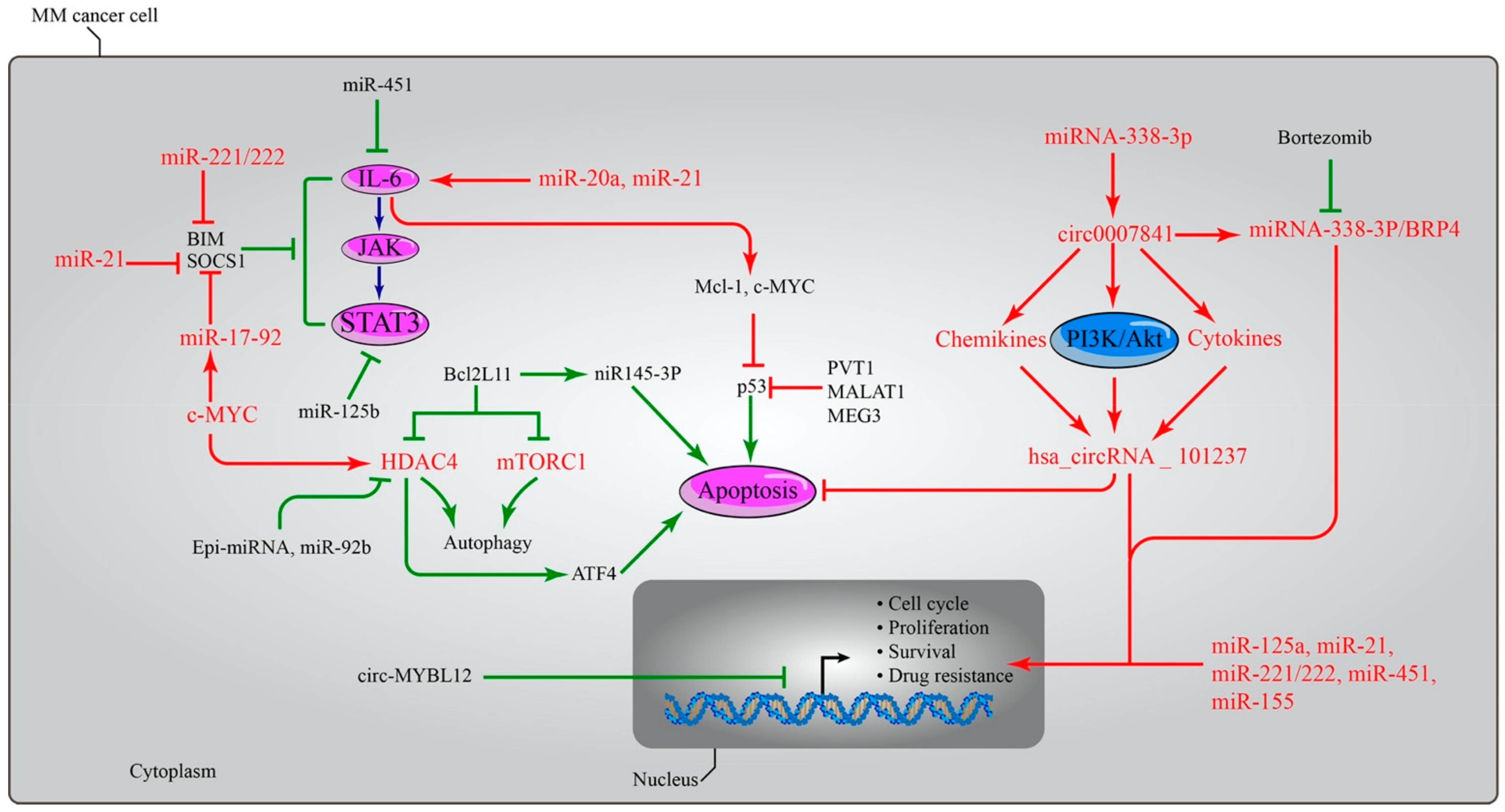
Figure 1. The dual roles of microRNAs in MM. microRNAs have the capacity to influence both the expressions of oncogenes and tumour suppressor genes. The upregulation of miR-21, miR-221/222, and miR-17-92 causes the deactivation of the BIM, SOCS1, and IL6-JAK-STAT3 pathways, which results in the activation of the Mcl-1, Bcl-XL, and c-Myc oncogenes and the suppression of the activity of the p53 gene through the LncRNAs PTV1, MALAT1, and MEG. In other pathways, the activation of miIR145-3p led to increased rates of the pro-apoptotic protein BCL2L11, and the inactivation of mTORC1 led to activation of the autophagic flux, which in turn led to increased autophagy and cell death. Both effects were caused by an increase in the rate of autophagy. miRNA-338-3p served as a substrate for circ_000784, which in turn activated the PI3K/AKT signalling pathway, as well as chemokines and cytokines, ultimately leading to the activation of hsa_circRNA_101237. Circ_0007841 revealed its oncogene activity via the miRNA-338-3p/BRD4 complex to boost cell proliferation and the processes of the cell cycle, hence bypassing the cell apoptosis and senescence pathway. Patients with multiple myeloma had significantly decreased levels of circ-MYBL2 expression in both bone marrow and serum, which is indicative of a poor prognosis owing to the advanced clinical stage of their disease and other variables. However, treatment with exogenous circ-MYBL2 resulted in a significant increase in the rate at which MM cells succumbed, as well as improved DNA synthesis and machinery for proliferation. Overexpression of miR-155 led to a dose- and time-dependent increase in drug-resistant multiple myeloma cells’ sensitivity to the anticancer drug bortezomib. In addition, the levels of miR-125a, miR-21, miR-221/222, miR-451, and miR-155 are increased in MM cells, which boosts the cell cycle, cell proliferation, and survival rate, as well as the likelihood of developing resistance to anti-myeloma therapy. miR: microRNA, BIM: Bcl-2-like protein 11, SOCS: Suppressors of cytokine signalling, IL6: Interleukin-6, JAK: Janus kinase, STAT: