 +1 credit
+1 credit
Video Upload Options
Ginseng is one of the most widely consumed herbs in the world and plays an important role in counteracting fatigue and alleviating stress. The main active substances of ginseng are its ginsenosides. Ocotillol-type triterpenoid is a remarkably effective ginsenoside from Vietnamese ginseng that has received attention because of its potential antibacterial, anticancer and anti-inflammatory properties, among others. The semisynthesis, modification and biological activities of ocotillol-type compounds have been extensively studied in recent years.
1. Introduction
From the 1940s to the middle of 2019, approximately 33.5% of approved drugs were either natural products or directly derived from them [1]. The development of new drug entities based on natural products as sources of novel structures is still an area of active research. Ginseng, including Asian ginseng (Panax vietnamensis HA et GRUSHV.) and American ginseng (Panax quinquefolium L.), is one of the most widely consumed herbs in the world and plays an important role in counteracting fatigue and alleviating stress [2][3]. Ginseng contains a variety of active ingredients, but its main active substances are attributed to its ginsenosides. The ginsenosides with the highest content in Vietnamese ginseng are protopanaxadiol, protopanaxatriol, oleanolic acid and 20,24-epoxydammarane (ocotillol) (Figure 1) [4][5].
Among the active components of ginseng, ocotillol-type compounds have received increasing attention because of their antibacterial, anticancer and anti-inflammatory properties [6][7]. Their different pharmacologic effects and potential molecular mechanisms have been gradually elucidated. Compared with the structure of dammarane ginsenosides (including the protopanaxadiol and protopanaxatriol types), ocotillol-type saponins are tetracyclic triterpenoid saponins containing a furan ring linked with aglycones.
Ocotillol-type saponins were first isolated from Fozrqwieria splendens Eliselm. in 1965 by Warnhoff et al. They were also found in Panax quinquefolium L, Panax vietnamensis HA et GRUSHV, and Panax japonicus var, to name a few [8][9][10][11][12][13][14][15][16][17]. However, because of the low content of ocotillol-type saponins in natural products, there were few studies on ocotillol-type derivatives in previous years [18][19] Fueled by the growing use of semisynthetic methods for the preparation of ocotillol-type derivatives, increased research of ocotillol-type derivatives has been recently observed. In 2016, Liu et al. published a review that focused on the discovery, semisynthesis, biological activities and metabolism of ocotillol-type saponins [6]. However, the structure of most derivatives and its structure–activity relationship (SAR) were not mentioned in the article. Compared with the previous review, this review summarized the semisynthesis, modification and pharmacological activities of ocotillol-type derivatives. All the structures of ocotillol-type derivatives and their SARs in antibacterial, anti-inflammatory and tumor multidrug resistance reversal were summarized. This review provides useful information for the development of ocotillol-type derivatives and gives a direction for further inspiration to enrich its structures with good pharmacological activities.
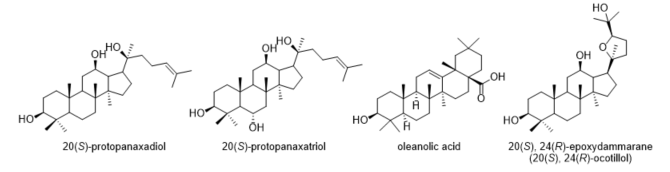
Figure 1. Ginsenosides are found in the highest abundance in Vietnamese ginseng.
2. Semisynthesis of Ocotillol-Type Compounds
Ocotillol-type sapogenins are less abundant in natural sources. Vietnamese ginseng contains higher amounts of ginseng saponins compared with other Panax genus species. The content of ocotillol-type saponins in Panax Vietnamese ginsengs is only 5.6%, while in Panax quinquefolius, it is less than 0.01% [20]. Additionally, 1 kg of fresh rhizome low-quality Vietnamese ginseng is about $1000 in 2019. These factors may have led to the slow development of ocotillol-type ginsenosides in previous years.
In 2005, 20(S)-protopanaxadiol (20(S)-PPD) was used as a raw material to obtain 4 and 5 by a semisynthetic method [21]. Yang et al. optimized and improved the synthetic process and achieved the industrial production of 4 and 5 [22].
Ocotillol-type sapogenins have been made using similar synthetic methods. 20(S)-PPD was used as the raw starting material and reacted with acetic anhydride, and then acetylated 20(S)-PPD was oxidized by m-CPBA. The molar ratio of the acetylated 20(S)-PPD to m-CPBA at −3 ℃ is approximately 1:4, 3 h. The ocotillol-type epimers (4, 5) were obtained by the hydrolysis of the oxidation products. The synthetic route is shown in Figure 2A [23].
After further research by Meng et al., the synthesis mechanism of ocotillol-type epimers was proposed as follows (Figure 2B). 20(S)-PPD or 20(R)-PPD is oxidized by m-CPBA to generate the 24,25-epoxy intermediates, and then an intramolecular ring-opening loop reaction is carried out according to Baldwin’s rule, and finally cyclization by a 5-exo-tet method forms a tetrahydrofuran ring [24][25][26].
Further research proved that the epimerization of C-24 could also result in remarkable differences in both the molecular conformation and the crystal packing arrangements. These remarkable differences may lead to diversity in both polarity and activity of the ocotillol-type epimers. The 24(S)-epimer (5) had two intramolecular hydrogen bonds, while the 24(R)-epimer (4) had one intramolecular hydrogen bond (Figure 3A,B) [27][28]. Crystal stacking showed that both the 20(S),24(R)-ocotillol and 20(S),24(S)-ocotillol generated an H-bonded tube, the 24(R)-epimer (4) generated a left-handed chiral channel, while the 24(S)-epimer (5) extended into the two-dimensional network with right-handed and left-handed chiral channels (Figure 3C–E) [29]. Additionally, the 24(R)-epimer (4) had weaker molecular polarity compared with the 24(S)-epimer (5). These differences in hydrogen bonding may contribute to the differences in the observed biological activity and molecular polarity of the 24(R)-epimer (4) compared with the 24(S)-epimer (5).
Ocotillol-type ginsenosides are rarely found in nature. Less than 20 naturally occurring ocotillol-type ginsenosides have been characterized and reported. The use of chemical methods to synthesize new ocotillol-type ginsenosides is a promising approach to generate structural diversity. Atopkina et al. reported the synthesis of ocotillol-type ginsenosides by coupling the acceptor 4 with α-acetobromoglucose and orthoester donors in the presence of mercury salts (Figure 2C) [30][31]. In 2016, Shen et al. used a gold-catalyzed glycosylation scheme to synthesize ocotillol-type ginsenosides under neutral conditions (Figure 2C) [32]. Many ocotillol-type ginsenosides can be synthesized by this method, and further investigations of ocotillol-type ginsenosides should be pursued.
