Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Samareh Younesian | -- | 3979 | 2022-11-14 20:01:01 | | | |
| 2 | Conner Chen | Meta information modification | 3979 | 2022-11-15 09:15:58 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Younesian, S.; Yousefi, A.; Momeny, M.; Ghaffari, S.H.; Bashash, D. DNA Methylation in Neurodegenerative Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/34498 (accessed on 26 July 2026).
Younesian S, Yousefi A, Momeny M, Ghaffari SH, Bashash D. DNA Methylation in Neurodegenerative Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/34498. Accessed July 26, 2026.
Younesian, Samareh, Amir-Mohammad Yousefi, Majid Momeny, Seyed H. Ghaffari, Davood Bashash. "DNA Methylation in Neurodegenerative Diseases" Encyclopedia, https://encyclopedia.pub/entry/34498 (accessed July 26, 2026).
Younesian, S., Yousefi, A., Momeny, M., Ghaffari, S.H., & Bashash, D. (2022, November 14). DNA Methylation in Neurodegenerative Diseases. In Encyclopedia. https://encyclopedia.pub/entry/34498
Younesian, Samareh, et al. "DNA Methylation in Neurodegenerative Diseases." Encyclopedia. Web. 14 November, 2022.
Copy Citation
DNA methylation is critical for the normal development and functioning of the human brain, such as the proliferation and differentiation of neural stem cells, synaptic plasticity, neuronal reparation, learning, and memory. Despite the physical stability of DNA and methylated DNA compared to other epigenetic modifications, some DNA methylation-based biomarkers have translated into clinical practice. Increasing reports indicate a strong association between DNA methylation profiles and various clinical outcomes in neurological diseases, such as neurodegenerative diseases.
DNA methylation
neurological disorders
Huntington’s disease
Alzheimer’s disease
1. Fragile X Syndrome (FXS)
Fragile X syndrome (FXS), the most common inherited mental retardation, is caused when the expression of the fragile X mental retardation protein (FMRP)—an essential factor for the maintenance of synapses—is downregulated in the brain tissue due to aberrant repeat of CGG trinucleotide in the 5′ UTR of the gene [1][2]. The presence of more than 200 copies of CGG trinucleotide attracts DNA methylation modifiers to the promotor of FMR1, thereby disrupting gene transcription. The results of in vitro analysis also confirmed that among different epigenetic modifications, this is DNA methylation has a significant role in the pathogenesis of the disease (Figure 1) [3]. FMRP, an RNA binding protein, has been represented as a molecular brake for the translation of several mRNAs, in particular, mRNAs linked to the activity of metabotropic glutamate receptor group I (mGluRI); hence, it possibly acts as an essential factor for maintaining normal synaptic plasticity. Loss of FMRP expression, in addition to activation of the mGluRI pathway, may lead to dysregulation of endocannabinoid signaling, ion channel dysfunction, reduction of GABA signaling, and increased actin polymerization [2]. Meanwhile, Liu et al. showed that enforcing the expression of TET1 in neural cells of FXS patients using the dCas9-TET1 CD could reactivate the expression of FMRP by eliminating the methyl groups from the promotor of the FMR1 gene [4].
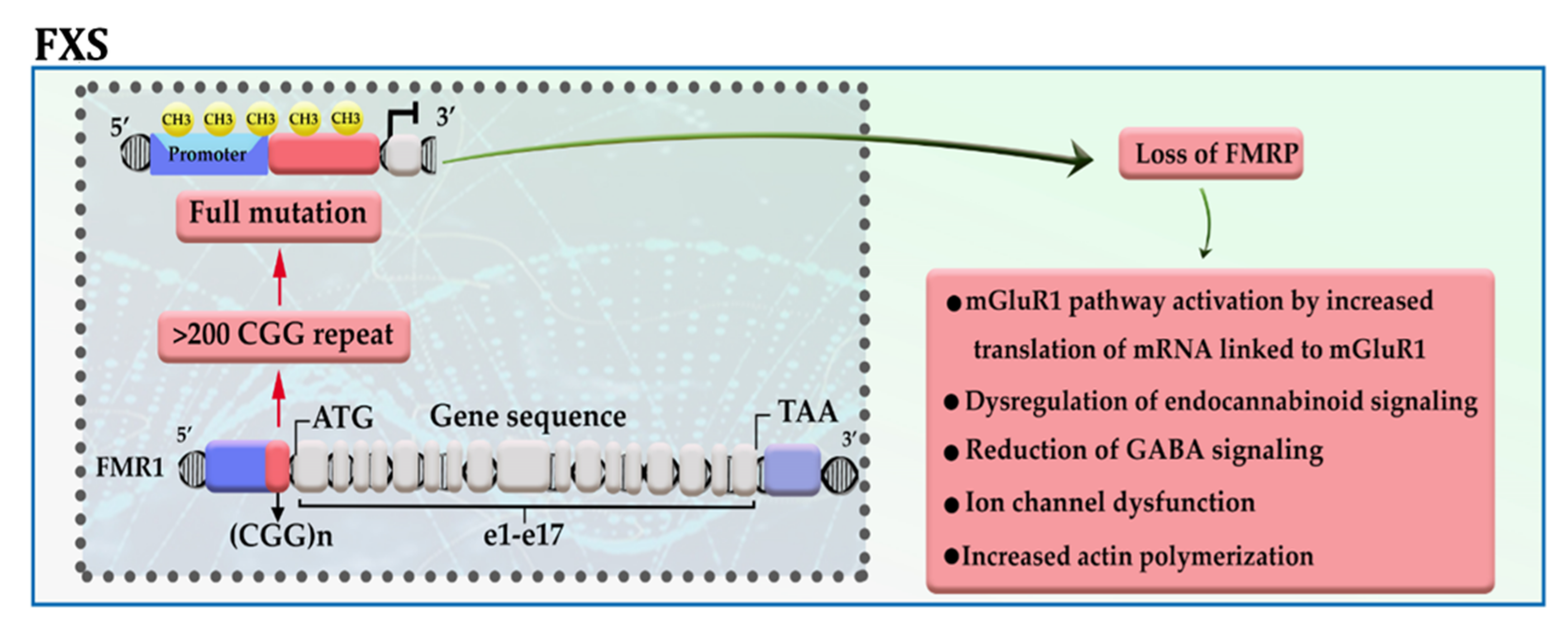
Figure 1. A schematic representation of the selected mechanisms in the pathogenesis of FXS. The presence of more than 200 copies of CGG repeat in the 5′ UTR region of the FMR1 gene leads to promoter hypermethylation, transcriptional silencing, and loss of FMR protein. mGluRI: group 1 metabotropic glutamate receptors; GABA: gamma-aminobutyric acid.
