 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Catarina Silva | + 2596 word(s) | 2596 | 2020-12-03 10:22:28 | | | |
| 2 | Nora Tang | -1 word(s) | 2595 | 2020-12-07 05:03:44 | | |
Video Upload Options
Tauopathies are neurodegenerative diseases characterized by the pathological accumulation of microtubule associated protein Tau (MAPT) in the form of neurofibrillary tangles and paired helical filaments in neurons and glia, leading to brain cell death. These diseases include frontotemporal dementia (FTD) and Alzheimer’s disease (AD) and can be sporadic or inherited when caused by mutations in the MAPT gene. Despite an incredibly high socio-economic burden worldwide, there are still no effective disease-modifying therapies and few Tau-focused experimental drugs have reached clinical trials. One major hindrance for therapeutic development is the knowledge gap in molecular mechanisms of Tau-mediated neuronal toxicity and death. For the promise of precision medicine for brain disorders to be fulfilled, it is necessary to integrate known genetic causes of disease, i.e., MAPT mutations, with understanding of the dysregulated molecular pathways that constitute potential therapeutic targets. Here, the growing understanding of known and proposed mechanisms of disease etiology will be reviewed, together with promising experimental Tau-directed therapeutics, such as recently developed Tau degraders. A particular focus will be given to the application of patient-specific stem cell models to study disease, and a number ofCurrent challenges faced by the fields of Tau research and drug discovery will alsobe addressed, including Tau pathophysiology unanswered questions, limitations of model systems and current challenges faced in developing cell-permeable small molecules that target Tau in disease.
1. Alzheimer’s Disease
Over a century after its first described case, Alzheimer’s disease (AD) is the most prevalent form of tauopathy and the most common cause of dementia (~60–80% of cases), and its frequency of incidence is rapidly increasing as the world’s population aged >65 continues to increase. Approximately 5.8 million Americans lived with AD in 2019, and this is predicted to double by 2050 [1][2], together with a financial burden predicted to increase from its current annual US $259 billion to more than $1 trillion by 2050. This trend is predicted to be global unless means of delaying, preventing, or treating AD are found [1][3].
2. MAPT
The microtubule-associated protein tau (MAPT) is a neuronal protein that regulates microtubule stability and dynamics as well as axonal transport [4][5]. Tau binds to microtubules via repeat microtubule-binding domains in the C-terminus, and this process is regulated by phosphorylation of sites within and adjacent the binding region (Figure 1a,b) [6]. The N-terminal projection region plays a role in signal transduction and membrane interactions (Figure 1a) [6]. Other tau physiological functions include interaction with the plasma membrane and scaffold proteins, signal transduction, DNA/RNA protection, and regulation of synaptic function [7][8]. In the human central nervous system (CNS), six tau isoforms are expressed by alternative splicing of the MAPT exons 2, 3, and 10, of which the longest isoform 2N4R tau (441 amino acids) contains two N-terminal inserts and four repeat domains in the C-terminus region (Figure 1a) [9]. This process is developmentally regulated and specific to each brain region based on physiological function [10][11]. Exons 2 and 3 are translated into the N1 and N2 domains, respectively, producing the 0N, 1N, and 2N tau isoforms of the N-terminal projection region (Figure 1a). In the human adult brain, the 2N isoform is the least expressed while the 1N isoform is the most abundant [10]. Exon 10 encodes the second microtubule-binding repeat domain in the C-terminal region (Figure 1a). Inclusion of exon 10 leads to the expression of three tau isoforms with four microtubule-binding domains (4R-Tau), whereas exclusion of exon 10 leads to expression