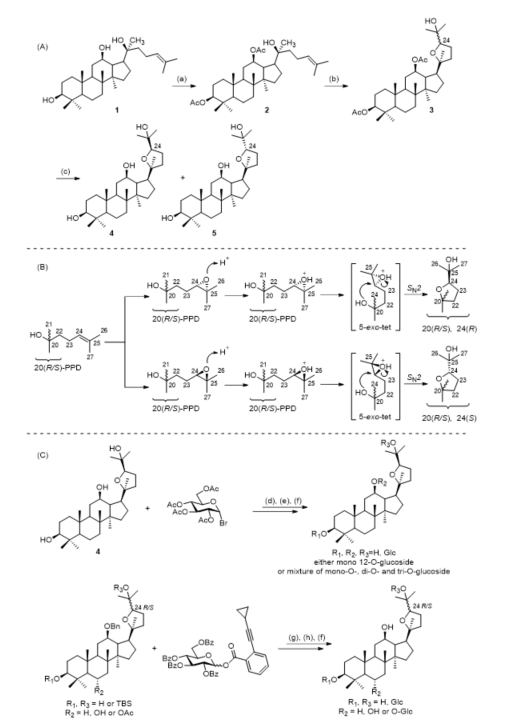
Figure 2. (A) Semisynthetic route for the preparation of ocotillol-type epimers. (B) Synthetic mechanism of ocotillol-type epimers. (C) Synthesis of ocotillol-type ginsenosides. Ac = acetyl; Bn = benzyl; Glc = β-d-glucopyranosyl; Bz = benzoyl. Reagents and conditions: (a) (CH3CO)2O, DMAP, pyridine, r.t.; (b) m-CPBA, CH2Cl2, r.t.; (c) NaOH, CH3OH, H2O, 65 °C; (d) alcohol, mercury cyanide, nitromethane, 90 °C, 1 h; (e) alcohol, α-acetobromoglucose, r.t.; (f) KOH/CH3OH, THF, r.t.; (g) Ph3PAuNTf2, CH2Cl2, r.t.; (h) H2, Pd(OH)2/C, CH3OH, r.t.
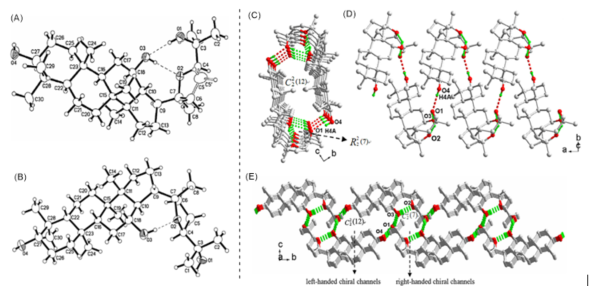
Figure 3. (A) The Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations (ORTEP) figure of 20(S),24(R)-ocotillol-type saponin (4). (B) The ORTEP figure of 20(S),24(S)-ocotillol-type saponin (5). Thermal ellipsoids shown at 30% probability. (C) and (D) view of the H-bonded 1D left-handed chiral channel in 20(S),24(R)-ocotillol-type saponin (4). (E) The 2D net with right-handed and left-handed chiral channels in 20(S),24(S)-ocotillol-type saponin (5).
3. Pharmacological Activities and Chemistry
3.1. Antibacterial Effects
Evidence has shown that ginseng has antibacterial properties, and its extract may be effective for treating bacterial infections in the future [33]. Compound 5 had strong antibacterial activities against Staphylococcus aureus (S. aureus) and Bacillus subtilis (B. subtilis) with minimum inhibitory concentration (MIC) values of 8 μg/mL [34]. Further research showed that 5 also had strong synergistic inhibition against community-associated methicillin-resistant S. aureus (MRSA; strain USA300), as 5 reduced the MIC of kanamycin (KAN) against MRSA USA300 from 1 μg/mL to 0.125 μg/mL giving a fractional inhibitory concentration index (FICI) of 0.14.
The furan ring, C-3 and C-12 are possible to explore in terms of chemical diversity as a modification of the furan ring, C-3, and C-12 significantly changed the antibacterial activity of ocotillol-type derivatives. Aromatic-substituted ocotillol-type derivatives 6–17 were synthesized by an esterification reaction, and their in vitro activity against Escherichia coli (E. coli), B. subtilis, S. aureus, Pseudomonas aeruginosa (P. aeruginosa) and Acinetobacter baumannii (A. baumannii) was determined (Figure 4) [35]. Compounds 6 and 7 exhibited excellent antibacterial activities with MIC values of 1 μg/mL against S. aureus and B. subtilis, while compounds 9, 10, 12 and 16 exhibited moderate antibacterial activities against S. aureus. Further research showed that 6 and 7 displayed good antibacterial activities against MRSA USA300 with MIC values of 4 μg/mL. Additionally, 6 and 7 combined with KAN and chloramphenicol had strong synergistic inhibition against MRSA USA300 and reduced the MICs of KAN against MRSA USA300 from 1 μg/mL to 0.0156 and 0.0625 μg/mL (FICI = 0.078 and 0.020, respectively).
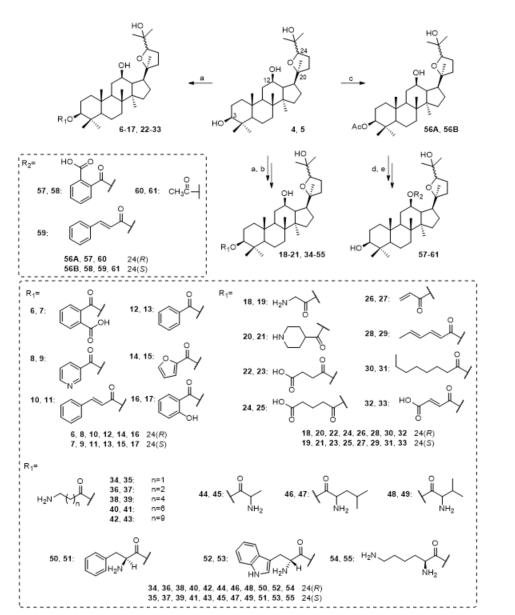
Figure 4. Synthesis of ocotillol-type derivatives 6–61. Reagents and conditions: (a) anhydrous CH2Cl2, anhydride or acids or Boc-amino acid, 1-ethyl-3(3-dimethylpropylamine) carbodiimide (EDCI), 4-dimethylaminopyridine (DMAP), r.t.; (b) trifluoroacetic acid (TFA), CH2Cl2, r.t.; (c) anhydrous pyridine, Ac2O, DMAP, r.t.; (d) anhydrous pyridine, anhydride or acid chloride, DMAP, ref.; (e) CH3OH, KOH, ref.
Bi et al. synthesized aliphatic ocotillol-type derivatives 18–33 (Figure 4) [36][37][38]. Compounds 18, 20–23, 25 and 30 showed good antibacterial activities against S. aureus and B. subtilis. Further screening results showed that 5, 18 and 19 had similar antibacterial activities against MRSA USA300 with MIC values of 8 μg/mL. Most ocotillol-type derivatives with an amino group at C-3 displayed excellent antibacterial activities, while those with a carboxylic group at C-3 showed moderate activities. A synergistic effect was observed for compound 19 as it reduced the MIC of KAN against MRSA USA300 from 1 µg/mL to 0.25 µg/mL with a FICI of 0.28.
