 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jayarama Gunaje | + 3515 word(s) | 3515 | 2020-12-09 04:33:19 | | | |
| 2 | Catherine Yang | -109 word(s) | 3406 | 2020-12-10 03:27:44 | | |
Video Upload Options
Aspirin is a non-steroidal anti-inflammatory drug that is routinely used in decreasing inflammation, pain, fever and cardiovascular diseases. Aspirin has recently been recommended for the prevention of colorectal cancers (CRC) in adults aged above 59 yrs by the United States Preventive Services Task Force (USPSTF). Though acknowledged for its ability to decrease incidences of CRC, a consensus has not yet been reached regarding the mechanisms involved in its chemopreventive actions. Here, we briefly describe the "metabolite hypothesis", discuss its strengths and limitations and provide our perspective.
1. Introduction
Colorectal cancer (CRC) is the second most common cause of cancer related deaths [1][2] and the third most common cancer diagnosis [3] in the United States of America. Every year in the United States, around 150,000 cases of CRC are predicted and 1/3 of the affected patients die from this disease. Colorectal cancer usually begins as a benign adenomatous polyp that develops into an advanced form of adenoma with high-grade dysplasia, which eventually leads to the development of invasive cancer [4]. Formation of benign precancerous polyps that is characterized by the aggregation of abnormally dividing cells in the intestinal lumen marks the initiation of CRC. Over time, these polyps gain numerous mutations and acquire the ability to invade the bowel wall and eventually spread to the adjacent lymph nodes, and distant metastatic sites. The risk of a polyp developing into CRC increases with increasing size of the polyps owing to increased genetic changes and accumulation of epigenetic factors. As genetic damage increases over time, features of high-grade dysplasia start to present, and if left untreated, the polyps will develop the ability to invade adjacent tissues and grow beyond the confinement of the colon and rectum [4].
Aspirin and other non-steroidal anti-inflammatory drugs (NSAIDs) have shown promising anti-cancer effects upon regular consumption for 5 or more years [5][6][7][8]. The first report linking aspirin use and reduction in CRC was published in 1988 by Kune et al. that showed a statistically significant reduction in cases taking aspirin-containing medication in both men and women [9]. Numerous epidemiological and clinical studies thereafter proved aspirins’ efficacy to reduce CRC. These include (1) observational case-control studies [9][10], (2) randomized control trials in subjects with sporadic colorectal adenomas [11], (3) randomized control trials in subjects with Lynch syndrome [12][13] and (4) individual patient data meta-analysis of random control trials in the prevention of vascular events [14]. These studies indicated a decrease in the occurrence of CRC upon aspirin consumption and the overall incidences of gastrointestinal (GI) cancers in individual cases highly correlated with that of randomized trials [10][14]. It was also suggested that daily use of aspirin was associated with a significant reduction in colorectal adenomas in patients with previous incidences of CRC [11]. The reduction in CRC reported in these studies varied between 20 and 40% [8][15][16]. Aspirin was also shown to decrease chemically induced carcinogenesis in colorectal tissues using animal models [17][18][19][20]. Interestingly, data from epidemiological and clinical studies indicated that low-dose aspirin (75–81 mg) is as effective in preventing CRC as higher doses (≥325 mg) [7][21]. Aspirin consumption has been shown to cause GI bleeding and renal toxicity in some individuals, with an incidence of less than 4 per 1000 cases [22][23]. However, due to the increasing body of evidence for aspirin’s efficacy against CRC, it is argued that the benefits outweigh the risk. Hence, the United States Preventive Services Task Force (USPSTF), recommended “initiating low dose aspirin use for the primary prevention of cardiovascular disease and colorectal cancer in adults aged 50–59 years” in 2016 [24]. Following this, numerous clinical trials have been launched to establish its clinical use against CRC [25][26][27][28].
2. Pharmacological Effects of Aspirin
Following oral consumption, aspirin is absorbed mainly in the stomach and upper small intestine [29]; in addition, lymphatic uptake of aspirin has also been recently reported [30]. Aspirin concentration maximally achieved in the plasma varies depending on the dose with anti-platelet (75 mg/day), analgesic (325–600 mg/4–6 h) and anti-inflammatory (1.2 g/4–6 h) doses reaching 7.31 μM, 25–80 μM, and 144 μM, respectively [16]. The half-life of aspirin is ~20 min and it undergoes hydrolysis by intestinal, plasma and liver esterases to generate salicylic acid [16][29]. The concentration of salicylic acid in the plasma has been estimated to be around 15 μM for anti-platelet doses, 500 μM for analgesic doses and 1.5–2.5 mM for anti-inflammatory doses. The half-life of salicylic acid is estimated to be 4–6 h in the plasma [16]. It is reported that the oral bioavailability of aspirin tablets is ~40–50%; however, some reports have estimated that aspirin’s oral bioavailability is ~68% [31]. It is important to note that the oral bioavailability of aspirin is significantly reduced in enteric coated and sustained release tablets [32].