2. Huntington’s Disease (HD)
The incidence of more than 40 CAG trinucleotide repeats in the huntingtin gene (HTT) located on chromosome 4 gives rise to one of the most dreadful neurodegenerative diseases, named Huntington’s disease (HD). The defective HTT protein is toxic to neural cells, especially those that are in subcortical basal ganglia (striatum) [5][6]. The mutant HTT (mHTT) produces two transcripts, full-length mHTT mRNA and mHTT exon 1 mRNA (the result of CAG repeat length-dependent aberrant splicing) that are translated into full mHTT protein and mHTT exon 1 protein. Full mHTT protein undergoing proteolysis (by caspases and calpains) is cleaved to generate mHTT exon 1 like protein and other products. mHTT exon 1 protein consists of HTTNT (an N-terminal segment of 17 amino acids), polyQ (a polyglutamine sequence encoded by the CAG repeat), and PRD (a proline-rich domain of 51 amino acids). mHTT exon 1 protein enters the nucleus. The mHTT exon 1 proteins can oligomerize; then, gradual aggregation of oligomers leads to the formation of mHTT fibrils and subsequently large inclusions in the cytoplasm and nucleus of neural cells. Intracytoplasmic inclusions (ICIs) have various toxic effects, including axonal transport impairment, glutamate excitotoxicity, and mitochondrial abnormalities as well as inhibition of proteasomes, chaperones, and autophagy, which can exacerbate the aggregation of mHTT exon 1 protein [5]. While the polyQ tract of intranuclear inclusions (INIs) can alter the interaction with transcription factors and epigenetic modifiers, thereby causing a shift from open to closed chromatin underlies the reduced transcriptional of essential genes, such as BDNF and PPARGC1A [7]. For example, the polyQ tract of mHTT increases the interaction of mHTT-MECP2 and the recruitment of MECP2 into the BDNF promoter, thereby reducing BDNF expression (Figure 2) [8].
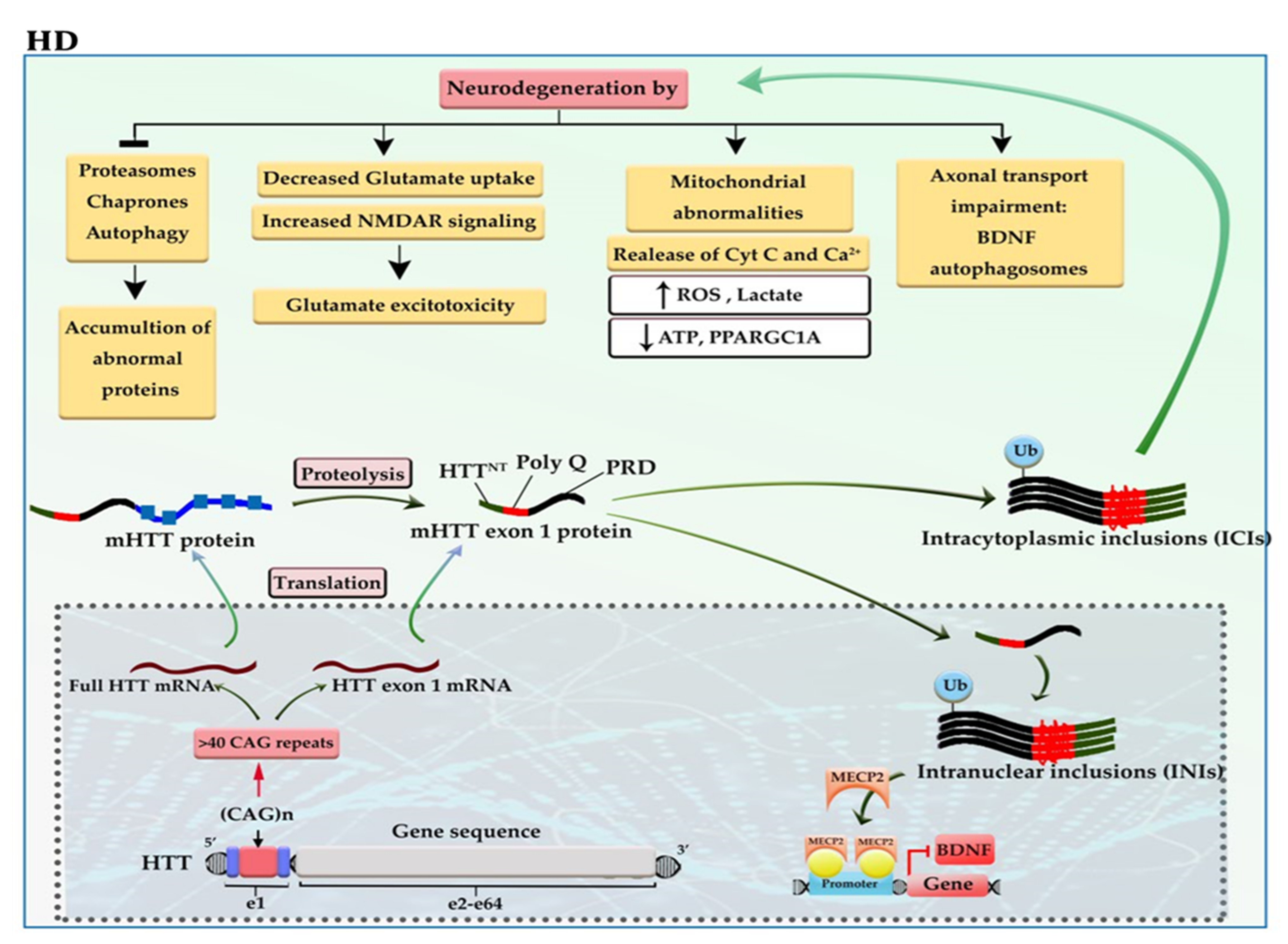
Figure 2. A schematic representation of the selected mechanisms in the pathogenesis of HD. HTT gene, located at 4p16, has 64 exons and a CAG trinucleotide repeat expansion in the exon 1 region. The CAG repeats in the coding sequence of mutant HTT (mHTT) produce mHTT exon 1 protein. mHTT exon 1 protein enters the nucleus. The gradual aggregation of mHTT exon 1 oligomers leads to the formation of large inclusions in the cytoplasm and nucleus of neural cells. The intracytoplasmic inclusions (ICIs) have various toxic effects on neural cells and can exacerbate the aggregation of mHTT exon 1 protein. Whereas the polyQ tract of intranuclear inclusions (INIs) recruits MECP2 to the promoter of BDNF, downregulating the expression of BDNF. BDNF: brain-derived neurotrophic factor; Cyt-c: cytochrome c; mHTT: mutant huntingtin; NMDAR: N-methyl-D-aspartate receptor; ROS: reactive oxygen species.
Upon alteration in methylation patterns such as gain of 5-methylcytosine (5mC), mHTT provides a platform for reducing the expression of genes, which are crucial for neurogenesis, neuronal activity, and survival like SOX2, PAX6, NES, and BDNF [9][10]. Moreover, the 5hmC reduction in the striatum and the cortex of transgenic HD mice can inhibit the progression of some essential signaling pathways for neurogenesis, neuronal function, and survival, including Wnt/β-catenin/SOX, NMDAR/calcium/CREB, and GABA type A receptor axes. Global loss of 5-hmC in the cortex may result from the lower expression of TET1, whereas global loss of 5-hmC in the striatum may result from downregulation of TETs and upregulation of the MECP2 [11]. It seems that DNA methylation abnormalities may also play a role in reducing the expression of adenosine A2a receptor (ADORA2A) protein, which is one of the earliest events in the pathogenesis of HD. The genome-wide methylation analysis reported the accumulation of methyl groups in exon 1 of the ADORA2A gene. OF note, ADORA2A is an essential G-coupled protein for the survival of neural cells in the subcortical basal ganglia [12]. Despite all observations and based on the monogenic nature of the disease, there is a reluctance about accepting the role of DNA methylation in the pathogenesis of HD; however, these conflicts have not affected the early diagnostic value of 5-hmC in HD patients.
3. Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD)
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are genetically and pathologically heterogeneous disorders to which multiple genetic factors contribute. The excessive expansion of hexanucleotide repeats GGGGCC (G4C2) in the first intron of the chromosome 9 open reading frame 72 (C9orf72) gene is the most frequent genetic cause of ALS and FTD [13]. Despite similar origins, the clinical manifestations of ALS and FTD are completely different; while ALS is a disease of motor neuron death [14], FTD is developed as a result of nerve cell loss in the frontal and anterior temporal lobes of the brain [15].
The C9orf72 gene has 11 exons and ≤11 hexanucleotide G4C2 repeats in the vast majority (>95%) of neurologically healthy individuals. This gene produces three transcript variants: Variant 1 encodes the short protein isoform, while Variants 2 and 3 produce the long protein isoform. The presence of pathogenic hexanucleotide G4C2 repeats expansion (pHRE) in intron 1, between non-coding exons 1a and 1b of the gene C9orf72, leads to the development of C9-ALS/FTD by loss-of-function and/or toxic gain-of-function mechanisms. When pHRE is located within the promoter region, it can impede transcription processes, leading to a reduction in C9orf72 protein levels and thereby C9orf72 haploinsufficiency. It has been reported that hypermethylation of G4C2 repeat expansion occurs in about 97% of C9-ALS/FTD patients with >50 repeats, which may explain how it triggers the loss of function of the C9orf72 protein [16]. When pHRE is located within intron 1, the pHRE is bidirectionally transcribed into sense and antisense RNAs containing G4C2 and G2C4 repeats. The pHRE causes the formation of secondary sense and antisense RNA structures and sequestration of RNA binding proteins (RBPs), leading to the accumulation of toxic RNA foci and impairing RNA processing. In the cytoplasm, sense and antisense RNAs undergo repeat-associated non-ATG (RAN) translation, producing potentially toxic dipeptide repeat proteins (DRPs) from the sense transcript (GA, GR, GP) and the antisense transcript (PA, PR, GP) (Figure 3). Both LoF and toxic GoF mechanisms can change the metabolism pathways and RNA processing. LoF mechanism changes the immune system and microglial function. GoF mechanisms impair proteostasis pathways, mitochondrial function, nucleocytoplasmic transport, transport granule function, and vesicular trafficking. GoF mechanisms can also cause nucleolar dysfunction and affect RNA splicing and transcription, resulting in DNA damage [17].
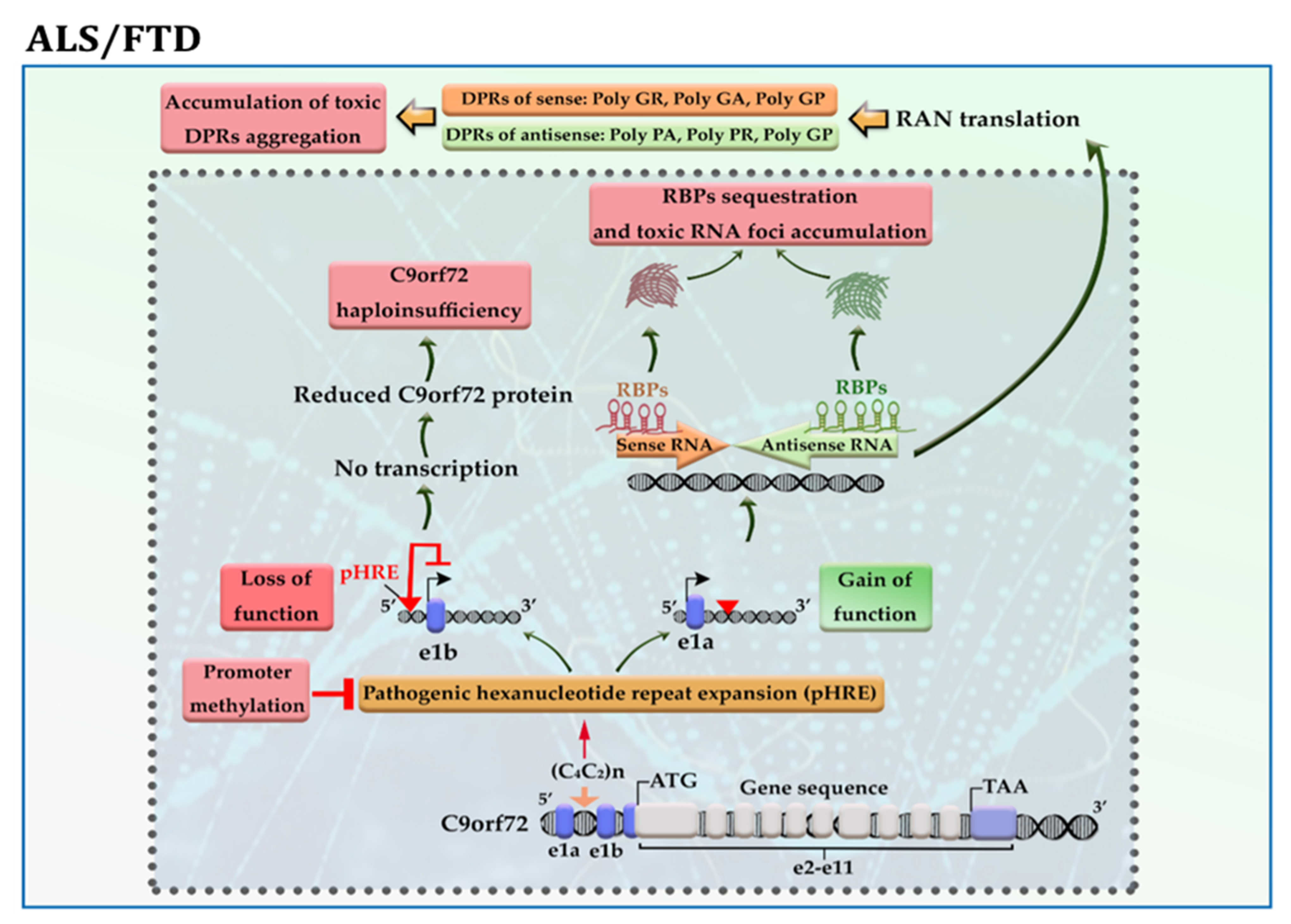
Figure 3. A schematic representation of the selected mechanisms in the pathogenesis of C9-ALS/FTD. Loss of function; the presence of pathogenic hexanucleotide G4C2 repeats expansion (pHRE) within the promoter region inhibits transcription processes and reduces C9orf72 protein levels. Gain of function; the presence of pHRE within intron 1 leads to the sequestration of RNA binding proteins (RBPs) and the accumulation of toxic RNA foci and dipeptide repeat proteins (DRPs). The promoter hypermethylation of the C9orf72 gene reduces the accumulation of RNA foci and/or DRPs aggregation in the neural cells. Amino acid abbreviations: A: alanine, R: arginine, G: glycine, and P: proline.