A series of ocotillol-type derivatives 34–55 with an amino group was also synthesized (Figure 4) [39][40]. The antibacterial activity results showed that most of the ocotillol-type derivatives with an amino group had moderate to good inhibitory activities against Gram-positive bacteria but had no effect on Gram-negative bacteria. Compounds 38, 40 and 51 had good inhibitory activities against MRSA USA 300 with MICs ≤ 4 µg/mL, while 51 had the same antibacterial activity as KAN. A synergistic effect was observed for 39 when it was combined with KAN as shown by the significant enhancement of the MIC from 4 µg/mL to 0.25 µg/mL (FICI < 0.0088) against MRSA USA300.
A series of derivatives 57–61 were synthesized and screened. Among them, compound 58 had the best antibacterial activity against MRSA USA300 with a MIC of 8 μg/mL, and 60 had a moderate inhibitory effect against both Gram-positive and Gram-negative bacteria (Figure 4). Additionally, 58 combined with KAN had strong synergistic inhibition against MRSA USA300 with a FICI of 0.008.
The synthetic approaches to prepare compounds 6–56B are only slightly different. Compound 6 was synthesized by the treatment of compound 4, DMAP and phthalic anhydride in dry dichloromethane over 6 h to obtain 6 with 73% yield at room temperature. 1-ethyl-3(3-dimethylpropylamine) carbodiimide (EDCI) is an excellent dehydrating agent that can accelerate the esterification reaction. Compound 20 was synthesized by the treatment of 4, DMAP, N-Boc-isonipecotic acid and EDCI in dry dichloromethane over 3 h to obtain the intermediate with 80% yield at room temperature. The use of EDCI can increase the speed and yield of the esterification reaction. It is noteworthy that the hydroxyl group at the C-12 does not easily react with anhydride or acid because of steric hindrance and the formation of hydrogens bond. After the addition of 56A, DMAP and phthalic anhydride to anhydrous pyridine at 120 °C for 25 h, the yield of the intermediate is only 50%.
Ocotillol ketone derivatives 62–69 were synthesized by Zhou et al. (Figure 5). Compound 4 (0.21 mmol) in dry dichloromethane (8 mL) was added to pyridinium chlorochromate (0.40 mmol), and the mixture was stirred at room temperature for 3 h to obtain compound 62 with 66% yield. While compound 64 was synthesized by combining 4 (0.33 mmol) and pyridinium chlorochromate (1.00 mmol) in dry dichloromethane (8 mL), the reaction takes about 8 h to obtain intermediate with 76% yield at room temperature. Compound 65 had excellent antibacterial activities against S. aureus with a MIC of 16 μg/mL, while compounds 67 and 69 had moderate inhibitory effects against S. aureus.
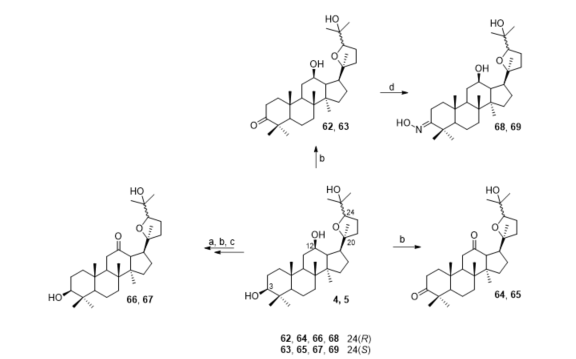
Figure 5. Synthesis of ocotillol-type derivatives 62–69. Reagents and conditions: (a) anhydrous pyridine, Ac2O, DMAP, r.t.; (b) anhydrous CH2Cl2, pyridinium chlorochromate (PCC), r.t.; (c) CH3OH, KOH, ref.; (d) anhydrous pyridine, NH2OH·HCl, 80 °C.
Ocotillol-type derivatives with a nitric oxide (NO) donor 70–91 were synthesized, their NO release ability and the antibacterial abilities of some derivatives were studied (Figure 6) [41]. Compounds 70–91 showed similar NO-releasing capability at 100 µM. Compounds 71, 75, 77, 83, 84, 86, and 91 showed better NO-releasing capability at 500 µM as after 30 min of reaction, they all released more than 0.2 M NO. Compounds 83 and 86 demonstrated good activities against Gram-positive bacteria (MIC = 16 μg/mL against B. subtilis 168 and S. aureus). Compound 86 displayed broad-spectrum activity against Gram-positive and Gram-negative bacteria. Compound 86 used with chloramphenicol also showed good synergistic effects with a FICI = 0.03 against MRSA USA300.
A series of ocotillol-type lactone derivatives 92–108 was designed by Zhang et al. (Figure 7) [42]. Compounds 96–98, 100 and 102 demonstrated good activities against S. aureus and B. subtilis with MIC values of 1 to 8 μg/mL. Compounds 96, 100, 101, 102, 105 and 107 showed good activities against MRSA USA 300. Compounds 96 and 102 also exhibited bactericidal activities with minimum bactericidal concentration (MBC) values of 4 and 8 μg/mL. Additionally, 102 reduced the MICs of KAN and chloramphenicol against MRSA USA300 from 1 and 4 μg/mL to 0.125 and 1 μg/mL (FICI = 0.141 and 0.375), respectively. Zhang et al. also analyzed the antibacterial effect of the ocotillol-type lactone derivative 102 by scanning electron microscopy, a cytoplasmic β-galactosidase leakage assay and UV-visible analysis [42]. The results showed that 102 might exert its antibacterial effect by damaging bacterial cell membranes and disrupting the function of DNA. The precise mechanism of its DNA antibacterial action is currently under investigation.