of three isoforms of 3R-Tau [10][12]. These four repeat domains (R1–R4, Figure 1a) are essential for tau ability to regulate stability of microtubules and support axonal transport. For this reason, relative 3R/4R expression is also developmentally regulated. During the fetal stage, 3R-Tau (0N3R) is the main isoform present, allowing for dynamic axonal properties conducive to synaptogenesis and formation of neural pathways, followed by postnatal expression of all isoforms. In the adult brain, 4R-Tau binds more tightly to microtubules and the overall 3R/4R ratio is maintained at 1:1 [10][11]. Despite its protein domains, tau’s native state defies the traditional ‘structure-function paradigm’ by lacking a well-defined three-dimensional structure, being classified as an intrinsically disordered protein. This is a characteristic of proteins that require rapid conformational changes and structural plasticity but is also a characteristic of proteins with high propensity for misfolding that play a role in the pathogenesis of neurodegenerative diseases [13][14]. Tau misfolding and aggregation into highly ordered β-sheet-rich paired helical filaments (PHFs) that subsequently deposit in the form of neurofibrillary tangles (NFTs) (Figure 1b) are implicated in a heterogeneous group of aging-related neurodegenerative disorders referred to as tauopathies, which include Alzheimer’s disease (AD), Pick’s disease (PiD), frontotemporal dementia (FTD), and progressive supranuclear palsy (PSP) (Table 1) [15][16][17][18][19][20][21][22][23][24][25][26][27][28][29]. While many MAPT mutations increase tau’s propensity for aggregation and toxicity, and are the cause of dominantly inherited tauopathies [30], the majority of tauopathies are sporadic with variable clinical and pathological presentations [15]. Tauopathies are mainly considered gain-of-function proteinopathies but, despite increasing understanding of tau physiology and role in disease, the mechanisms of tau aggregation with disruption of molecular pathways leading to neuronal death are still poorly understood [31][32][33]. Evidence indicates that native tau is highly soluble, contains several charged and hydrophilic residues, and shows little tendency for aggregation. Thus, for tau to become aggregation competent, it must undergo conformational and post-translational modifications (PTMs) within and near the hexapeptide motifs in the C-terminal repeat domain (Figure 1b,c) [34][35], which also makes 4R-Tau more aggregation prone [36][37]. Little is known about the consequences of tau loss-of-function, but reduced binding of hyperphosphorylated tau to axonal microtubules may alter their structure and/or function, disrupting axonal transport, driving synaptic dysfunction and loss, and promoting neurotoxicity.
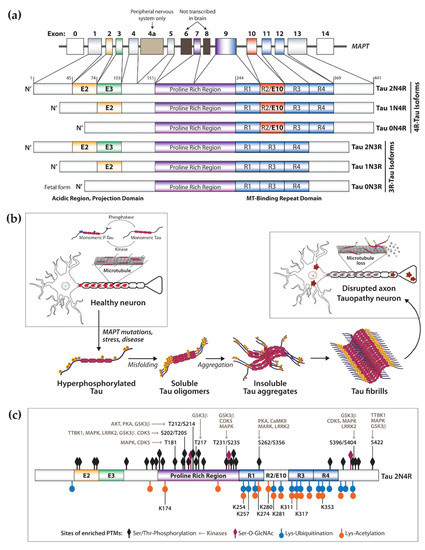
Figure 1. Human microtubule associated protein Tau physiological function and in disease. (a) Alternative splicing of the MAPT gene leads to developmentally regulated expression of six Tau isoforms, containing three (3R) or four (4R) microtubule (MT)-binding domains in the C-terminus, and zero, one or two N-terminus domains. (b) Simplified representation of Tau function as a regulator of microtubule stability and dynamics in human neurons. Tau binding is regulated by phosphorylation via the concerted action of kinases and phosphatases. In disease Tau becomes hyperphosphorylated and no longer binds microtubules, contributing to axonal dysfunction. Together with post-translational modification, Tau misfolding drives oligomerization and aggregation into larger order insoluble fibrils such as NFTs and PHFs found in the somatodendritic space and processes of CNS neurons. (c) Tau undergoes extensive post-translational modification (PTMs), which are exacerbated in disease. Indicated in the 2N4R Tau isoform are the locations of highest PTM density, including phosphorylation, acetylation, O-GlcNAcylation and ubiquitination. Also indicated are sites of phosphorylation prevalent in tauopathies and key regulatory kinases.