The promoter hypermethylation and associated silencing of the C9orf72 gene occur in about 30% of C9-ALS/FTD patients with a favorable prognosis. Further investigations revealed that hypermethylation of the C9orf72 promoter protects neural cells from cell death by reducing the accumulation of RNA foci and/or DRPs aggregation in the cells, thereby inhibiting their toxic downstream effects [18]. In addition, hypermethylation of C9orf72 is related to longer survival in patients with C9-FTD [19], while it is related to reduced disease duration before death in patients with C9-ALS [20]. As described above, some phenotypes are likely dependent on the loss of function and others on the gain of function, so hypermethylation could have pleiotropic effects.
Other alterations in DNA methylation reported in both diseases include hypomethylation of the mitochondrial displacement loop (D-loop) region together with increased mitochondrial DNA (mtDNA) copy number in SOD1-mutant and sporadic ALS [21]; and hypermethylation of the GRN gene in FTD [22]. In ALS, overexpression of DNMTs such as DNMT1 and DNMT3A, which participate in the apoptotic death of motor neurons, was observed. Recent evidence also demonstrates an association between overexpression of DNMTs and the propagation of mitochondrial-dependent apoptosis in neural cells of spinal cord lesions in ALS mice models [23]. Furthermore, DNA methylation analysis of ALS patients determined that increased DNA methylation (DNAm) age acceleration was associated with shorter survival and earlier age of onset [24].
4. Alzheimer’s Disease (AD)
Alzheimer’s disease (AD) is the most common form of late-onset neurodegenerative disorder and is characterized by progressive cognitive decline and neuronal death. Evidence demonstrates that mutations in APP and presenilin genes (PSEN1 and PSEN2) are found in patients with early-onset AD, while APOE polymorphism is limited to patients with late-onset AD [25]. In addition to genetic risk factors, analysis of AD brain tissue has taken the veil off the incidence of DNA hypomethylation in low levels of SAM [26]; it not only leads to the reduction of folic acid coupled with an increase in homocysteine in the PB samples of patients [27] but also elevates the expression of genes involved in the amyloid beta (Aβ) pathway, such as APP, PSEN1, and BACE1 in the brain [28]. The evidence of DNA hypomethylation in AD patients has also been confirmed in another study, as the number of 5-mC and 5-hmC reduced more significantly in the brain tissue of AD patients compared to healthy individuals [29]. Apart from DNA hypomethylation, the hypermethylation of some genes may also lead to AD. These genes include ANK1, RPL13, RHBDF2, DUSP22, and SORL1 [30][31][32][33]. As shown in Figure 4, the aberrant expression of these genes contributes to the progression of the Aβ and tau pathways. An imbalance in the production and clearance of Aβ and tau aggregates promotes the extracellular accumulation of amyloid plaques and the intracellular aggregation of neurofibrillary tangles (NFT). The forms of Aβ and tau aggregates have various toxic effects on neural cells. The Aβ aggregates lead to activated glial cells induced-neuroinflammation, synaptic toxicity (long-term potentiation (LTP) impairment and long-term depression (LTD) enhancement), mitochondrial dysregulation, and ion channel dysfunction. In addition, Aβ can activate the kinases involved in the tau pathway, leading to tau hyperphosphorylation [25].
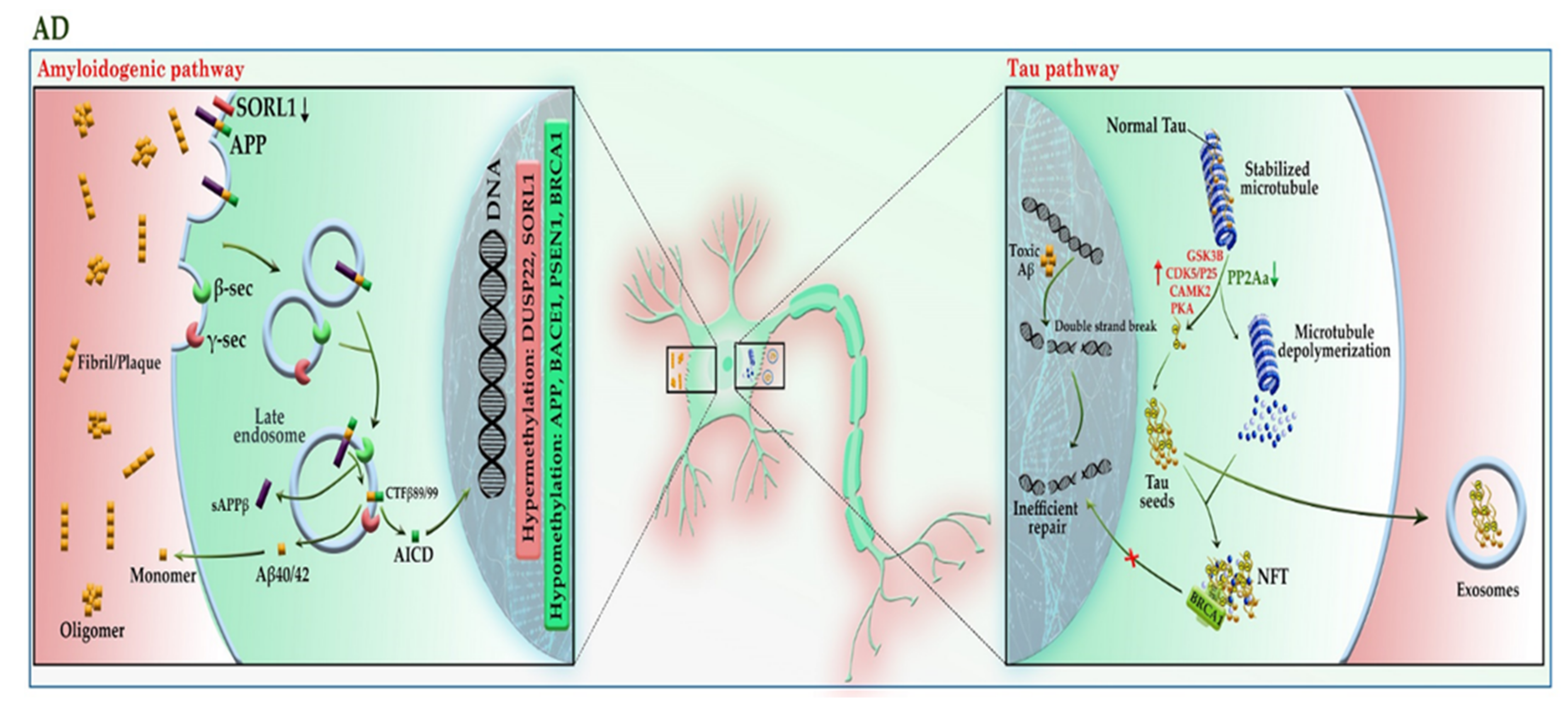
Figure 4. A schematic representation of the selected mechanisms in the pathogenesis of AD. Under physiological conditions, APP is cleaved in the non-amyloidogenic pathway (not shown). In the absence of SORL1 due to epigenetic silencing or mutation, APP is shunted into the late endosomal pathway. In the amyloidogenic pathway, APP enters the late endosome, where it is cleaved by the β-secretase (BACE1), and then by γ-secretase. AICD enters the nucleus and acts as a transcription factor, whereas the Aβ40/42 peptides and sAPPβ are secreted to the extracellular space. An imbalance of Aβ production and its clearance from the brain promotes Aβ aggregation and deposition. The Aβ aggregates can activate the kinases involved in the tau pathway, leading to tau hyperphosphorylation. The aberrant hyperphosphorylation of tau causes p-tau to be separated from microtubules (MTs), leading to MTs depolymerization and axonal degeneration. The disruption of the tau pathway leads to the accumulation of tau aggregates to form oligomers and neurofibrillary tangles (NFTs) within neurons. The p-tau oligomers (tau seeds) can be released into the extracellular space and taken up by unaffected neurons. The tau aggregates sequester BRCA1 protein in the cytoplasm and prevent it from executing its physiological function, leading to the accumulation of DNA damage induced by Aβ. Red and green colors highlighted hypermethylation and hypomethylation, respectively. AICD: amyloid precursor protein (APP) intracellular domain; APP: amyloid precursor protein; BRCA1: breast cancer type 1; CAMK2: calcium/calmodulin-dependent protein kinase II; CDK5: cyclin-dependent kinase 5; CTF-β89/99: β-C-terminal fragment 88/99; DUSP22: dual-specificity phosphatase 22; GSK-3B: glycogen synthase kinase-3B; PKA: protein kinase A; PP2A: protein phosphatase 2; sAAPβ: soluble amyloid precursor protein β; SAM: S-adenosyl methionine; SORL1: sortilin related receptor 1; β-sec: beta-secretase 1; γ-sec: γ-secretase.