Aspirin like all other NSAIDs works through the inhibition of cyclooxygenase (COX; COX-1 and COX-2) enzymes in the body. COX-1 is the only isoform present in mature platelets and is responsible for platelet aggregation. COX-2 is not expressed under normal conditions; however, it is expressed in many tissues during inflammation, wound healing, and neoplasia [33][34]. The molecular mechanism of aspirin involves its ability to inhibit arachidonic acid metabolism through prostaglandin H (PGH) synthase or the COX pathway. Aspirin inhibits COX-1 by acetylating Ser530, and acetylates COX-2 at Ser516, thereby inactivating them [35–37]. Depending on the cell type, inhibition of COX activity affects synthesis of different prostaglandins (PGs) including PGD2, PGE2 and PGF2α, prostacyclin (PGI2) and thromboxane A2 (TXA2). TXA2 is the major metabolite that promotes the activation and aggregation of platelets [38]. It is to be noted that aspirin is more selective to COX-1 as compared to COX-2, with IC50 for COX-1 inhibition observed at 1.7 μM compared to the >100 μM required for COX-2 inhibition [35][36].
3. Inhibition of Cyclin Dependent Kinases (CDKs) by Aspirin Metabolites: The Metabolite Hypothesis
According to this recently proposed hypothesis, metabolites of aspirin/salicylic acid (2-3-dihydroxybenzoic acid (2,3-DHBA/pyrocatechuic acid) and 2,5-dihydroxybenzoic acid (2,5-DHBA/gentisic acid)) are key contributors to its anti-cancer effects against CRC (Figure 1) [37]. This is supported by the observations in our laboratory that have demonstrated that both 2,3-DHBA and 2,5-DHBA are capable of inhibiting cancer cell growth in vitro [38]. While 2,5-DHBA was universally effective in arresting cancer cell growth in colon cancer cell lines (HCT-116 and HT-29) and breast cancer cell lines (MDA-MB-231), 2,3-DHBA was more selective against MDA-MB-231 cell line; the concentration of the metabolites required to inhibit colony formation varied with each cell type. In HT-29 and HCT-116 cells, the inhibition with 2,5-DHBA was observed between 250 and 500 μM while in MDA-MB-231 cells, it required 50–100 μM of 2,5-DHBA. In contrast, 2,3-DHBA showed effective inhibition at ~500 μM in MDA-MB-231 cells. Our studies also indicated that CDKs are likely to be cellular targets of 2,3-DHBA and 2,5-DHBA. Both 2,3-DHBA and 2,5-DHBA were capable of inhibiting CDKs, particularly CDK1 and CDK6 [38][39].
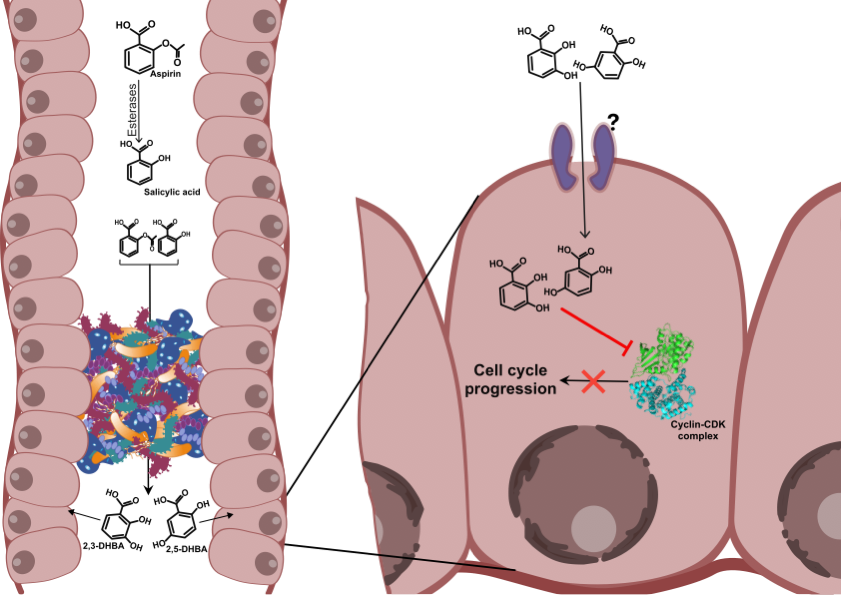
It is hypothesized that 2,3-DHBA and 2,5-DHBA are generated in the body due to microbial transformation of aspirin and salicylic acid that are left unabsorbed in the intestine. Previous studies have reported that only 40–50% of the orally consumed aspirin is bioavailable [16], and that aspirin undergoes rapid hydrolysis in the intestine to generate salicylic acid [31]. With enteric coated tablets, the bioavailability of aspirin is further reduced [16][32] and therefore more aspirin may be left unabsorbed in the intestine. Aspirin and salicylic acid remaining in the intestine may then be subjected to biotransformation by the resident microbiota. In support of this, Kim et al. demonstrated the conversion of aspirin and salicylic acid to hydroxy salicylic acids, mediated by the human gut microflora. Additionally, when rats were administered with ampicillin and aspirin, these authors showed that the aspirin metabolizing activity by the bacteria was significantly reduced as evidenced from fecal samples, and the levels of aspirin in the plasma doubled [40]. In a recent report it was also demonstrated that administration of aspirin to rats along with amoxicillin decreased aspirin metabolism in the intestine as compared to treatment with aspirin alone [41]. These two reports suggest that the biotransformation of aspirin by the gut microflora may be a source of 2,3-DHBA and 2,5-DHBA. Although aspirin and salicylic acid are also metabolized in the liver to generate 2,3-DHBA and 2,5-DHBA, they are reported to be minor metabolites of liver biotransformation [42]. Therefore, it is unlikely that these liver generated DHBAs, with low concentrations in the plasma, contribute to the anti-cancer effects of low-dose aspirin on colorectal tissues.