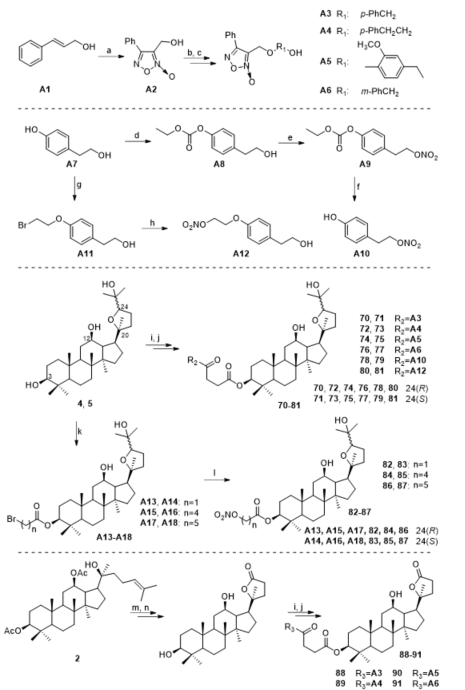
Figure 6. Synthesis of ocotillol-type derivatives 70–91. Reagents and conditions: (a) NaNO2, HOAc, r.t., 1 h; (b) SOCl2, pyridine, CH2Cl2, r.t., 8 h; (c) HOR1OH, K2CO3, KI, CH3CN, r.t., 3 h; (d) 1 M NaOH, ethyl chlorocarbonate, −5 ℃; (e) Acetic anhydride, nitrosonitric acid, CH2CI2, 0 ℃; (f) cholamine, ethyl alcohol, r.t.; (g) K2CO3, 1,2-dibromoethane, THF, r.t.; (h) AgNO3, CH3CN, 70 ℃, protection from light; (i) succinic anhydride, DMAP, CHCl3, 42 °C, 6 h; (j) A3-6, A10, A12, DMAP, EDCI, 25 °C, CH2Cl2, 6 h; (k) Bromoacetic acid, 5-bromopentanoic acid or 6-bromohexanoic acid, Et3N, DMAP, EDCI, dry CHCl3, r.t.; (l) AgNO3, CH3CN, 70 °C, protection from light; (m) CrO3, CH3COOH, H2O, r.t., 3 h; (n) NaOH, H2O, CH3OH, reflux, 6 h.
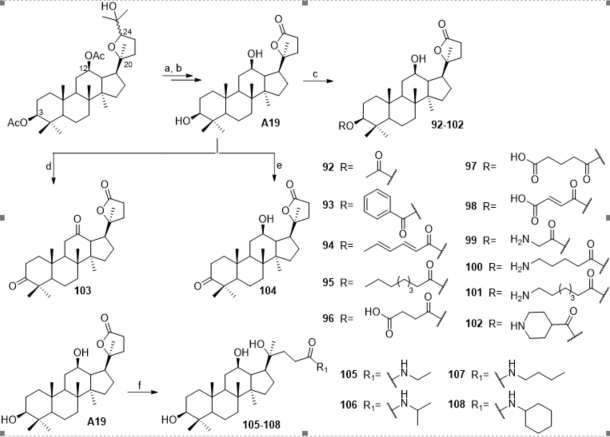
Figure 7. Synthesis of ocotillol-type derivatives 92–108. Reagents and conditions: (a) Jones reagent, acetone, r.t.; (b) (1) KOH, MeOH, H2O, 60 °C; (2) 50% H2SO4; (c) 1) corresponding acid, anhydride or Boc-amino acid, DMAP, EDCI, CH2Cl2, r.t.; (2) CH2Cl2, TFA, r.t.; (d) excess of PCC, CH2Cl2, r.t.; (e) 1 M of PCC, CH2Cl2, r.t.; (f) NaOH, THF, R1NH2, r.t.
At present, the antibacterial target of ocotillol-type derivatives is still not clear. Bi et al. synthesized the ocotillol-type probe 109A, which had a MIC of 8 μg/mL against B. subtilis. An epifluorescent microscopy study showed that 109A was mainly distributed on the bacterial cell membrane rather than within the nucleoid (Figure 8). On this basis, Bi et al. synthesized the ocotillol-type probe 109B, which had a MIC of 1 μg/mL against MRSA 18–19 (Hospital-acquired methicillin-resistant Staphylococcus aureus, collected in Chengdu, China from 2018) [43]. The antibacterial mechanism of 109B against MRSA 18–19 is currently underway. The number of ocotillol-type probes is small, which limits the discovery of their antibacterial target. In 2017, 28-hydroxy protopanaxadiol was synthesized as a novel probe template [44]. The synthesis of new ocotillol-type probes employing 28-hydroxy protopanaxadiol may provide an effective means to enrich the structure of ocotillol-type probes. Additionally, functional probes that target the cell membrane are needed. Further research of ocotillol-type probes will promote the discovery of the target protein and provide a reference for the development of more effective drugs.
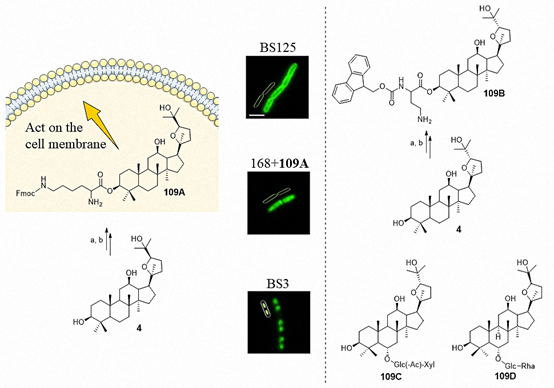
Figure 8. Structure of the ocotillol-type derivative 109A-109D and the epifluorescent microscopy images of B. subtilis strain BS125 (top), strain 168 with compound 109A treatment (middle), and strain BS3 (bottom). Scale bar: 4 μm. Reagents and conditions: (a) DMAP, EDCI, N-Boc-N'-Fmoc-l-Lysine, CH2Cl2, r.t.; (b) TFA, CH2Cl2, r.t.
Based on the present research of the ocotillol-type derivatives, a preliminary SAR of their antibacterial activities is summarized in Figure 9. The 24(S)-configuration is preferred, while substitution at the 3-OH changes the conformation to render the 24(R)-compound bioactive. A hydrogen donor at C-3 and C-12 are preferred to maintain the activity against Gram-positive bacteria. Decreased activity was observed when the functional groups at C-3 and C-12 were a ketone. When R2 is an ester, mild activity against Gram-negative bacteria was observed.
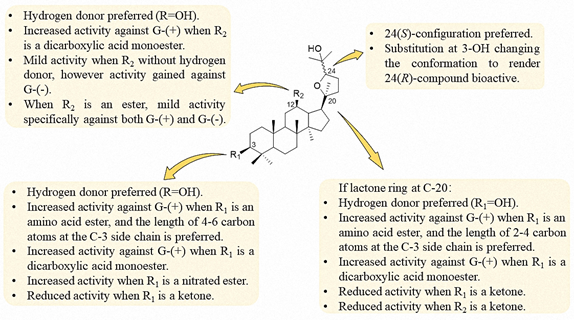
Figure 9. Structure–activity relationship (SAR) of the antibacterial activity of ocotillol-type derivatives.