Table 1. Summary and key features of primary and secondary tauopathies categorization.
|
Clinical |
Symptomology |
Tau |
Neuronal Pathology |
Glia Pathology |
Affected Brain Regions |
|
|---|---|---|---|---|---|---|
|
Primary Tauopathy |
Pick’s disease (PiD) |
Behavioral change, social disinhibition, eating disorder, absent/late parkinsonism. |
3R |
Round cytoplasmic inclusions (Pick bodies), rare NFTs. |
Ramified astrocytes. |
Dentate gyrus of the hippocampus, frontal and temporal neocortical layers II, IV. Frontal, insular and anterior temporal cortices. |
|
Behavioral variant of FTD (bvFTD) |
Behavioral disinhibition, apathy, empathy loss, compulsiveness, executive and cognitive dysfunction. |
3R > 4R |
Cytoplasmic NFTs, short dystrophic neurites. |
Orbitofrontal, dorsolateral prefrontal, medial prefrontal cortices. Subcortical brain nuclei. Temporal-parietal lobes. |
||
|
Progressive supranuclear palsy (PSP) |
Apathy, anxiety, sleep disturbance. Spectrum from pure motor to pure cognitive presentations. |
4R |
NFTs, neuropile threads. |
Tufted astrocytes, somatodendritic coiled bodies. |
Subthalamic nucleus, basal ganglia, brainstem. Posterior mesencephalic cortex. |
|
|
Corticobasal syndrome (CBS) |
Asymmetric motor symptoms, apraxia, sensory impairment. Spectrum from pure motor to pure cognitive presentation. |
4R |
NFTs, neuropile threads, ballooned neurons, pleomorphic inclusions (pre-tangles). |
Annular clusters of short fuzzy cell processes, astrocytic Tau plaques, argyrophilic inclusions. |
Frontoparietal cortex, striatum, substantia nigra. |
|
|
Argyrophilic grain disease (AGD) |
Personality change, emotional imbalance, memory failure. |
4R |
Argyrophilic grains, dendritic straight filaments and smooth tubules. |
Thorn-shaped astrocytes, coiled bodies. |
Medial temporal lobe, entorhinal cortex, hippocampus, amygdala. |
|
|
Aging-related Tau astrogliopathy (ARTAG) |
Cognitive decline. |
4R |
- |
Thorn-shaped and granular-fuzzy astrocytes. |
Medial temporal lobe, lobar (frontal, parietal, occipital, lateral temporal), subcortical, brainstem. |
|
|
Globular glial tauopathy (GGT) |
Behavior change, cognitive decline, motor neuron disease (Parkinsonism). |
4R |
- |
Globular inclusions in astrocytes and oligodendrocytes. |
White matter, limbic and isocortical regions. Hippocampus. |
|
|
Primary progressive aphasia (PPA) |
Language deterioration, loss of semantic memory. |
3R, 4R |
NFTs, amyloid plaques |
Globular astrocytic inclusions. |
Anterior and temporal lobes, parietal lobe. Frontoinsular cortex |
|
|
Primary age-related tauopathy (PART) |
Cognitive impairment. |
3R, 4R |
NFTs, neuropile threads |
Medial temporal lobe. |
Medial temporal lobe. |
|
|
Tangle-only dementia (TOD) |
Late-onset dementia. |
3R, 4R |
Intracellular PHFs, NFTs and neuropil threads. |
Hippocampus. |
||
|
Secondary Tauopathy |
Alzheimer’s disease (AD) |
Memory loss, cognitive dysfunction, social behavior changes. |
3R, 4R |
NFTs, neuropile threads, neuritic plaques. |
Entorhinal cortex, hippocampus, cerebral cortex. |
|
|
Chronic traumatic encephalop-athy (CTE) |
Memory loss, confusion, personality/behavior changes. Motor decline. |
3R, 4R |
P-Tau aggregates around small vessels, TDP-43 cytoplasmic inclusions. |
P-Tau aggregates around small vessels. |
Cortical sulci, isocortex layers II–III, hippocampus, subcortical nuclei. |
Diseases where tau has a direct and predominant causal effect on neurodegeneration are referred to as ‘primary tauopathies’, which include progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick’s disease (PiD), aging-related Tau astrogliopathy (ARTAG), argyrophilic grain disease (AGD), primary age-related tauopathy (PART), and tangle-only dementia (TOD) (Table 1) [38][39]. Other amyloidosis that are also associated with the formation of tau inclusions, but where tau is not the primary or unique pathological feature, such as AD and chronic traumatic encephalopathy (CTE), are referred to as ‘secondary tauopathies’ (Table 1) [39][40]. The distinction between the two categories does not imply that there is not an equally important role for tau in the pathophysiology and relevance for tau-directed therapeutics. Although beyond the scope of this review, Table 1 summarizes key aspects that highlight the complexity and diversity of disorders associated with tau pathology. To date, there is no cure or disease-effective treatment that targets the cause of any tauopathy [41].