The in vitro studies which demonstrated the inhibitory effect of 2,3-DHBA and 2,5-DHBA on colon cancer cell growth suggested that the metabolites are effective at a concentration ranging from 100–500 μM [38]. We believe that these concentrations are pharmacologically achievable in the human intestine. It is reported that the intestinal fluid volume ranges between 160 and 750 mL depending on fasting or fed conditions respectively [43]. Considering that 50% of the aspirin/enteric-coated aspirin remains in the intestine/colon after oral ingestion, a dose of 81 mg aspirin will reach concentrations between 0.3 and 1.4 mM depending on the intestinal fluid volume. Hence, the concentration of 2,3-DHBA and 2,5-DHBA required to exert anti-cancer effects would be achievable from the biotransformation of aspirin remaining in the intestine.
Interestingly, Hydroxybenzoic acids (HBAs) are also generated from flavonoid degradation and are also present in fruits and vegetables [44][45][46]. The metabolite hypothesis thus proposes that HBAs generated from the degradation of aspirin/salicylic acid, flavonoids and those HBAs present in the diet may contribute to their chemopreventive properties [37]. This attractive theory requires further validation and in vivo experiments using germ free mice, which would provide information on the role of HBAs in aspirin’s anti-cancer effects against CRC. Additionally, a quantitative estimation of these metabolites generated in the intestine from aspirin/flavonoid degradation will establish whether pharmacologically relevant concentrations of HBAs are achievable in situ.
4. Potential Role of the Gut Microbiota in Aspirin’s Effect against CRC
In many studies, variability has been reported in aspirin’s ability to decrease the occurrences of cancers. While genetic reasons may account for some of this variability, it may also depend on the levels of HBAs generated in the intestine though cytochrome P450 (CYP450)-mediated metabolism within the cells or microbial degradation in the lumen. In this regard, a report showed that in patients carrying a CYP2C9 variant allele, there was a decreased risk reduction in colorectal adenomas, following aspirin administration. They suggested that this may be because of the inability of CYP2C9 to generate 2,5-DHBA [47], which again highlights the importance of aspirin metabolites in cancer prevention. On the other hand, microorganisms may also significantly differ with respect to their ability to biotransform aspirin and salicylic acid to generate HBAs. The intestinal microbiota may vary depending on the diet and therefore both diet and microbial composition in the intestine may play a role in HBA generation, accounting for the observed variability in epidemiological studies. Several reports have documented that diet influences the composition of the microbiota [90–92] and if HBAs are indeed important contributors to the cancer preventive properties of aspirin, newer strategies are required to achieve the desired chemopreventive effects in all population. This may include adjusting to a healthier diet that incorporates more fruits and vegetables along with aspirin, or consumption of appropriate probiotics along with aspirin in populations where dietary adjustment is difficult.
Interestingly, in one randomized clinical study, aspirin was shown to increase the abundance of Prevotella, Akkermansia, and Ruminococcaceae species while decreasing the levels of Bacteroides, Parabacteroides and Dorea species, that have collectively been associated with reduced CRC risk [48]. Another in vivo experiment with germ-free and conventionalized mice also demonstrated that aspirin was able to increase the probiotic bacteria Bifidobacterium and Lactobacillus, suggestive of a role in CRC prevention. However, this study also demonstrated that the aerobic microbe Lysinibacillus sphaericus was capable of degrading aspirin in the lumen; supplementation with antibiotics prevented such microbe-mediated aspirin degradation. Their study also pointed out the ability of aspirin to inhibit tumor formation in germ-free mice, raising the possibility that aspirin may prevent CRC through a microbiome-independent mechanism, and suggested that this may involve inhibition of COX-2 and PGE2 synthesis [94]. This raises the possibility that aspirin may exert anti-cancer effects independent of the formation of metabolites. A role for aspirin in increasing the beneficial bacteria which may contribute to human gut health and cancer prevention should be explored in future investigations. Under normal conditions, a portion of aspirin taken orally may be left unabsorbed, and therefore has the potential to act directly on colorectal tissues. However, aspirin may also be subjected to metabolism, generating 2,3-DHBA and 2,5-DHBA, that in turn act on the colorectal tissues to prevent cancer. As 2,5-DHBA and 2,3-DHBA have been shown to inhibit cancer cell growth [38][49], it would be interesting to determine if these aspirin metabolites can potentiate the cancer preventive properties of aspirin when administered to germ-free mice. Such studies should validate the significance of both intact aspirin and its metabolites collectively contributing to CRC prevention.