Signal transducer and activator of transcription, p53: tumour protein p53, Mcl-1: Myeloid cell leukemia-1, Bcl-XL: B-cell lymphoma-extra-large, c-Myc: c-myelocytomatosis oncogene product, mTOR: mammalian target of rapamycin complex 1, LncRNAs: Long Non-coding RNA, PTV1: plasmacytoma variant translocation 1, MALAT1: metastasis-associated lung adenocarcinoma transcript 1, MEG: maternally expressed gene, BCL2L11: Bcl-2-like protein 11, PI3K/AKT: Phosphatidylinositol-3-Kinase and Protein Kinase B, and BRD4: Bromodomain Containing 4.
References
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62.
- Jabbour, E.; Short, N.J.; Montalban-Bravo, G.; Huang, X.; Bueso-Ramos, C.; Qiao, W.; Yang, H.; Zhao, C.; Kadia, T.; Borthakur, G.; et al. Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood J. Am. Soc. Hematol. 2017, 130, 1514–1522.
- Cai, B.; Lyu, H.; Huang, J.; Wang, S.; Lee, C.K.; Gao, C.; Liu, B. Combination of bendamustine and entinostat synergistically inhibits proliferation of multiple myeloma cells via induction of apoptosis and DNA damage response. Cancer Lett. 2013, 335, 343–350.
- Richon, V. Cancer biology: Mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br. J. Cancer 2006, 95, S2–S6.
- Maes, K.; De Smedt, E.; Kassambara, A.; Hose, D.; Seckinger, A.; Van Valckenborgh, E.; Menu, E.; Klein, B.; Vanderkerken, K.; Moreaux, J.; et al. In vivo treatment with epigenetic modulating agents induces transcriptional alterations associated with prognosis and immunomodulation in multiple myeloma. Oncotarget 2015, 6, 3319.
- Lavelle, D.; DeSimone, J.; Hankewych, M.; Kousnetzova, T.; Chen, Y.-H. Decitabine induces cell cycle arrest at the G1 phase via p21WAF1 and the G2/M phase via the p38 MAP kinase pathway. Leuk. Res. 2003, 27, 999–1007.
- Kiziltepe, T.; Hideshima, T.; Catley, L.; Raje, N.; Yasui, H.; Shiraishi, N.; Okawa, Y.; Ikeda, H.; Vallet, S.; Pozzi, S.; et al. 5-Azacytidine, a DNA methyltransferase inhibitor, induces ATR-mediated DNA double-strand break responses, apoptosis, and synergistic cytotoxicity with doxorubicin and bortezomib against multiple myeloma cells. Mol. Cancer Ther. 2007, 6, 1718–1727.