3.2. Anti-Inflammatory Activities of Ocotillol-Type Derivatives
lipopolysaccharide-stimulated RAW 264.7 cells can release the inflammatory mediator NO, prostaglandin E2 (PGE2), tumor necrosis factor (TNF-α), interleukin-6 (IL-6) and anti-inflammatory mediator interleukin-10 (IL-10). The anti-inflammatory activity of 20(S)-ocotillol (4, 5) and 20(R)-ocotillol (150, 151) was evaluated in RAW 264.7 cells. The results showed that both 20(S)-ocotillol and 20(R)-ocotillol inhibited the release of the inflammatory cytokines NO and interleukin-6 (IL-6). However, the 20(S)-epimers mainly inhibited the release of PGE2 and primarily increased the release of the anti-inflammatory mediator IL-10. The 20(R)-epimers inhibited the release of the inflammatory cytokine TNF-α [45][46]. Oral ocotillol-type ginsenosides such as 109C (Figure 8) are metabolized to ocotillol-type sapogenin in the gut by gut microbiota [47]. Ocotillol-type sapogenin showed the highest inhibitory effect. In vitro studies demonstrated that 20(R),24(R)-ocotillol might ameliorate colitis by inhibiting the expression of the proinflammatory cytokines TNF-α, interleukin-1β, IL-6, interleukin-17 (IL-17), interleukin-23 (IL-23) and interferon-γ (IFN-γ). Additionally, 20(R),24(R)-ocotillol strongly ameliorated Trinitro-benzene-sulfonic acid-induced iNOS and cyclooxygenase-2 (COX-2) expression, as well as activation of their transcription factors NF-κB and MAPKs in mice [47][48]. In 2019, Wang et al. found that 109D (Figure 8) could attenuate lipopolysaccharide (LPS)-induced acute lung injury. A further mechanistic study indicated that 109D reversed the LPS-induced increases of mRNA expression and protein levels of macrophage inflammatory protein-2 (MIP-2) and intercellular adhesion molecule-1 (ICAM-1) [49]. Compound 109D also possessed neuroprotective activity by inhibiting the TLR4-mediated transforming growth factor-β-activated kinase-1(TAK1)/ nuclear factor kappa-B kinase 2 (IKK) /NF-κB, MAPKs, and Akt signaling pathways to exert anti-neuroinflammatory effects on LPS-activated microglia. In vivo experiments demonstrated that 109D significantly inhibited microglial activation and proinflammatory factor expression in the mouse cortex and hippocampus after the LPS injection [50].
Ocotillol-type derivatives with NO-inhibitory activity were further studied (Figure 10) [51][52][53][54]. Derivatives 6, 46, 110, 112, 113, 121, 132 and 136 showed significant NO-inhibitory activities, while 115, 116 and 119 had no obvious NO-inhibitory activities. Derivatives 46 and 136 exhibited the most potent NO-inhibitory activities and were even comparable to a steroid drug. Additionally, 46 and 136 significantly decreased LPS-induced TNF-α and IL-6 synthesis and iNOS and COX-2 expression via the NF-κB pathway.
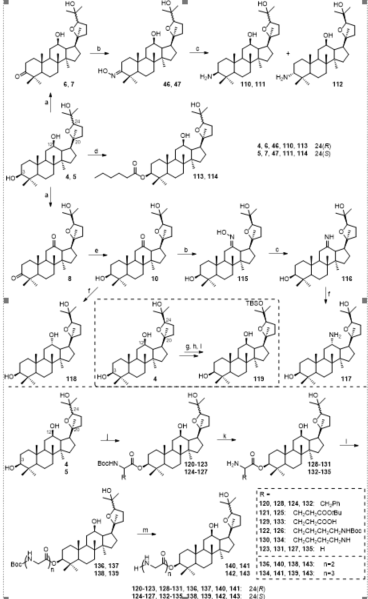
Figure 10. Synthesis of ocotillol-type derivatives 110–143. Reagents and conditions: (a) PCC, CH2Cl2, r.t.; (b) Hydroxylamine hydrochloride, pyridine, 80 °C; (c) NaCNBH3, TiCl3, AcONH4, MeOH, r.t.; (d) n-Hexanoic acid, EDCI, DMAP, r.t.; (e) NaBH4, i-PrOH, r.t.; (f) NaBH4, MeOH, r.t.; (g) Ac2O, CH2Cl2, r.t.; (h) trifluoromethanesulfonic acid tert-butyldimethylsilyl ester (TBS-OTF), lutidine, r.t.; (i) KOH, MeOH, THF, r.t.; (j) Boc-amino acid, EDCI, DMAP, CH2Cl2, r.t.; (k) TFA, CH2Cl2, r.t.; (l) O-benzotriazole-N,N,N',N'-tetraMethyl-uroniuM-hexafluorophosphate (HBTU), NEt3, DMF, r.t.; (m) TFA, CH2Cl2, r.t.
Wang et al. synthesized a series of ocotillol-type derivatives (Figure 11) [55][56][57]. Compound 144 had a protective effect on the lung function of experimental model mice with hormone-resistant asthma caused by non-typeable Hemophilus influenzae and improved their hormone resistance. Compounds 58 and 145–149 had inhibitory effects on the IL-6 expression and promoting effects on the IL-10 expression in the serum of rats induced by chronic obstructive pulmonary disease (COPD) caused by cigarette smoking.
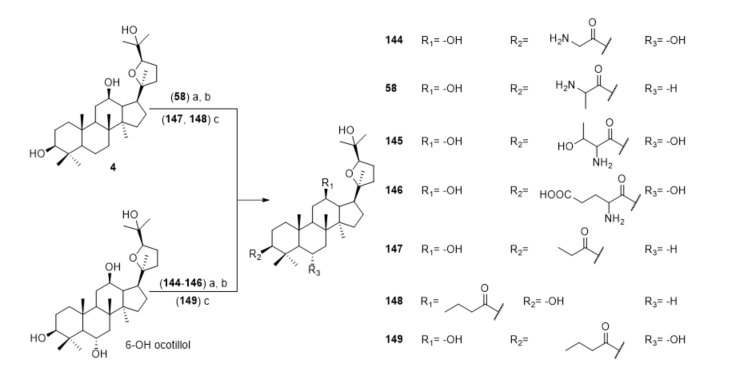
Figure 11. Synthesis of ocotillol-type derivatives 144–149. Reagents and conditions: (a) Boc-amino acid, DMAP, EDCI, CH2Cl2, r.t.; (b) TFA, CH2Cl2, r.t.; (c) aliphatic acid, DCC, EDCI, CH2Cl2, r.t.
Based on the present research of ocotillol-type derivatives, a preliminary SAR of their anti-inflammatory effects is summarized, as shown in Figure 12. The 24(R)-configuration is preferred for the anti-inflammatory activity. An oxime at C-3 is preferred for good inhibitory activity of LPS-induced NO synthesis. Boc-amino groups seem to be preferred to inhibit the activity of LPS-induced NO synthesis than amino groups at C-3. A hydrogen donor at C-12 is preferred to inhibit LPS-induced NO synthesis. A fatty acid or amino acid group at C-3 has inhibitory effects on the expression of IL-6 and promotes the expression of IL-10 in the serum of a rat model of COPD induced by cigarettes.
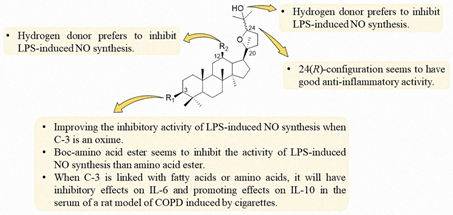
Figure 12. SAR of ocotillol-type derivatives with anti-inflammatory activity.