5. Perspective and Future Studies
Cancer is a genetic disease which arises due to mutations in protooncogenes and tumor suppressor genes whose imbalances in expression lead to uncontrolled cell proliferation. Although traditionally viewed as a disorder of cell proliferation, recent evidence suggests that cancer should also be considered as a metabolic disease owing to reprogramming of the metabolic pathways to meet increased cellular demands for energy and growth [50]. Therefore, effective preventive measures for cancer require identification of compounds that not only target proteins that regulate cell proliferation, but also those that affect cell metabolism. In this regard, aspirin holds great promise for the prevention of cancer; however, despite considerable efforts in identifying the mechanism of cancer prevention by aspirin, a consensus has not yet been reached. It appears that aspirin and its primary metabolite salicylic acid affect many enzymes, proteins, transcription factors, and signaling pathways involved in cell proliferation and cancer development; thus, the wide range of targets recognized for aspirin has made the identification of its primary target difficult.
Broadly speaking, researchers have classified the known mechanisms for aspirin’s cancer prevention under two categories—COX-dependent and COX-independent pathways. The platelet hypothesis (COX-dependent pathway) proposes sequential inhibition of COX-1 enzyme activity in platelets and COX-2 expression in nucleated cells of the colonic mucosa [6][33]. While it is an interesting and attractive hypothesis, it has not been conclusively proven. One major issue is that the hypothesis proposed is indirect and requires the involvement of multiple steps and factors to execute the observed anti-cancer effects. Additionally, the primary target (platelet COX-1) and subsequent downstream effector (epithelial/COX-2) are in different cell types. The platelet hypothesis also does not explain the preferential protective effects of aspirin against CRC as compared to other cancers. If platelet COX-1 inhibition is primarily responsible for cancer prevention following its absorption into circulation, it is reasonable to expect that aspirin should equally be effective against all cancers, which is not the case. Moreover, according to this hypothesis, the chemopreventive effects of aspirin are applicable against CRC that is developed after mucosal injury led platelet activation; it does not explain how low-dose aspirin works against cancers due to sporadic mutations where platelets may not be involved. This hypothesis was proposed to accommodate the inability of low-dose aspirin to directly inhibit COX-2, an important COX isoform implicated in the development of many cancers. Since low-dose aspirin was as effective as higher doses in preventing CRC, and the only known effect of low dose aspirin was COX-1 inhibition in platelets, the proponents of the platelet hypothesis opined that the inhibition of COX-1 in platelets may be central to the prevention of tumorigenesis. It is important to note that although COX-2 expression is implicated in the development of CRC, aspirin has been shown to be effective in inhibiting cancer cell growth even in cells that do not overexpress COX-2 [51]. Additionally, it has also been observed that some CRC cell lines do not produce COX-2 or they express an inactive form of COX-2 [52]. Another report also showed that cancers affecting the proximal colon, that generally do not over-express COX-2 [53], were more responsive to aspirin use than those cancers of the distal colon [7]. These observations raise the possibility that alternate pathways, independent of COX inhibition, may also play a substantial role in aspirin’s chemopreventive effects. However, prevention of platelet aggregation by aspirin may be important in curtailing the metastatic spread of cancer cells as platelet aggregation and activation has previously been implicated in cancer cell growth, cancer cell survival in circulation, protection from immune destruction and angiogenesis at sites of metastasis [54][55][56][57].
Aspirin’s preferential effects against CRC compared to cancers of other tissues raises the possibility that it may act directly on colorectal tissues before its absorption into circulation. Such an effect requires substantial concentrations of aspirin in the intestinal milieu which we believe is achievable. Since the bioavailability of aspirin is ~50% [58] and there is significant hydrolysis of aspirin occurring in the intestine [31], colorectal tissues are likely to be exposed to millimolar concentrations of aspirin and salicylic acid. This will be particularly true following consumption of enteric-coated tablets as its absorption is significantly lower than regular aspirin [32][59] making it more available in the lumen of the intestine. In such an environment, both aspirin and salicylic acid can target specific proteins in the colonic cells to modulate their function and bring about changes required for effective chemoprevention. In this context, intact aspirin has the potential to target proteins through acetylation, and along with salicylic acid, may also target specific transcription factors such as NF-κB or c-myc in colorectal tissues. Alternatively, other signaling pathways like Wnt/β-catenin or Akt/mTOR pathways may also be regulated as described above by aspirin and salicylic acid under these conditions.