Approved symptomatic therapeutics include acetylcholinesterase inhibitors and memantine for AD to treat cognitive and behavioral symptoms, levodopa or dopamine agonists for FTD-associated parkinsonism motor dysfunction, and antidepressants (e.g., selective serotonin reuptake inhibitors). Current research has shown progress on different strategies to mitigate tau accumulation, prevent aggregation, and promote clearance [41][42]. Growing evidence suggests that early Tau PTMs, misfolding and oligomerization, impaired protein degradation, and Tau relocalization have higher impact on toxicity than late-stage PHFs and NFTs. Based on this, multiple experimental therapeutic approaches focus on targeting early forms of toxic tau and in promoting enhancement of protein clearance. So far, the strategy showing the greatest progress as measured by advancement into clinical trials is tau immunotherapy, where humanized tau antibodies have reached clinical trials for AD, PSP, and PPA. Even so, a major roadblock for therapeutic development is the still incomplete understanding of the molecular mechanisms and pathways involved in tau-mediated neuronal toxicity and death, which constitute probable therapeutic targets. In an effort to connect the knowledge from these two research fields, here we will review the current and ever-evolving understanding of the mechanisms of tau pathogenicity and respective approaches to therapeutics development.
3. Molecular Mechanisms of Tau Pathology
There are two non-mutually exclusive principal models for the mechanism of tau-induced neuronal pathology [43][44]. One model focuses on tau’s propensity for misfolding, oligomerization and fibril formation, and toxic gain-of-function, which is exacerbated by tau PTMs and inefficient clearance (Figure 2) [45][46]. The other model focuses on tau loss-of-function as a result of abnormal PTMs and sequestration into aggregates, leading to disruption of axonal integrity and transport (Figure 2) [43][47]. Here, we will review several molecular mechanisms and protein modifications associated with tau-mediated toxicity in the CNS, which represent potential therapeutic targets of tauopathy.
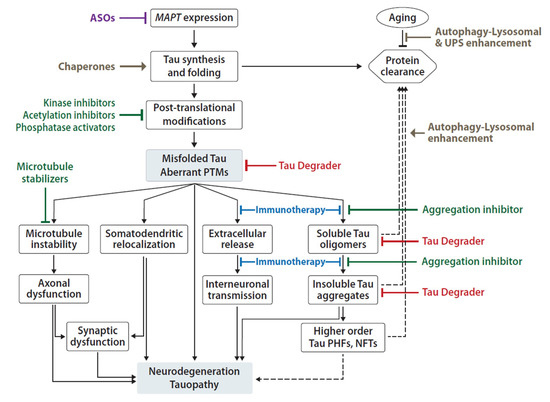
Figure 2. Summary of proposed mechanisms of Tau pathogenicity and corresponding experimental therapeutic approaches. Tau toxicity can be driven by loss-of-function leading to microtubule depolymerization and axonal transport disruption; and it can be driven by gain-of-function of aberrant Tau oligomers, aggregates and fibrils associated with neuronal toxicity, pathology spread and ultimately death. Current development of therapeutic agents include reduction of MAPT expression by ASOs (purple), small molecule (green) inhibitors of PTMs and aggregation, enhancement of Tau folding and/or clearance mechanisms (brown), Tau-specific degraders (red) and anti-Tau immunotherapies (blue). Solid arrows represent known and/or direct effects; dashed arrows represent indirect/proposed mechanisms; flat-ended connections represent inhibitory effect.