Physiologically, tau protein can bind to and stabilize microtubules (MTs). This attachment regulates by the phosphorylation level of tau. The aberrant hyperphosphorylation of tau as a result of hyperactivation of tau kinases or downregulation of tau phosphatases causes phosphorylated-tau (p-tau) to be separated from MTs, leading to MTs depolymerization and axonal degeneration. The hyperactivity of tau kinases may be the result of the effect of Aβ or the epigenetic silencing of their inhibitors, such as DUSP22, which inhibits PKA. In addition, PP2A, a major tau phosphatase, is activated by the addition of a methyl (CH3) group. Under diminished SAM in the brain cells, PP2A inactivation leads to tau hyperphosphorylation. Hyperphosphorylated tau can accumulate to form oligomers, β-sheet-containing structure paired helical filaments (PHFs), and ultimately NFT inside neurons. Apart from axonal degeneration, tau aggregates may impair kinesin-dependent transport, mitochondrial function, and pre- and postsynaptic functions, leading to neuronal cell death. In addition, p-tau oligomers (tau seeds) can be released into the extracellular space via exosomes or directly from the plasma membrane, taken up by unaffected neurons, and tau pathology gradually engages more brain regions as the disease progresses.
Aβ and tau interact in many neuronal compartments, especially at the level of mitochondrial function, leading to impairment of oxidative phosphorylation, mitochondrial transportation, mitophagy, and mitochondrial fission and fusion [25]). In addition to the toxic effects mentioned above, tau aggregates can sequester BRCA1 protein in the cytoplasm and prevent it from executing its physiological function, leading to the accumulation of DNA damage induced by Aβ. It seems that neurons try to cope with this condition by upregulating the expression of the BRCA1 gene through demethylation of the promoter region [34].
Moreover, it has been shown that identifying methylation quantitative trait locus (mQTL) in the approximate PM20D1 gene is associated with AD development [35]. Recently, many efforts are being accomplished to use methylation patterns as a reliable marker for predicting disease prognosis. For example, greater methylation levels at specific CpG sites of the BACE1 gene promoter were associated with lower Aβ load and higher tangle density in AD patients with dementia compared to subjects without cognitive impairment (NCI) or mild cognitive impairment (MCI) [28]. PIN1 hypermethylation can serve as a useful predictive biomarker to distinguish frontotemporal dementia from AD [36]. Moreover, the levels of DNA methylation in the critical genes involved in AD pathogenesis, such as APP, BACE1, LRP1, and SORL1 may be considered in obese individuals as a sign of AD development in the following years (NCT02868905). These findings together indicate that DNA methylation plays an essential role in AD development even before the onset of the disease.
5. Parkinson’s Disease (PD)
Parkinson’s disease, the second most common neurodegenerative disorder, is characterized by a widespread intracellular accumulation of the α-synuclein proteins, accompanied by a loss of dopaminergic neurons, both of which occur in the substantia nigra pars compacta of the midbrain. The proper regulation of the α-synuclein (SNCA) is critical for neural health because its higher levels can accumulate to form oligomers, protofibrils, insoluble fibrils, and ultimately “Lewy bodies” in the cytoplasm of neurons, triggering apoptosis within the cells [37]. The existence of mutations or the hypomethylation of CpG islands in intron 1 of the SNCA gene enforces the expression of this protein in PD patients [38][39]. Interestingly, α-synuclein can relocate DNMT1 from the nucleus into the cytoplasm of neural cells and depletes the nuclear reservoir of this DNMT, thereby leading to the hypomethylation and associated with the upregulation of many PD-related genes, including SNCA and CYP2E1 (Figure 5).