Equally important are also the role of aspirin metabolites 2,3-DHBA and 2,5-DHBA in its chemopreventive effects. Both DHBAs have been demonstrated to inhibit CDK enzyme activity and colony formation in cancer cells, although 2,5-DHBA was more potent when compared to 2,3-DHBA [38][39]. In view of the suggestion that intestinal bacteria are capable of degrading aspirin and salicylic acid to DHBAs, their contribution to the chemopreventive effect of aspirin should also be considered. Except for a few reports, a role for HBAs in cancer prevention has not been investigated in depth [38][39][49]. Exploring the role played by HBAs in aspirin’s cancer prevention should take precedence in future investigations as they are also generated following degradation of flavonoids [60][61][62][63], which are known to prevent cancer. Interestingly, the aspirin metabolite 2,5-DHBA is also produced from the degradation of flavonoids in the human gut, and this has been detected both in the plasma and urine samples from test subjects [44][46]. In addition, there is plenty of evidence in the literature that increased consumption of fruits and vegetables, that are rich in HBAs, is inversely correlated to the occurrence of cancer [64][44][45][46]; an interesting target proposed for HBAs are CDKs present in colonic epithelial cells [37][38][39][65]. Some reports also indicate that the phenolic acid content in the human gut can collectively reach millimolar concentrations [46][46]. If one considers HBAs as possible common mediators of CRC prevention, it provides the simplest explanation through which all three compounds (aspirin, flavonoids and a diet rich in HBAs) can exert anti-cancer effects. Thus, the metabolite hypothesis borne out of the finding that HBAs inhibit cancer cell growth [37][38][39][65] needs to be explored in future research with regards to its targets and mechanisms of action.
The literature is immense on the mechanisms of cancer prevention by aspirin, yet they are unclear and complex even after two decades of research. Going back to the question—“how does low-dose aspirin preferentially act to protect against CRC?”—neither a consensus has been reached with regard to the mechanisms responsible for its cancer preventive actions nor has a single target been defined as solely responsible for its protective effect. This preferential preventive effect may mainly be attributed to the fact that colorectal tissues are the first to get exposed to aspirin/salicylic acid or its metabolites following oral consumption. In our view, aspirin or its metabolites acting locally on colorectal tissues to target specific proteins appear to be the most simplistic explanation as compared to aspirin targeting platelet COX-1 in the circulation, leading to prevention of tumorigenesis. However, aspirin through inhibition of platelet COX-1 may play a role in preventing cancer metastasis, which is also important in the overall anti-cancer effects of aspirin. We believe that in view of aspirin’s reported ability to prevent CRC, efforts should be put-forth to resolve this important dilemma. In this regard, the recently described metabolite hypothesis along with other targets of aspirin, acting directly on colorectal tissues should be further explored. We hope that a resolution would be reached in the near future on aspirin’s mode of action against CRC through scientific research and open debate.
References from : https://www.mdpi.com/1422-0067/21/23/9018/htm
References
- Bardhan, K.; Liu, K. Epigenetics and colorectal cancer pathogenesis. Cancers 2013, 5, 676–713.
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164.
- Ahmed, M. Colon Cancer: A Clinician’s Perspective in 2019. Gastroenterol. Res. 2020, 13, 1–10.
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460.
- Chan, A.T.; Giovannucci, E.L.; Meyerhardt, J.A.; Schernhammer, E.S.; Curhan, G.C.; Fuchs, C.S. Long-term use of aspirin and nonsteroidal anti-inflammatory drugs and risk of colorectal cancer. JAMA 2005, 294, 914–923.
- Thun, M.J.; Jacobs, E.J.; Patrono, C. The role of aspirin in cancer prevention. Nat. Rev. Clin. Oncol. 2012, 9, 259–267.
- Rothwell, P.M.; Wilson, M.; Elwin, C.E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750.
- Rothwell, P.M.; Wilson, M.; Price, J.F.; Belch, J.F.; Meade, T.W.; Mehta, Z. Effect of daily aspirin on risk of cancer metastasis: A study of incident cancers during randomised controlled trials. Lancet 2012, 379, 1591–1601.
- Kune, G.A.; Kune, S.; Watson, L.F. Colorectal cancer risk, chronic illnesses, operations, and medications: Case control results from the Melbourne Colorectal Cancer Study. Cancer Res. 1988, 48, 4399–4404.
- Algra, A.M.; Rothwell, P.M. Effects of regular aspirin on long-term cancer incidence and metastasis: A systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012, 13, 518–527.
- Cole, B.F.; Logan, R.F.; Halabi, S.; Benamouzig, R.; Sandler, R.S.; Grainge, M.J.; Chaussade, S.; Baron, J.A. Aspirin for the chemoprevention of colorectal adenomas: Meta-analysis of the randomized trials. J. Natl. Cancer Inst. 2009, 101, 256–266.