3.3. Anticancer Effects of Ocotillol-Type Derivatives
The antitumor effect of ocotillol-type derivatives is mainly concentrated in the ocotillol monomer or in substances directly extracted from plants; thus, there are few reports on its structural modification [58][59]. 172 (Figure 13A) showed effective antitumor-promoting activity on a mouse hepatic tumor and mouse skin [60][61]. A series of ocotillol-type derivatives had been studied for their cytotoxic activity against HeLa, A549 and MCF-7 cancer cells. Pharmacological experiments on HeLa cells showed that ocotillol-type derivatives had cytotoxicity. Among them, compounds 5, 152 and 173 (Figure 13A) possessed good activities with IC50 values of 11.53 ± 0.49 μM, 4.58 ± 0.66 μM and 19.84 ± 1.10 μM toward HeLa cells, respectively [62]. Compounds 162, 163, 167 and 166 showed reduced cell viabilities toward HeLa cells at 48.59%, 47.39%, 52.82% and 59.02% at 100 μg/mL, respectively [63].
Pharmacological results indicated that ocotillol-type derivatives had anticancer potential, and the configurations at C-20 or C-24 and the number of glycosyl units at C-3 could have an important influence on the cytotoxicity in vitro. There are only a small number of studies on ocotillol-type derivatives with anticancer activity, and thus, there is an opportunity to increase the number of ocotillol-type derivatives with anticancer activity.
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803.
- Pan, Y.X.; Yang, M.H.; Zhang, M.; Jia, M. Rapid discrimination of commercial American ginseng and Asian ginseng according to diols composition using a colorimetric sensor array. Sens. Actuators B Chem. 2019, 294, 48–54.
- Williamson, E.M.; Liu, X.; Izzo AA. Trends in use, pharmacology and clinical applications of emerging herbal nutraceuticals. Br. J. Pharmacol. 2019, 177, 1227–1240.
- Bi, Y.; Yang, X.; Zhang, T.T.; Liu, Z.Y.; Zhang, X.C.; Lu, J.; Cheng, K.G.; Xu, J.Y.; Wang, H.B.; Lv, G.Y. Design, synthesis, nitric oxide release and antibacterial evaluation of novel nitrated ocotillol-type derivatives. Eur. J. Med. Chem. 2015, 101, 71–80.
- Qi, L.W.; Wang, C.Z.; Yuan, C.S. Ginsenosides from american ginseng: Chemical and pharmacological diversity. Phytochemistry 2011, 72, 689–699.
- Liu, J.; Xu, Y.R.; Yang, J.J.; Wang, W.Z.; Zhang, J.Q.; Zhang, R.M.; Meng, Q.G. Discovery, semisynthesis, biological activities, and metabolism of ocotillol-type saponins. J. Ginseng. Res. 2017, 41, 373–378.
- Zhao, M.; Fu, L.W.; Li, N.; Tang, W.X.; Li, M.J.; Hasegawa, T.; Ogura, H.; Kataoka, T.; Hirose, K.; Ando, M. The ursane-, oleanane-, dammarane-, lupene-, and taraxasterane-type triterpenes isolated from Nerium oleander and their biological activities. Phytomedicines 2006, 6, 83–107.
- Warnhoff, E.W.; Halls, C.M.M. Desert plant constituents: Li. ocotillol: An intermediate in the oxidation of hydroxy isoctenyl side chains. Can. J. Chem. 1965, 43, 3311–3321.
- Tian, X. Studies on ocotillol-type ginsenoside and its related compounds. Ph.D. Thesis, Jilin University, Jilin, China, 2012.
- Tanaka, O.; Morita, T.; Kasai, R.; Kinouchi, J.; Sanada, S.; Ida, Y.; Shoji, J. Study on saponins of rhizomes of panax pseudo-ginseng subsp. himalaicus collected at tzatogang and Pari-la, Bhutan-Himalaya. Chem. Pharm. Bull. 1985, 33, 2323–2330.
- Liu, J.P.; Wang, F.; Li, P.Y.; Lu, D. A new ocotillol-type triterpenoid saponin from red American ginseng. Nat. Prod. Res. 2012, 26, 731–735.
- Namba, T.; Matsushige, K.; Morita, T.; Tian, Z. Saponins of plants of Panax species collected in central nepal and their chemotaxonomical significance. I. Chem. Pharm. Bull. 1986, 34, 730–738.
- Han, L.; Lin, M.Y.; Zheng, Q.; Liu, H.Y.; Liu, H.Y.; Dong, G.; Liu, J.P.; Li, P.Y. A new epimer of ocotillol from stems and leaves of american ginseng. Nat. Prod. Res. 2014, 28, 935–939.
- Duc, N.M.; Nham, N.T.; Kasai, R.; Ito, A.; Osamu, T. Saponins from vietnamese ginseng, Panax vietnamensis Ha et Grushv. collected in central Vietnam. I. Chem. Pharm. Bull. 1993, 41, 2010–2014.
- Zou, K.; Zhu, S.; Tohda, C.; Komatsu, K. Dammarane-type triterpene saponins from Panax j aponicus. J. Nat. Prod. 2002, 65, 346–351.
- Yamasaki, K.; Tanaka, O.; Bulletin, P. Saponins from vietnamese ginseng, panax vietnamensis HA et grushv. collected in central Vietnam. II. Chem. Pharm. Bull. 1994, 42, 115–122.
- Morita, T.; Kasai, R.; Tanaka, O.; Tian, Z.; Zhou, J.; Yang, T.R. Saponins of Zu-Tziseng, rhizomes of panax japonicus C.A. MEYER var. major (BURK.) C.Y. Wu et K.M. FENG, collected in Yunnan, China. Chem. Pharm. Bull. 1982, 30, 4341–4345.
- Schlag, E.M.; McIntosh, M.S. Ginsenoside content and variation among and within American ginseng (Panax quinquefolius L.) populations. Phytochemistry 2006, 67, 1510–1529.
- Li, B.; Wang, C.Z.; He, T.C.; Yuan, C.S.; Du, W. Antioxidants potentiate American ginseng-induced killing of colorectal cancer cells. Cancer. Lett. 2010, 289, 62–70.
- Le, Q.U.; Lay, H.L.; Wu, M.C.; Nguyen, T.H.H.; Nguyen, D.L. Phytoconstituents and biological activities of panax vietnamensis (Vietnamese Ginseng): A precious ginseng and call for further research-a systematic review. Nat. Prod. Commun. 2018, 13, 1801301–1934578.
- Li, P.Y.; Liu, J.P. Pseudoginsenoside Pdq and Its Semi-Synthesis Process and Medicine Use. ZL200510016774.4, 14 December 2005.
- Yang, G.Q.; Li, Y.; Yang, Q.; Yue, X.; Jiang, Y.T. Simple and efficient synthesis of Pseudoginsenoside HQ. Chin. J. Org. Chem. 2017, 37, 1530–1536.