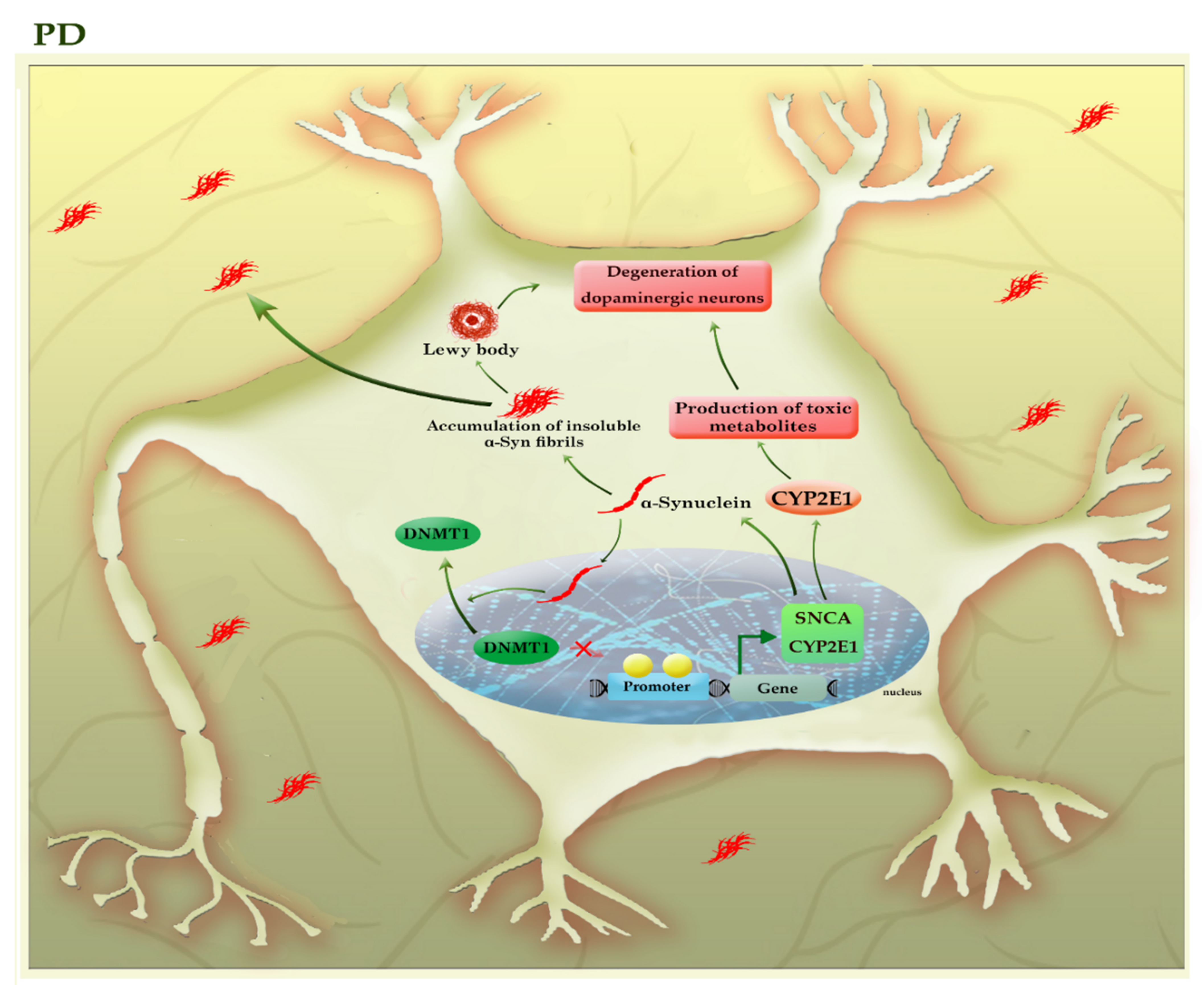
Figure 5. A schematic representation of the selected mechanisms in the pathogenesis of PD. The higher levels of α-synuclein (SNCA) can accumulate to form oligomers, insoluble fibrils, and ultimately “Lewy bodies”, triggering apoptosis within the neural cells. α-synuclein can relocate DNMT1 from the nucleus into the cytoplasm of neural cells and depletes the nuclear reservoir of this DNMT, thereby leading to the hypomethylation and associated with the upregulation of many PD-related genes, including SNCA and CYP2E1. The increased activity of the CYP2E1 gene may contribute to the degeneration of dopaminergic neurons by the formation of toxic metabolites. The light green colors highlighted hypomethylated genes. CYP2E1: cytochrome P450 2E1; SNCA (α-Syn): synuclein alpha.
The impact of DNMT1 on the SNCA gene is reversible, as DNMT1 re-expression in the nucleus could partially diminish the expression of SNCA in the neural cells of transgenic mouse brains [40]. On the other hand, α-synuclein aggregates can be released into the extracellular space and taken up by unaffected neurons, so α-synuclein pathology gradually engages more brain regions as the disease progresses. Toxic effects of various forms of α-synuclein aggregates include activated microglia induced-neuroinflammation and mitochondrial abnormalities, as well as inhibition of the lysosomal autophagy system (LAS) and ubiquitin-proteasome system, exacerbating the aggregation of α-synuclein [37]. Since the disease is silent until after a significant neuronal loss occurs, so identification of early diagnostic biomarkers would be essential to timely diagnosis and treatment. Given the importance of DNA methylation in SCNA regulation and based on the fact that epigenetic alterations may occur before the onset of the disease, it has been suggested that the analysis of CpG methylation of the SNCA gene in blood samples can use as an early diagnostic method for PD [38].
In addition, promoter hypomethylation and increased activity of the CYP2E1 gene may contribute to the degeneration of dopaminergic neurons by the formation of toxic metabolites [41]. In addition, it seems that levodopa owes its success in treating PD to its impact on regulating the DNA methylation process [42]. Thus, DNA methylation plays an essential role in the pathogenesis and progression of PD. Table 1 summarizes the results of various studies that highlighted the role of DNA methylation in neurodegenerative diseases.
Table 1. Potential DNA methylation markers in neurodegenerative diseases.
| Cell/Tissue Type | Main Findings | Ref. | |
|---|---|---|---|
| Neurodegenerative Diseases | |||
| HD | Brain tissues from HD patients and HCs | Increased 5mC, together with reduced 5hmC levels, were detected in the 5’UTR region of the ADORA2A gene in the putamen of HD patients compared to HCs. | [12] |
| ALS | Postmortem brain and spinal cord samples from sporadic ALS and HCs | DNMT1 and DNMT3A were upregulated in the motor cortex and spinal cord motor neurons of patients with sporadic ALS compared to HCs. 5-mC was detected in the motor cortex of ALS but not in HCs. | [23] |
| Postmortem frozen spinal cord samples and WB from sporadic ALS and HCs | Global 5-mC and 5-hmC were increased in the spinal cord, but not WB of patients with sporadic ALS compared to HCs. | [43] | |
| DNA blood from patients with ALS and HCs | C9orf72 promoter hypermethylation was associated with reduced disease duration before death in patients with C9-ALS. | [20] | |
| PBMCs from sporadic ALS patients, HCs, and familial ALS patients with SOD1- or C9orf72-mutant | The hypomethylation of the mitochondrial D-loop region, together with increased mtDNA copy number, could represent compensatory mechanisms to counteract mitochondrial impairment in SOD1-mutant and sporadic ALS patients. | [21] | |
| Blood and CNS tissues from sporadic ALS patients | Blood/CNS-based DNAm-age acceleration may be used as a marker to predict the age of onset and survival in ALS patients. | [24] | |
| ALS, FTD | Brain or blood samples from C9-ALS/FTD patients and HCs | The hypermethylation of G4C2 repeat expansion occurs in about 97% of C9-ALS/FTD patients with >50 repeats. It was found in both blood and brain tissues for the same individual, suggesting its potential use as a biomarker. | [16] |
| FTD | Brain or blood samples from C9-FTD patients and non-carrier family members | C9orf72 promoter hypermethylation was associated with longer survival in patients with C9-FTD. | [19] |
| Brain samples from HCs, FTD, AD, and PD patients | Promoter hypermethylation and associated silencing of the GRN gene were detected in patients with FTD compared to HCs, AD, and PD samples. | [22] | |
| PBMCs from FTD patients and HCs | Promoter hypermethylation associated with a reduced mRNA expression of the GRN gene was found in PB of patients with FTD compared to HCs. | [44] | |
| AD | Postmortem human brains from AD patients, donors with NCI and MCI | The greater methylation levels at specific CpG sites of the BACE1 gene promoter were associated with higher tangle density and lower β-amyloid load among persons with AD dementia than subjects with NCI or MCI. | [28] |
| Neurons of postmortem brain samples from AD patients and HCs | Promoter hypomethylation and increased mRNA and protein expression of BRCA1 was detected in the neurons of hippocampal and entorhinal cortex from AD patients compared to HCs. | [34] | |