- Burn, J.; Bishop, D.T.; Mecklin, J.P.; Macrae, F.; Moslein, G.; Olschwang, S.; Bisgaard, M.L.; Ramesar, R.; Eccles, D.; Maher, E.R.; et al. Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N. Engl. J. Med. 2008, 359, 2567–2578.
- Burn, J.; Gerdes, A.M.; Macrae, F.; Mecklin, J.P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Lancet 2011, 378, 2081–2087.
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.; Wilson, M.; Mehta, Z.; et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612.
- Cuzick, J.; Thorat, M.A.; Bosetti, C.; Brown, P.H.; Burn, J.; Cook, N.R.; Ford, L.G.; Jacobs, E.J.; Jankowski, J.A.; La Vecchia, C.; et al. Estimates of benefits and harms of prophylactic use of aspirin in the general population. Ann. Oncol. 2015, 26, 47–57.
- Dovizio, M.; Tacconelli, S.; Sostres, C.; Ricciotti, E.; Patrignani, P. Mechanistic and pharmacological issues of aspirin as an anticancer agent. Pharmaceuticals 2012, 5, 1346–1371.
- Shpitz, B.; Bomstein, Y.; Kariv, N.; Shalev, M.; Buklan, G.; Bernheim, J. Chemopreventive effect of aspirin on growth of aberrant crypt foci in rats. Int. J. Colorectal Dis. 1998, 13, 169–172.
- Rohwer, N.; Kuhl, A.A.; Ostermann, A.I.; Hartung, N.M.; Schebb, N.H.; Zopf, D.; McDonald, F.M.; Weylandt, K.H. Effects of chronic low-dose aspirin treatment on tumor prevention in three mouse models of intestinal tumorigenesis. Cancer Med. 2020, 9, 2535–2550.
- Tian, Y.; Ye, Y.; Gao, W.; Chen, H.; Song, T.; Wang, D.; Mao, X.; Ren, C. Aspirin promotes apoptosis in a murine model of colorectal cancer by mechanisms involving downregulation of IL-6-STAT3 signaling pathway. Int. J. Colorectal Dis. 2011, 26, 13–22.
- Reddy, B.S.; Rao, C.V.; Rivenson, A.; Kelloff, G. Inhibitory effect of aspirin on azoxymethane-induced colon carcinogenesis in F344 rats. Carcinogenesis 1993, 14, 1493–1497.
- Garcia-Albeniz, X.; Chan, A.T. Aspirin for the prevention of colorectal cancer. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 461–472.
- Garcia Rodriguez, L.A.; Martin-Perez, M.; Hennekens, C.H.; Rothwell, P.M.; Lanas, A. Bleeding Risk with Long-Term Low-Dose Aspirin: A Systematic Review of Observational Studies. PLoS ONE 2016, 11, e0160046.
- Cea Soriano, L.; Lanas, A.; Soriano-Gabarro, M.; Garcia Rodriguez, L.A. Incidence of Upper and Lower Gastrointestinal Bleeding in New Users of Low-Dose Aspirin. Clin. Gastroenterol. Hepatol. 2019, 17, 887–895 e886.
- Bibbins-Domingo, K.; U.S. Preventive Services Task Force. Aspirin Use for the Primary Prevention of Cardiovascular Disease and Colorectal Cancer: U.S. Preventive Services Task Force Recommendation StatementAspirin Use for the Primary Prevention of CVD and CRC. Ann. Intern. Med. 2016, 164, 836–845.
- Bosetti, C.; Santucci, C.; Gallus, S.; Martinetti, M.; La Vecchia, C. Aspirin and the risk of colorectal and other digestive tract cancers: An updated meta-analysis through 2019. Ann. Oncol. 2020, 31, 558–568.
- Drew, D.A.; Chin, S.M.; Gilpin, K.K.; Parziale, M.; Pond, E.; Schuck, M.M.; Stewart, K.; Flagg, M.; Rawlings, C.A.; Backman, V.; et al. ASPirin Intervention for the REDuction of colorectal cancer risk (ASPIRED): A study protocol for a randomized controlled trial. Trials 2017, 18, 50.
- Roy, H.K.; Turzhitsky, V.; Wali, R.; Radosevich, A.J.; Jovanovic, B.; Della’Zanna, G.; Umar, A.; Rubin, D.T.; Goldberg, M.J.; Bianchi, L.; et al. Spectral biomarkers for chemoprevention of colonic neoplasia: A placebo-controlled double-blinded trial with aspirin. Gut 2017, 66, 285–292.
- Cao, Y.; Nishihara, R.; Wu, K.; Wang, M.; Ogino, S.; Willett, W.C.; Spiegelman, D.; Fuchs, C.S.; Giovannucci, E.L.; Chan, A.T. Population-wide Impact of Long-term Use of Aspirin and the Risk for Cancer. JAMA Oncol. 2016, 2, 762–769.