- Meng, Q.G.; Bi, Y.; Wang, L.; Jiang, N.; Jiang, Y.T.; Zhang, J.F.; Yi, S.T.; Sun, H.J. Synthesis, structural determination of a new ocotillol derivative and its epimer. Lett. Org. Chem. 2011, 8, 682–685.
- Xu, Y.R.; Wang, W.Z.; Yang, J.; Li, X.; Meng, Q.G. Advances in the synthesis and biological activities of ocotillol-type saponins. Chin. J. Org. Chem. 2016, 36, 724–735.
- Yang, J.J.; Xu, Y.R.; Wang, W.Z.; Yang, J.; Meng, Q.G. Synthesis and formation mechanism of ocotillol and its epimer. J. Yantai. Univ. 2016, 29, 181–186.
- Wang, W.Z.; Xu, Y.R.; Li, X.L.; Yang, J.; Meng, Q.G. Synthesis and formation of ocotillol and its empimer. J. China. Pharm. Univ. 2016, 47, 282–287.
- Xu, Y.R.; Yang, J.J.; Liu, J.; Hou, G.G.; Meng, Q.G. Synthesis and crystal structures of C24-epimeric 20(R)-ocotillol-type saponins. Acta Crystallogr. Sect. C: Struct. Chem. 2016, 72, 498–503.
- Liu, J.; Xu, Y.R.; An, X.S.; Hou, G.G.; Meng, Q.G. Synthesis and crystal structures of a 3-acetylated (20S,24S)-ocotillol-type saponin and its C-24 epimer. Acta Crystallogr. Sect. C: Struct. Chem. 2017, 73, 464–469.
- Meng, Q.G.; Tan, W.J.; Hou, G.G.; Zhang, X.Y.; Hu, X.Y.; Yang, F.; Bai, G.J.; Zhu, W.W.; Cai, Y.; Bi, Y. Synthesis and structural characterization of two epimers driven from 20(S)-protopanaxadiol. J. Mol. Struct. 2013, 1054-1055, 1-5.
- Atopkina, L.N.; Novikov, V.L.; Denisenko, V.A.; Uvarova, N.I. Glycosylation of triterpenes of the dammarane series. III. Rregio- and stereoselective synthesis of 12-O-β-D-glucopyranosides of 20(S),24(R)-epoxydammarane-3-12β,25-triols under Helfer-ich’s conditions. Chem. Nat. Compd. 1985, 21, 674–675.
- Atopkina, L.N.; Malinovskaya, G.V.; Elyakov, G.B.; Uvarova, N.I.; Woerdenbag, H.I.; Koulman, A.; Pras, N.; Potier, P. Cytotoxicity of natural ginseng glycosides and semisynthetic analogues. Planta Med. 1999, 65, 030–034.
- Shen, R.Z.; Cao, X.; Laval, S.; Sun, J.S.; Yu, B. Synthesis of ocotillol-type ginsenosides. J. Org. Chem. 2016, 81, 10279–10294.
- Nguyen, N.H.; Nguyen, C.T. Pharmacological effects of ginseng on infectious diseases. Inflammopharmacology 2019, 27, 871–883.
- Zhou, Z.W.; Ma, C.; Zhang, H.Y.; Bi, Y.; Chen, X.; Tian, H.; Xie, X.N.; Meng, Q.G.; Lewis, P.J.; Xu, J.Y. Synthesis and biological evaluation of novel ocotillol-type triterpenoid derivatives as antibacterial agents. Eur. J. Med. Chem. 2013, 68, 444–453.
- Bi, Y.; Ma, C.; Zhang, H.Y.; Zhou, Z.W.; Xu, J.Y. Novel 3-substituted ocotillol-type triterpenoid derivatives as antibacterial candidates. Chem. Biol. Drug. Des. 2014, 84, 489–496.
- Bi, Y.; Ma, C.; Zhou, Z.W.; Zhang, T.T.; Zhang, H.Y.; Zhang, X.C.; Lu, J.; Meng, Q.G.; Lewis, P.J.; Xu, J.Y. Synthesis and antibacterial evaluation of novel hydrophilic ocotillol-type triterpenoid derivatives from 20(S)-protopanaxadio. Rec. Nat. Prod. 2015, 9, 356–368.
- Bi, Y.; Xu, J.Y.; Zhou, Z.W.; Zhang, H.Y.; Peter, J.L.; Ma, C.; Chen, X.; Yang, J.; Zhang, T.T. (20S, 24R)-Ocotillol type Ginsenoside Derivative Having Antibacterial Activity and Preparation Method and Application Thereof. ZL201210433920.3, 30 October 2012.
- Xu, J.Y.; Bi YI.; Zhang, H.Y.; Zhou, Z.W.; Peter, J.L.; Ma, C.; Chen, X.; Zhang, D.; Tian, H.; Xie, X.N.; Huang, W.W.; et al. (20S,24S)-Ocotillol Ginsenoside Derivatives with Antibacterial activity, and Preparation Method and Application Thereof. ZL201210422186.0, 30 October 2012.
- Bi, Y.; Liu, X.X.; Zhang, H.Y.; Yang, X.; Liu, Z.Y.; Lu, J.; Lewis, P.; Wang, C.Z.; Xu, J.Y.; Meng, Q.G.; et al. Synthesis and antibacterial evaluation of novel 3-substituted ocotillol-type derivatives as leads. Molecules 2017, 22, 590.
- Wang, K.Y.; Zhou, Z.W.; Zhang, H.Y.; Cao, Y.C.; Xu, J.Y.; Ma, C.; Meng, Q.G.; Bi, Y. Design, synthesis and antibacterial evaluation of 3-substituted ocotillol-type derivatives. Molecules 2018, 23, 3320.
- Bi, Y.; Yang, J.; Ma, C.; Liu, Z.Y.; Meng, Q.G. Design, synthesis and in vitro NO-releasing activities of ocotillol-type furoxans. Pharmazie 2015, 70, 213–218.
- Zhang, Z.Y.; Chen, Z.G.; Zhang, S.Y.; Shao, X.; Zhou, Z.W. Antibacterial activity of the structurally novel ocotillol-type lactone and its analogues. Fitoterapia 2020, 144, 104597.
- Bi, Y.; Cao, Y.C.; Wang, K.Y.; Meng, Q.G. Synthesis and Application of Ocotillol-Type Ginsengenin with Dansylamide Group or Fluorenylmethoxycarbonyl Group. CN202010548873.1, 15 September 2020.
- Liu, Z.Y.; Zhang, H.Y.; Bi, Y.; Liu, X.X.; Lu, J.; Zhang, X.C.; Xu, J.Y.; Wang, C.Z.; Yuan, C.S. Design and synthesis of 28-hydroxy protopanaxadiol as a novel probe template. Nat. Prod. Res. 2017, 31, 1523–1528.