- Nojima, M.; Maruyama, R.; Yasui, H.; Suzuki, H.; Maruyama, Y.; Tarasawa, I.; Sasaki, Y.; Asaoku, H.; Sakai, H.; Hayashi, T.; et al. Genomic screening for genes silenced by DNA methylation revealed an association between RASD1 inactivation and dexamethasone resistance in multiple myeloma. Clin. Cancer Res. 2009, 15, 4356–4364.
- Ewald, B.; Sampath, D.; Plunkett, W. Nucleoside analogs: Molecular mechanisms signaling cell death. Oncogene 2008, 27, 6522–6537.
- Cao, Y.; Yang, H.; Jin, L.; Du, J.; Fan, Z. Genome-Wide DNA Methylation Analysis during Osteogenic Differentiation of Human Bone Marrow Mesenchymal Stem Cells. Stem Cells Int. 2018, 2018, 8238496.
- New, M.; Olzscha, H.; La Thangue, N.B. HDAC inhibitor-based therapies: Can we interpret the code? Mol. Oncol. 2012, 6, 637–656.
- Maes, K.; Menu, E.; Van Valckenborgh, E.; Van Riet, I.; Vanderkerken, K.; De Bruyne, E. Epigenetic modulating agents as a new therapeutic approach in multiple myeloma. Cancers 2013, 5, 430–461.
- Hideshima, T.; Qi, J.; Paranal, R.M.; Tang, W.; Greenberg, E.; West, N.; Colling, M.E.; Estiu, G.; Mazitschek, R.; Perry, J.A.; et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. USA 2016, 113, 13162–13167.
- Robinson, R.M.; Basar, A.P.; Reyes, L.; Duncan, R.M.; Li, H.; Dolloff, N.G. PDI inhibitor LTI6426 enhances panobinostat efficacy in preclinical models of multiple myeloma. Cancer Chemother. Pharmacol. 2022, 89, 643–653.
- Prince, H.M.; Bishton, M.J.; Johnstone, R.W. Panobinostat (LBH589): A potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol. 2009, 5, 601–612.
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241.
- Catley, L.; Weisberg, E.; Kiziltepe, T.; Tai, Y.T.; Hideshima, T.; Neri, P.; Tassone, P.; Atadja, P.; Chauhan, D.; Munshi, N.C.; et al. Aggresome induction by proteasome inhibitor bortezomib and α-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006, 108, 3441–3449.
- Richardson, P.G.; Schlossman, R.L.; Alsina, M.; Weber, D.M.; Coutre, S.E.; Gasparetto, C.; Mukhopadhyay, S.; Ondovik, M.S.; Khan, M.; Paley, C.S.; et al. PANORAMA 2: Panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013, 122, 2331–2337.
- Mishima, Y.; Santo, L.; Eda, H.; Cirstea, D.; Nemani, N.; Yee, A.J.; O’Donnell, E.; Selig, M.K.; Quayle, S.N.; Arastu-Kapur, S.; et al. Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br. J. Haematol. 2015, 169, 423–434.
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, N.E.; Burke, J.N.; et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: A multicentre phase 1b trial. Lancet Oncol. 2016, 17, 1569–1578.
- Taniguchi, Y. The bromodomain and extra-terminal domain (BET) family: Functional anatomy of BET paralogous proteins. Int. J. Mol. Sci. 2016, 17, 1849.
- Alzrigat, M.; Jernberg-Wiklund, H.; Licht, J.D. Targeting EZH2 in Multiple Myeloma-Multifaceted Anti-Tumor Activity. Epigenomes 2018, 2, 16.
- Ishiguro, K.; Kitajima, H.; Niinuma, T.; Maruyama, R.; Nishiyama, N.; Ohtani, H.; Sudo, G.; Toyota, M.; Sasaki, H.; Yamamoto, E.; et al. Dual EZH2 and G9a inhibition suppresses multiple myeloma cell proliferation by regulating the interferon signal and IRF4-MYC axis. Cell Death Discov. 2021, 7, 7.
- Mozzetta, C.; Boyarchuk, E.; Pontis, J.; Ait-Si-Ali, S. Sound of silence: The properties and functions of repressive Lys methyltransferases. Nat. Rev. Mol. Cell Biol. 2015, 16, 499–513.
- Yap, T.A.; Winter, J.N.; Giulino-Roth, L.; Longley, J.; Lopez, J.; Michot, J.M.; Leonard, J.P.; Ribrag, V.; McCabe, M.T.; Creasy, C.L.; et al. Phase I study of the novel enhancer of zeste homolog 2 (EZH2) inhibitor GSK2816126 in patients with advanced hematologic and solid tumors. Clin. Cancer Res. 2019, 25, 7331–7339.