- Needs, C.J.; Brooks, P.M. Clinical pharmacokinetics of the salicylates. Clin. Pharmacokinet. 1985, 10, 164–177.
- Lichtenberger, L.M.; Phan, T.; Fang, D.; Edler, S.; Philip, J.; Li-Geng, T.; Dial, E.J. Bioavailability of aspirin in rats comparing the drug’s uptake into gastrointestinal tissue and vascular and lymphatic systems: Implications on aspirin’s chemopreventive action. J. Physiol. Pharmacol. 2016, 67, 635–642.
- Rowland, M.; Riegelman, S.; Harris, P.A.; Sholkoff, S.D. Absorption kinetics of aspirin in man following oral administration of an aqueous solution. J. Pharm. Sci. 1972, 61, 379–385.
- Cox, D.; Maree, A.O.; Dooley, M.; Conroy, R.; Byrne, M.F.; Fitzgerald, D.J. Effect of enteric coating on antiplatelet activity of low-dose aspirin in healthy volunteers. Stroke 2006, 37, 2153–2158.
- Patrignani, P.; Patrono, C. Aspirin and Cancer. J. Am. Coll. Cardiol. 2016, 68, 967–976.
- Dovizio, M.; Alberti, S.; Guillem-Llobat, P.; Patrignani, P. Role of platelets in inflammation and cancer: Novel therapeutic strategies. Basic Clin. Pharmacol. Toxicol. 2014, 114, 118–127.
- Warner, T.D.; Giuliano, F.; Vojnovic, I.; Bukasa, A.; Mitchell, J.A.; Vane, J.R. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: A full in vitro analysis. Proc. Natl. Acad. Sci. USA 1999, 96, 7563–7568.
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97–120.
- Sankaranarayanan, R.; Kumar, D.R.; Patel, J.; Bhat, G.J. Do Aspirin and Flavonoids Prevent Cancer through a Common Mechanism Involving Hydroxybenzoic Acids? The Metabolite Hypothesis. Molecules 2020, 25, 2243.
- Sankaranarayanan, R.; Valiveti, C.K.; Dachineni, R.; Kumar, D.R.; Lick, T.; Bhat, G.J. Aspirin metabolites 2,3DHBA and 2,5DHBA inhibit cancer cell growth: Implications in colorectal cancer prevention. Mol. Med. Rep. 2020, 21, 20–34.
- Dachineni, R.; Kumar, D.R.; Callegari, E.; Kesharwani, S.S.; Sankaranarayanan, R.; Seefeldt, T.; Tummala, H.; Bhat, G.J. Salicylic acid metabolites and derivatives inhibit CDK activity: Novel insights into aspirin’s chemopreventive effects against colorectal cancer. Int. J. Oncol. 2017, 51, 1661–1673.
- Kim, I.S.; Yoo, D.H.; Jung, I.H.; Lim, S.; Jeong, J.J.; Kim, K.A.; Bae, O.N.; Yoo, H.H.; Kim, D.H. Reduced metabolic activity of gut microbiota by antibiotics can potentiate the antithrombotic effect of aspirin. Biochem. Pharmacol. 2016, 122, 72–79.
- Zhang, J.; Sun, Y.; Wang, R.; Zhang, J. Gut Microbiota-Mediated Drug-Drug Interaction between Amoxicillin and Aspirin. Sci. Rep. 2019, 9, 16194.
- Hutt, A.J.; Caldwell, J.; Smith, R.L. The metabolism of aspirin in man: A population study. Xenobiotica 1986, 16, 239–249.
- Schiller, C.; Frohlich, C.P.; Giessmann, T.; Siegmund, W.; Monnikes, H.; Hosten, N.; Weitschies, W. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Aliment. Pharmacol. Ther. 2005, 22, 971–979.
- Russell, W.R.; Scobbie, L.; Labat, A.; Duthie, G.G. Selective bio-availability of phenolic acids from Scottish strawberries. Mol. Nutr. Food Res. 2009, 53 (Suppl. S1), S85–S91.
- Tomás-Barberán, F.A.; Clifford, M.N. Dietary hydroxybenzoic acid derivatives—Nature, occurrence and dietary burden. J. Sci. Food Agric. 2000, 80, 1024–1032.
- Williamson, G.; Clifford, M.N. Colonic metabolites of berry polyphenols: The missing link to biological activity? Br. J. Nutr. 2010, 104 (Suppl. S3), S48–S66.
- Bigler, J.; Whitton, J.; Lampe, J.W.; Fosdick, L.; Bostick, R.M.; Potter, J.D. CYP2C9 and UGT1A6 genotypes modulate the protective effect of aspirin on colon adenoma risk. Cancer Res. 2001, 61, 3566–3569.