4. Tau Directed Therapeutics
The rate of failure in drug development for tauopathies is relatively high. Promising preclinical data has supported the development of multiple experimental therapies for tauopathy, but showing a pharmacodynamic effect in transgenic animal models has had poor translatability into clinical efficacy. For AD, 99% of all drug trials have failed partly due to limited bioavailability, poor blood-brain barrier (BBB) penetration, low cell permeability, and reduced drug half-life. The BBB still constitutes, and for good reason, a formidable obstacle to ensuring that any drug reaches its target in the CNS and therefore, an effective therapy will have to have a super affinity for Tau and/or be administered at high doses, which becomes exceptionally difficult and expensive. Alternatives include intrathecal approaches, which can circumvent the BBB but have an immensely high cost of treatment and other medical implications associated that will render the medicinal application worldwide almost prohibitive. Using small molecules that cross the BBB is an attractive and possibly optimal approach, but so far, the majority of experimental Tau drugs with positive pre-clinical data have clear limitations of BBB permeability. Another significant challenge for Tau therapeutics is the demonstration of selective target engagement and unvalidated biomarkers for pharmacodynamics and modifier effect on disease progression of primary tauopathies. Although robust assays can measure some forms of Tau found in CSF and blood, these Tau species do not necessarily represent the relevant pathological Tau found in the brain parenchyma. Several PET ligands are being developed and tested, but not without challenges. All these aspects highlight the need for profound changes in drug development approaches. An effective therapeutic agent is the result of a multi-variable effort that combines the right target, drug, biomarker, participant and clinical trial. The right target represents the biologic process most relevant for tauopathy, with proven function in disease pathophysiology and greater representation in disease than in normal physiological function and is a non-redundant process necessary for neuronal survival. The main proposed targets and pathways relevant for tauopathies were described in the section above. So far, apart from the use of combination therapy in AD to target the cholinergic system that has some effect on cognitive function, no other target has been shown to have patient benefit. A challenge in developing small molecules that target Tau and lead to effective disease-modifying therapeutics is the insufficient understanding of disease mechanisms, as well as the lack of a well-defined Tau fold for active molecular binding in disease. Moreover, a drug for tauopathy must have appropriate pharmacokinetic (PK) and pharmacodynamic (PD) parameters, ability to penetrate the BBB, efficacy demonstrated in animals and, importantly, in patient-derived ex vivo neurons, and acceptable toxicity. Achieving appropriate BBB penetration is a major hurdle for many experimental therapeutics. The human BBB has p-glycoprotein transporters and other multidrug resistance channels (MDRs) that may not be present in the animal models employed pre-clinically and that for this reason do not adequately predict human CNS entry. Despite the still critical limitations from incomplete understanding of disease mechanisms, lack of reliable clinical biomarkers, variable drug effects on less-then optimal mouse models, and limited implementation of humanized cellular models of tauopathy, the field has seen groundbreaking advances in multiple fronts for development of Tau therapeutics. At the time of this review, 24 therapeutics towards Tau have been tested in clinical trials Phase 1 or later, with 15 agents currently in active development based on publicly available data. Here, we will highlight some of the main current approaches showing the mostprogress in experimental Tau-directed therapeutics (Figure 2).
References
- Cornutiu, G. The Epidemiological Scale of Alzheimer’s Disease. J. Clin. Med. Res. 2015, 7, 657–666.
- Association, A.S. Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2019, 15, 321–387.
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71.
- Gigant, B.; Landrieu, I.; Fauquant, C.; Barbier, P.; Huvent, I.; Wieruszeski, J.M.; Knossow, M.; Lippens, G. Mechanism of Tau-promoted microtubule assembly as probed by NMR spectroscopy. J. Am. Chem. Soc. 2014, 136, 12615–12623.
- Trinczek, B.; Ebneth, A.; Mandelkow, E.M.; Mandelkow, E. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J. Cell Sci. 1999, 112 Pt 14, 2355–2367.