- Marango, J.; Shimoyama, M.; Nishio, H.; Meyer, J.A.; Min, D.-J.; Sirulnik, A.; Martinez-Martinez, Y.; Chesi, M.; Bergsagel, P.L.; Zhou, M.-M.; et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood J. Am. Soc. Hematol. 2008, 111, 3145–3154.
- Di Luccio, E. Inhibition of nuclear receptor binding SET domain 2/multiple myeloma SET domain by LEM-06 implication for epigenetic cancer therapies. J. Cancer Prev. 2015, 20, 113.
- Oka, S.; Ono, K.; Nohgawa, M. Successful treatment with azacitidine for the simultaneous occurrence of multiple myeloma and acute myeloid leukemia with concomitant del (5q) and the JAK2 V617F mutation. Ann. Hematol. 2017, 96, 1411–1413.
- Tian, E.; Tang, H.; Xu, R.; Liu, C.; Deng, H.; Wang, Q. Azacytidine induces necrosis of multiple myeloma cells through oxidative stress. Proteome Sci. 2013, 11, 24.
- Maes, K.; De Smedt, E.; Lemaire, M.; De Raeve, H.; Menu, E.; Van Valckenborgh, E.; McClue, S.; Vanderkerken, K.; De Bruyne, E. The role of DNA damage and repair in decitabine-mediated apoptosis in multiple myeloma. Oncotarget 2014, 5, 3115.
- Wang, B.; Lyu, H.; Pei, S.; Song, D.; Ni, J.; Liu, B. Cladribine in combination with entinostat synergistically elicits anti-proliferative/anti-survival effects on multiple myeloma cells. Cell Cycle 2018, 17, 985–996.
- Huang, X.; Gao, L.; Wang, S.; Lee, C.K.; Ordentlich, P.; Liu, B. HDAC inhibitor SNDX-275 induces apoptosis in erbB2-overexpressing breast cancer cells via down-regulation of erbB3 expression. Cancer Res. 2009, 69, 8403–8411.
- Banik, D.; Moufarrij, S.; Villagra, A. Immunoepigenetics combination therapies: An overview of the role of HDACs in cancer immunotherapy. Int. J. Mol. Sci. 2019, 20, 2241.
- Mithraprabhu, S.; Kalff, A.; Chow, A.; Khong, T.; Spencer, A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014, 9, 1511–1520.
- Raje, N.; Yee, A.J. Do We Need Another HDAC Inhibitor in Multiple Myeloma? Hematologist 2015, 12, 4155.
- Siegel, D.; Hussein, M.; Belani, C.; Robert, F.; Galanis, E.; Richon, V.M.; Garcia-Vargas, J.; Sanz-Rodriguez, C.; Rizvi, S. Vorinostat in solid and hematologic malignancies. J. Hematol. Oncol. 2009, 2, 31.
- Kikuchi, J.; Wada, T.; Shimizu, R.; Izumi, T.; Akutsu, M.; Mitsunaga, K.; Noborio-Hatano, K.; Nobuyoshi, M.; Ozawa, K.; Kano, Y.; et al. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood J. Am. Soc. Hematol. 2010, 116, 406–417.
- Mitsiades, N.; Mitsiades, C.S.; Richardson, P.G.; McMullan, C.; Poulaki, V.; Fanourakis, G.; Schlossman, R.; Chauhan, D.; Munshi, N.C.; Hideshima, T.; et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood J. Am. Soc. Hematol. 2003, 101, 4055–4062.
- Dickinson, M.; Johnstone, R.W.; Prince, H.M. Histone deacetylase inhibitors: Potential targets responsible for their anti-cancer effect. Investig. New Drugs 2010, 28, 3–20.
- Khan, S.B.; Maududi, T.; Barton, K.; Ayers, J.; Alkan, S. Analysis of histone deacetylase inhibitor, depsipeptide (FR901228), effect on multiple myeloma. Br. J. Haematol. 2004, 125, 156–161.
- Ocio, E.M.; San Miguel, J.F. The DAC system and associations with multiple myeloma. Investig. New Drugs 2010, 28, 28–35.
- North, B.J.; Almeciga-Pinto, I.; Tamang, D.; Yang, M.; Jones, S.S.; Quayle, S.N. Enhancement of pomalidomide anti-tumor response with ACY-241, a selective HDAC6 inhibitor. PLoS ONE 2017, 12, e0173507.