| PFC neurons of Postmortem human brains from AD patients and HCs | Hypomethylation of the enhancers in the DSCAML1 gene, which targets the BACE1 promoter, caused the overexpression of BACE1 in AD patients; and was correlated with an increase in Aβ plaques, NFTs, and cognitive decline. | [45] | |
| PBMCs from AD patients, FTD donors, and HCs | PIN1 hypermethylation can serve as a useful predictive biomarker to distinguish AD from FTD. | [36] | |
| PBMCs from HCs, young obese females, or AD donors | Methylation levels of genes involved in AD pathogenesis, such as APP, BACE1, LRP1, and SORL1, can serve as prognostic biomarkers in obese individuals. | NCT02868905 | |
| PD | PBMCs from sporadic PD patients and HCs | The methylation status of SNCA intron-1 can be used as an early diagnostic marker for PD. | [38] |
| Postmortem human brains from PD patients and HCs | Promoter hypomethylation and the increased activity of CYP2E1 may contribute to the degeneration of dopaminergic neurons by the formation of toxic metabolites. | [41] | |
AD: Alzheimer’s disease; Aβ: amyloid β; ALS: amyotrophic lateral sclerosis; APP: amyloid beta precursor protein; ADORA2A: adenosine A2a receptor; BACE1: beta-site amyloid precursor protein cleaving enzyme 1; BRCA1: breast cancer type 1 susceptibility protein; CB: cerebellum; C9-ALS: C9orf72-associated ALS; CNS: central nervous system; C9-ALS/FTD: C9orf72-associated ALS/FTD; C9-FTD: C9orf72-associated FTD; CYP2E1: cytochrome P450 family 2 subfamily E member 1; C9orf72: chromosome 9 open reading frame 72; DNMT: DNA (cytosine-5)-methyltransferase; D-loop: displacement loop; DNAm: DNA methylation; DSCAML1: Down syndrome cell adhesion molecules like 1; FTD: frontotemporal dementia; GRN: granulin precursor; HD: Huntington’s disease; HCs: healthy controls; 5-hmC: 5-hydroxymethylcytosine; LRP1: low-density lipoprotein receptor-related protein 1; mtDNA: mitochondrial DNA; MCI: mild cognitive impairment; 5-mC: 5-methylcytosine; NFTs: neurofibrillary tangles; NCI: no cognitive impairment; PD: Parkinson’s disease; PFC: prefrontal cortex; PB: peripheral blood; PBMNs: peripheral blood mononuclear cells; PIN1: peptidyl-prolyl cis-trans isomerase NIMA-interacting 1; SOD1: superoxide dismutase 1; SORL1: sortilin related receptor 1; SNCA: synuclein alpha; WB: whole blood.
References
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914.
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065.
- De Esch, C.E.; Ghazvini, M.; Loos, F.; Schelling-Kazaryan, N.; Widagdo, W.; Munshi, S.T.; van der Wal, E.; Douben, H.; Gunhanlar, N.; Kushner, S.; et al. Epigenetic Characterization of the FMR1 Promoter in Induced Pluripotent Stem Cells from Human Fibroblasts Carrying an Unmethylated Full Mutation. Stem Cell Rep. 2014, 3, 548–555.
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992.e6.
- Caron, N.S.; Dorsey, E.R.; Hayden, M. Therapeutic approaches to Huntington disease: From the bench to the clinic. Nat. Rev. Drug Discov. 2018, 17, 729–750.
- Yang, S.; Yang, H.; Huang, L.; Chen, L.; Qin, Z.; Li, S.; Li, X.-J. Lack of RAN-mediated toxicity in Huntington’s disease knock-in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 4411–4417.
- Moumné, L.; Betuing, S.; Caboche, J. Multiple Aspects of Gene Dysregulation in Huntington’s Disease. Front. Neurol. 2013, 4, 127.
- McFarland, K.N.; Huizenga, M.N.; Darnell, S.B.; Sangrey, G.R.; Berezovska, O.; Cha, J.-H.J.; Outeiro, T.F.; Sadri-Vakili, G. MeCP2: A novel Huntingtin interactor. Hum. Mol. Genet. 2013, 23, 1036–1044.
- Ng, C.W.; Yildirim, F.; Yap, Y.S.; Dalin, S.; Matthews, B.J.; Velez, P.J.; Labadorf, A.; Housman, D.E.; Fraenkel, E. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc. Natl. Acad. Sci. USA 2013, 110, 2354–2359.
- Pan, Y.; Daito, T.; Sasaki, Y.; Chung, Y.H.; Xing, X.; Pondugula, S.; Swamidass, S.J.; Wang, T.; Kim, A.H.; Yano, H. Inhibition of DNA Methyltransferases Blocks Mutant Huntingtin-Induced Neurotoxicity. Sci. Rep. 2016, 6, 31022.
- Wang, F.; Yang, Y.; Lin, X.; Wang, J.-Q.; Wu, Y.-S.; Xie, W.; Wang, D.; Zhu, S.; Liao, Y.-Q.; Sun, Q.; et al. Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington’s disease. Hum. Mol. Genet. 2013, 22, 3641–3653.
- Villar-Menéndez, I.; Blanch, M.; Tyebji, S.; Pereira-Veiga, T.; Albasanz, J.L.; Martín, M.; Ferrer, I.; Perez-Navarro, E.; Barrachina, M. Increased 5-Methylcytosine and Decreased 5-Hydroxymethylcytosine Levels are Associated with Reduced Striatal A2AR Levels in Huntington’s Disease. NeuroMolecular Med. 2013, 15, 295–309.
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256.
- Hardiman, O.; van den Berg, L.H.; Kiernan, M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 639–649.
- Bang, J.; Spina, S.; Miller, B.L. Frontotemporal dementia. Lancet 2015, 386, 1672–1682.
- Xi, Z.; Zhang, M.; Bruni, A.; Maletta, R.G.; Colao, R.; Fratta, P.; Polke, J.M.; Sweeney, M.G.; Mudanohwo, E.; Nacmias, B.; et al. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol. 2015, 129, 715–727.
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558.
- Liu, E.Y.; Russ, J.; Wu, K.; Neal, D.; Suh, E.; McNally, A.G.; Irwin, D.; Van Deerlin, V.M.; Lee, E.B. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol. 2014, 128, 525–541.
- Russ, J.; Liu, E.Y.; Wu, K.; Neal, D.; Suh, E.; Irwin, D.; McMillan, C.; Harms, M.B.; Cairns, N.J.; Wood, E.M.; et al. Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathol. 2014, 129, 39–52.
- Xi, Z.; Zinman, L.; Moreno, D.; Schymick, J.; Liang, Y.; Sato, C.; Zheng, Y.; Ghani, M.; Dib, S.; Keith, J.; et al. Hypermethylation of the CpG Island Near the G4C2 Repeat in ALS with a C9orf72 Expansion. Am. J. Hum. Genet. 2013, 92, 981–989.