- Zhang, J.Q.; Zhang, Q.; Xu, Y.R.; Li, H.X.; Zhao, F.L.; Wang, C.M.; Liu, Z.; Liu, P.; Liu, Y.N.; Meng, Q.G.; et al. Synthesis and in vitro anti-inflammatory activity of C20 epimeric ocotillol-type triterpenes and protopanaxadiol. Planta Med. 2019, 85, 292–301.
- Zhao, F.;Meng, Q.G.; Yang, J.J.; Zhang, Q.; Li, H.X.; Liu, P.; Liu, Y.A.; Xu, Y.R.; Wang, W.Z.; Li, X.L.; et al. Dammarane-Type Ginsenoside/Ginsengenin and Anti-Inflammatory Application of Ocotillol-Type Derivative of Dammarane-Type Ginsenoside/Ginsengenin. CN201710708593.0, 19 December 2017.
- Jeong, J.J.; Van, T.H.L.; Lee, S.Y.; Eun, S.H.; Nguyen, M.D.; Park, J.H.; Kim, D.H. Anti-inflammatory effects of vina-ginsenoside R2 and majonoside R2 isolated from Panax vietnamensis and their metabolites in lipopolysaccharide-stimulated macrophages. Int. Immunopharmacol. 2015, 28, 700–706.
- Lee, S.Y.; Jeong, J.J.; Le, T.H.; Eun, S.H.; Nguyen, M.D.; Park, J.H.; Kim, D.H. Ocotillol, a majonoside R2 metabolite, ameliorates 2,4,6-trinitrobenzenesulfonic acid-induced colitis in mice by restoring the balance of Th17/Treg cells. J. Agric. Food. Chem. 2015, 63, 7024–7031.
- Wang, P.W.; Hou, Y.; Zhang, W.; Zhang, H.T.; Che, X.H.; Gao, Y.F.; Liu, G.G.; Liu, Y.L.; Yang, D.P.; Wang, J.M.; et al. Pseudoginsenoside-F11 attenuates lipopolysaccharide-induced acute lung injury by suppressing neutrophil infiltration and accelerating neutrophil clearance. Inflammation 2019, 42, 1857–1868.
- Wang, X.; Wang, C.;Wang, J.; Zhao, S.; Zhang, K.; Wang, J.; Zhang, W.; Wu, C.; Yang, J. Pseudoginsenoside-F11 (PF11) exerts anti-neuroinflammatory effects on LPS-activated microglial cells by inhibiting TLR4-mediated TAK1/IKK/NF-κB, MAPKs and Akt signaling pathways. Neuropharmacology 2014, 79, 642–656.
- Sun, Y.X.; Fang, X.J.; Gao, M.; Wang, C.H.; Gao, H.Y.; Bi, W.J.; Tang, H.H.; Cui, Y.T.; Zhang, L.M.; Fan, H.Y.; et al. Synthesis and structure-activity relationship of pyxinol derivatives as novel anti-Inflammatory agents. ACS Med. Chem. Lett. 2020, 11, 457–463.
- Yang, G.Q.; Gao, M.; Sun, Y.X.; Wang, C.H.; Fang, X.J.; Gao, H.Y.; Diao, W.S.; Yu, H. Design, synthesis and anti-inflammatory activity of 3-amino acid derivatives of ocotillol-type sapogenins. Eur. J. Med. Chem. 2020, 202, 112507.
- Yang, G.Q.; Zhang, C.; Gao, M.; Gao, H.Y.; Zou, Z.J.; Yuan, Z. Ocotillol Type Derivative Having Anti-Inflammatory Activity and Preparation Method and Application Thereof. CN202010367674.0, 11 September 2020.
- Yang, G.Q.; Gao, M.; Wang, C.H.; Ren, R.Y.; Zou, Z.J.; Qiao, X. The Use of Des Martin’s Reagents in the Synthesis of Key Intermediates of Ocotillol-Type Derivatives. CN202010367823.3, 25 September 2020.
- Wang, F.; Zhang, J.; Liu, J.P.; Wang, C.Z.; Zheng, J.T.; Liu, J.L.; Zhang, J.R.; Wang, G.Q.; Guan, X.W.; Dong, B. (20S,24R)-Ocotillol Type Ginsenoside Glycine Derivatives and Preparation Method and Application Thereof. CN201911112761.5, 5 May 2020.
- Liu, J.L.; Liu, Y.H.; Wang, Z.Y.; Yang, N.; Si, Y.; Jiao, Y.F.; Zhang, Y.; Lin, H.Q.; Li, P.Y.; Liu, J.P. (20S,24R)-Ocotillol Type Ginsenoside Fatty Acid Derivatives and Preparation Method and Application Thereof. CN201911008914.1, 6 December 2019.
- Liu, J.L.; Zhang, Y.; Wang, Z.Y.; Liu, Y.H.; Jiao, Y.F.; Si, Y.; Zhou, B.S.; Zhang, J.; Li, P.Y.; Liu, J.P. (20S,24R)-Ocotillol Ginsenoside Amino-Acid Derivative, Preparation Method and Use. CN201911008913.7, 3 January 2020.
- Le, T.H.V.; Lee, S.Y.; Lee, G.J.; Nguyen, N.K.; Park, J.H.; Nguyen, M.D. Effects of steaming on saponin compositions and antiproliferative activity of Vietnamese ginseng. J. Ginseng. Res. 2015, 39, 274–278.
- Le, T.H.V.; Lee, S.Y.; Kim, T.R.; Kim, J.Y.; Kwon, S.W.; Nguyen, N.K.; Park, J.H.; Nguyen, M.D. Processed Vietnamese ginseng: Preliminary results in chemistry and biological activity. J. Ginseng. Res. 2014, 38, 154–159.
- Takao, K.; Midori, T.; Eiichiro, I.; Murakami, T.; Tokuda, H.; Nishino, H.; Duc, N.M.; Kasai, R.; Yamasaki, K. Cancer chemopreventive activity of majonoside-R2 from Vietnamese ginseng, Panax vietnamensis. Cancer Lett. 1999, 147, 11–16.
- Tran, Q.L. Triterpene saponins from Vietnamese ginseng (Panax vietnamensis) and their hepatocytoprotective activity. J. Nat. Prod. 2001, 64, 456–461.
- Yang, J.; Li, X.W.; Sun, T.; Gao, Y.; Chen, Y.X.; Jin, Y.R.; Li, Y. Semisynthesis and bioactive evaluation of oxidized products from 20(S)-ginsenoside Rg3, Rh2, protopanaxadiol (PPD) and their 20(R)-epimers as cytotoxic agents. Steroids 2016, 106, 26–34.
- Yang, J.; Yu, X.; Cai, X.; Chen, Y.X.; Zang, H.M.; Li, X.W.; Jin, Y.R. Semisynthesis and cytotoxicity evaluation of a series of ocotillol type saponins and aglycones from 20(S)-ginsenoside Rg2, Rh1, protopanaxatriol and their 20(R)-epimers. Chem. Res. Chin. Univ. 2016, 32, 35–40.