- Amengual, J.E.; Lue, J.K.; Ma, H.; Lichtenstein, R.; Shah, B.; Cremers, S.; Jones, S.; Sawas, A. First-in-Class Selective HDAC6 Inhibitor (ACY-1215) Has a Highly Favorable Safety Profile in Patients with Relapsed and Refractory Lymphoma. Oncologist 2021, 26, e184–e366.
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple MyelomaRicolinostat, Bortezomib, and Dexamethasone for Myeloma. Clin. Cancer Res. 2017, 23, 3307–3315.
- Moreaux, J.; Reme, T.; Leonard, W.; Veyrune, J.L.; Requirand, G.; Goldschmidt, H.; Hose, D.; Klein, B. Gene expression-based prediction of myeloma cell sensitivity to histone deacetylase inhibitors. Br. J. Cancer 2013, 109, 676–685.
- Fandy, T.E.; Srivastava, R.K. Trichostatin A sensitizes TRAIL-resistant myeloma cells by downregulation of the antiapoptotic Bcl-2 proteins. Cancer Chemother. Pharmacol. 2006, 58, 471–477.
- Heller, G.; Schmidt, W.M.; Ziegler, B.; Holzer, S.; Müllauer, L.; Bilban, M.; Zielinski, C.C.; Drach, J.; Zöchbauer-Müller, S. Genome-wide transcriptional response to 5-aza-2′-deoxycytidine and trichostatin a in multiple myeloma cells. Cancer Res. 2008, 68, 44–54.
- Chen, D.; Yang, X.; Liu, M.; Zhang, Z.; Xing, E. Roles of miRNA dysregulation in the pathogenesis of multiple myeloma. Cancer Gene Ther. 2021, 28, 1256–1268.
- Kumar, D.; Gokhale, P.; Broustas, C.; Chakravarty, D.; Ahmad, I.; Kasid, U. Expression of SCC-S2, an antiapoptotic molecule, correlates with enhanced proliferation and tumorigenicity of MDA-MB 435 cells. Oncogene 2004, 23, 612–616.
- Kumar, D.; Whiteside, T.L.; Kasid, U. Identification of a novel tumor necrosis factor-α-inducible gene, SCC-S2, containing the consensus sequence of a death effector domain of fas-associated death domain-like interleukin-1β-converting enzyme-inhibitory protein. J. Biol. Chem. 2000, 275, 2973–2978.
- Rastgoo, N.; Wu, J.; Liu, A.; Pourabdollah, M.; Atenafu, E.G.; Reece, D.; Chen, W.; Chang, H. Targeting CD47/TNFAIP8 by miR-155 overcomes drug resistance and inhibits tumor growth through induction of phagocytosis and apoptosis in multiple myeloma. Haematologica 2020, 105, 2813.
- Liu, X.; Tang, H.; Liu, J.; Wang, X. hsa_circRNA_101237: A novel diagnostic and prognostic biomarker and potential therapeutic target for multiple myeloma. Cancer Manag. Res. 2020, 12, 2109.
- Wu, H.; Liu, C.; Yang, Q.; Xin, C.; Du, J.; Sun, F.; Zhou, L. MIR145-3p promotes autophagy and enhances bortezomib sensitivity in multiple myeloma by targeting HDAC4. Autophagy 2020, 16, 683–697.
- Yu, S.; Ai, L.; Wei, W.; Pan, J. circRNA circ-MYBL2 is a novel tumor suppressor and potential biomarker in multiple myeloma. Hum. Cell 2021, 34, 219–228.
- Kawano, Y.; Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 2003, 116, 2627–2634.
- Wang, Y.; Lin, Q.; Song, C.; Ma, R.; Li, X. Circ_0007841 promotes the progression of multiple myeloma through targeting miR-338-3p/BRD4 signaling cascade. Cancer Cell Int. 2020, 20, 383.
- Petersen, S.; Wilson, A.J.; Hirst, J.; Roby, K.F.; Fadare, O.; Crispens, M.A.; Beeghly-Fadiel, A.; Khabele, D. CCNE1 and BRD4 co-amplification in high-grade serous ovarian cancer is associated with poor clinical outcomes. Gynecol. Oncol. 2020, 157, 405–410.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
907
Revisions:
2 times
(View History)
Update Date:
21 Nov 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No