- Stoccoro, A.; Smith, A.R.; Mosca, L.; Marocchi, A.; Gerardi, F.; Lunetta, C.; Cereda, C.; Gagliardi, S.; Lunnon, K.; Migliore, L.; et al. Reduced mitochondrial D-loop methylation levels in sporadic amyotrophic lateral sclerosis. Clin. Epigenetics 2020, 12, 137.
- Banzhaf-Strathmann, J.; Claus, R.; Mücke, O.; Rentzsch, K.; Van Der Zee, J.; Engelborghs, S.; De Deyn, P.P.; Cruts, M.; Van Broeckhoven, C.; Plass, C.; et al. Promoter DNA methylation regulates progranulin expression and is altered in FTLD. Acta Neuropathol. Commun. 2013, 1, 16.
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic Regulation of Motor Neuron Cell Death through DNA Methylation. J. Neurosci. 2011, 31, 16619–16636.
- Zhang, M.; McKeever, P.M.; Xi, Z.; Moreno, D.; Sato, C.; Bergsma, T.; McGoldrick, P.; Keith, J.; Robertson, J.; Zinman, L.; et al. DNA methylation age acceleration is associated with ALS age of onset and survival. Acta Neuropathol. 2020, 139, 943–946.
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40.
- Morrison, L.D.; Smith, D.D.; Kish, S.J. Brain S-Adenosylmethionine Levels Are Severely Decreased in Alzheimer’s Disease. J. Neurochem. 2002, 67, 1328–1331.
- Coppedè, F.; Tannorella, P.; Pezzini, I.; Migheli, F.; Ricci, G.; Ienco, E.C.; Piaceri, I.; Polini, A.; Nacmias, B.; Monzani, F.; et al. Folate, Homocysteine, Vitamin B12, and Polymorphisms of Genes Participating in One-Carbon Metabolism in Late-Onset Alzheimer’s Disease Patients and Healthy Controls. Antioxidants Redox Signal. 2012, 17, 195–204.
- Carmo, S.D.; Hanzel, C.E.; Jacobs, M.L.; Machnes, Z.; Iulita, M.F.; Yang, J.; Yu, L.; Ducatenzeiler, A.; Danik, M.; Breuillaud, L.S.; et al. Rescue of Early bace-1 and Global DNA Demethylation by S-Adenosylmethionine Reduces Amyloid Pathology and Improves Cognition in an Alzheimer’s Model. Sci. Rep. 2016, 6, 34051.
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.; Coleman, P.D.; Rutten, B.P.; Hove, D.L.V.D. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099.
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163.
- Smith, R.G.; Pishva, E.; Shireby, G.; Smith, A.R.; Roubroeks, J.A.Y.; Hannon, E.; Wheildon, G.; Mastroeni, D.; Gasparoni, G.; Riemenschneider, M.; et al. A meta-analysis of epigenome-wide association studies in Alzheimer’s disease highlights novel differentially methylated loci across cortex. Nat. Commun. 2021, 12, 3517.
- Sanchez-Mut, J.V.; Aso, E.; Heyn, H.; Matsuda, T.; Bock, C.; Ferrer, I.; Esteller, M. Promoter hypermethylation of the phosphatase DUSP22 mediates PKA-dependent TAU phosphorylation and CREB activation in Alzheimer’s disease. Hippocampus 2014, 24, 363–368.
- Yu, L.; Chibnik, L.B.; Srivastava, G.P.; Pochet, N.; Yang, J.; Xu, J.; Kozubek, J.; Obholzer, N.; Leurgans, S.E.; Schneider, J.A.; et al. Association of Brain DNA Methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with Pathological Diagnosis of Alzheimer Disease. JAMA Neurol. 2015, 72, 15–24.
- Mano, T.; Nagata, K.; Nonaka, T.; Tarutani, A.; Imamura, T.; Hashimoto, T.; Bannai, T.; Koshi-Mano, K.; Tsuchida, T.; Ohtomo, R.; et al. Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E9645–E9654.
- Sanchez-Mut, J.V.; Heyn, H.; Silva, B.A.; Dixsaut, L.; Esparcia, P.G.; Vidal, E.; Sayols, S.; Glauser, L.; Monteagudo-Sánchez, A.; Perez-Tur, J.; et al. PM20D1 is a quantitative trait locus associated with Alzheimer’s disease. Nat. Med. 2018, 24, 598–603.
- Ferri, E.; Arosio, B.; D’Addario, C.; Galimberti, D.; Gussago, C.; Pucci, M.; Casati, M.; Fenoglio, C.; Abbate, C.; Rossi, P.D.; et al. Gene promoter methylation and expression of Pin1 differ between patients with frontotemporal dementia and Alzheimer’s disease. J. Neurol. Sci. 2016, 362, 283–286.
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013.
- Ai, S.-X.; Xu, Q.; Hu, Y.-C.; Song, C.-Y.; Guo, J.-F.; Shen, L.; Wang, C.-R.; Yu, R.-L.; Yan, X.-X.; Tang, B.-S. Hypomethylation of SNCA in blood of patients with sporadic Parkinson’s disease. J. Neurol. Sci. 2013, 337, 123–128.
- Moran, S.; Martínez-Cardús, A.; Sayols, S.; Musulén, E.; Balañá, C.; Estival-Gonzalez, A.; Moutinho, C.; Heyn, H.; Diaz-Lagares, A.; de Moura, M.C.; et al. Epigenetic profiling to classify cancer of unknown primary: A multicentre, retrospective analysis. Lancet Oncol. 2016, 17, 1386–1395.
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. α-Synuclein Sequesters Dnmt1 from the Nucleus. J. Biol. Chem. 2011, 286, 9031–9037.
- Kaut, O.; Schmitt, I.; Wüllner, U. Genome-scale methylation analysis of Parkinson’s disease patients’ brains reveals DNA hypomethylation and increased mRNA expression of cytochrome P450 2E1. Neurogenetics 2012, 13, 87–91.
- Schmitt, I.; Kaut, O.; Khazneh, H.; Deboni, L.; Ahmad, A.; Berg, D.; Klein, C.; Fröhlich, H.; Wüllner, U. L-dopa increases α-synuclein DNA methylation in Parkinson’s disease patients in vivo and in vitro. Mov. Disord. 2015, 30, 1794–1801.
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of Epigenetically Altered Genes in Sporadic Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e52672.
- Galimberti, D.; D’Addario, C.; Dell’Osso, B.; Fenoglio, C.; Marcone, A.; Cerami, C.; Cappa, S.; Palazzo, M.C.; Arosio, B.; Mari, D.; et al. Progranulin gene (GRN) promoter methylation is increased in patients with sporadic frontotemporal lobar degeneration. Neurol. Sci. 2012, 34, 899–903.
- Li, P.; Marshall, L.; Oh, G.; Jakubowski, J.L.; Groot, D.; He, Y.; Wang, T.; Petronis, A.; Labrie, V. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 2019, 10, 2246.
More
Information
Subjects:
Genetics & Heredity
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
945
Revisions:
2 times
(View History)
Update Date:
15 Nov 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No