- Prizment, A.E.; Staley, C.; Onyeaghala, G.C.; Vivek, S.; Thyagarajan, B.; Straka, R.J.; Demmer, R.T.; Knights, D.; Meyer, K.A.; Shaukat, A.; et al. Randomised clinical study: Oral aspirin 325 mg daily vs. placebo alters gut microbial composition and bacterial taxa associated with colorectal cancer risk. Aliment. Pharmacol. Ther. 2020.
- Altinoz, M.A.; Elmaci, I.; Cengiz, S.; Emekli-Alturfan, E.; Ozpinar, A. From epidemiology to treatment: Aspirin’s prevention of brain and breast-cancer and cardioprotection may associate with its metabolite gentisic acid. Chem. Biol. Interact. 2018, 291, 29–39.
- Coller, H.A. Is cancer a metabolic disease? Am. J. Pathol. 2014, 184, 4–17.
- Hanif, R.; Pittas, A.; Feng, Y.; Koutsos, M.I.; Qiao, L.; Staiano-Coico, L.; Shiff, S.I.; Rigas, B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem. Pharmacol. 1996, 52, 237–245.
- Hsi, L.C.; Baek, S.J.; Eling, T.E. Lack of cyclooxygenase-2 activity in HT-29 human colorectal carcinoma cells. Exp. Cell. Res. 2000, 256, 563–570.
- Birkenkamp-Demtroder, K.; Olesen, S.H.; Sorensen, F.B.; Laurberg, S.; Laiho, P.; Aaltonen, L.A.; Orntoft, T.F. Differential gene expression in colon cancer of the caecum versus the sigmoid and rectosigmoid. Gut 2005, 54, 374–384.
- Ornelas, A.; Zacharias-Millward, N.; Menter, D.G.; Davis, J.S.; Lichtenberger, L.; Hawke, D.; Hawk, E.; Vilar, E.; Bhattacharya, P.; Millward, S. Beyond COX-1: The effects of aspirin on platelet biology and potential mechanisms of chemoprevention. Cancer Metastasis Rev. 2017, 36, 289–303.
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134.
- Lichtenberger, L.M.; Vijayan, K.V. Are Platelets the Primary Target of Aspirin’s Remarkable Anticancer Activity? Cancer Res. 2019, 79, 3820–3823.
- Lichtenberger, L.M.; Fang, D.; Bick, R.J.; Poindexter, B.J.; Phan, T.; Bergeron, A.L.; Pradhan, S.; Dial, E.J.; Vijayan, K.V. Unlocking Aspirin’s Chemopreventive Activity: Role of Irreversibly Inhibiting Platelet Cyclooxygenase-1. Cancer Prev. Res. 2017, 10, 142–152.
- Pedersen, A.K.; FitzGerald, G.A. Dose-related kinetics of aspirin. Presystemic acetylation of platelet cyclooxygenase. N. Engl. J. Med. 1984, 311, 1206–1211.
- Sagar, K.A.; Smyth, M.R. A comparative bioavailability study of different aspirin formulations using on-line multidimensional chromatography. J. Pharm. Biomed. Anal. 1999, 21, 383–392.
- De Ferrars, R.M.; Czank, C.; Zhang, Q.; Botting, N.P.; Kroon, P.A.; Cassidy, A.; Kay, C.D. The pharmacokinetics of anthocyanins and their metabolites in humans. Br. J. Pharmacol. 2014, 171, 3268–3282.
- Hanske, L.; Engst, W.; Loh, G.; Sczesny, S.; Blaut, M.; Braune, A. Contribution of gut bacteria to the metabolism of cyanidin 3-glucoside in human microbiota-associated rats. Br. J. Nutr. 2013, 109, 1433–1441.
- Thilakarathna, W.; Rupasinghe, H.P.V. Microbial metabolites of proanthocyanidins reduce chemical carcinogen-induced DNA damage in human lung epithelial and fetal hepatic cells in vitro. Food Chem. Toxicol. 2019, 125, 479–493.
- Pace, E.; Jiang, Y.; Clemens, A.; Crossman, T.; Rupasinghe, H.P.V. Impact of Thermal Degradation of Cyanidin-3-O-Glucoside of Haskap Berry on Cytotoxicity of Hepatocellular Carcinoma HepG2 and Breast Cancer MDA-MB-231 Cells. Antioxidants 2018, 7, 24.
- Altinoz, M.A.; Elmaci, I.; Ozpinar, A. Gentisic Acid, a Quinonoid Aspirin Metabolite in Cancer Prevention and Treatment. New Horizons in Management of Brain Tumors and Systemic Cancers. J. Cancer Res. Oncobiol. 2018, 1, 109.
- Sankaranarayanan, R.; Valiveti, C.K.; Kumar, D.R.; Van Slambrouck, S.; Kesharwani, S.S.; Seefeldt, T.; Scaria, J.; Tummala, H.; Bhat, G.J. The Flavonoid Metabolite 2,4,6-Trihydroxybenzoic Acid Is a CDK Inhibitor and an Anti-Proliferative Agent: A Potential Role in Cancer Prevention. Cancers 2019, 11, 427.