- Preuss, U.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. The ‘jaws’ model of tau-microtubule interaction examined in CHO cells. J. Cell Sci. 1997, 110 Pt 6, 789–800.
- Dorostkar, M.M.; Zou, C.; Blazquez-Llorca, L.; Herms, J. Analyzing dendritic spine pathology in Alzheimer’s disease: Problems and opportunities. Acta Neuropathol. 2015, 130, 1–19.
- Marciniak, E.; Leboucher, A.; Caron, E.; Ahmed, T.; Tailleux, A.; Dumont, J.; Issad, T.; Gerhardt, E.; Pagesy, P.; Vileno, M.; et al. Tau deletion promotes brain insulin resistance. J. Exp. Med. 2017, 214, 2257–2269.
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526.
- Trabzuni, D.; Wray, S.; Vandrovcova, J.; Ramasamy, A.; Walker, R.; Smith, C.; Luk, C.; Gibbs, J.R.; Dillman, A.; Hernandez, D.G.; et al. MAPT expression and splicing is differentially regulated by brain region: Relation to genotype and implication for tauopathies. Hum. Mol. Genet. 2012, 21, 4094–4103.
- Kosik, K.S.; Orecchio, L.D.; Bakalis, S.; Neve, R.L. Developmentally regulated expression of specific tau sequences. Neuron 1989, 2, 1389–1397.
- Park, S.A.; Ahn, S.I.; Gallo, J.M. Tau mis-splicing in the pathogenesis of neurodegenerative disorders. BMB Rep. 2016, 49, 405–413.
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764.
- Kovacech, B.; Skrabana, R.; Novak, M. Transition of tau protein from disordered to misordered in Alzheimer’s disease. Neurodegener. Dis. 2010, 7, 24–27.
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159.
- Josephs, K.A.; Hodges, J.R.; Snowden, J.S.; Mackenzie, I.R.; Neumann, M.; Mann, D.M.; Dickson, D.W. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 2011, 122, 137–153.
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766.
- Boxer, A.L.; Yu, J.T.; Golbe, L.I.; Litvan, I.; Lang, A.E.; Hoglinger, G.U. Advances in progressive supranuclear palsy: New diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. 2017, 16, 552–563.
- Irwin, D.J.; Brettschneider, J.; McMillan, C.T.; Cooper, F.; Olm, C.; Arnold, S.E.; Van Deerlin, V.M.; Seeley, W.W.; Miller, B.L.; Lee, E.B.; et al. Deep clinical and neuropathological phenotyping of Pick disease. Ann. Neurol. 2016, 79, 272–287.
- Rankin, K.P.; Mayo, M.C.; Seeley, W.W.; Lee, S.; Rabinovici, G.; Gorno-Tempini, M.L.; Boxer, A.L.; Weiner, M.W.; Trojanowski, J.Q.; DeArmond, S.J.; et al. Behavioral variant frontotemporal dementia with corticobasal degeneration pathology: Phenotypic comparison to bvFTD with Pick’s disease. J. Mol. Neurosci. 2011, 45, 594–608.
- Dickson, D.W.; Yen, S.H.; Suzuki, K.I.; Davies, P.; Garcia, J.H.; Hirano, A. Ballooned neurons in select neurodegenerative diseases contain phosphorylated neurofilament epitopes. Acta Neuropathol. 1986, 71, 216–223.
- Braak, H.; Braak, E. Argyrophilic grain disease: Frequency of occurrence in different age categories and neuropathological diagnostic criteria. J. Neural Transm. 1998, 105, 801–819.
- Armstrong, M.J.; Litvan, I.; Lang, A.E.; Bak, T.H.; Bhatia, K.P.; Borroni, B.; Boxer, A.L.; Dickson, D.W.; Grossman, M.; Hallett, M.; et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013, 80, 496–503.
- Day, G.S.; Lim, T.S.; Hassenstab, J.; Goate, A.M.; Grant, E.A.; Roe, C.M.; Cairns, N.J.; Morris, J.C. Differentiating cognitive impairment due to corticobasal degeneration and Alzheimer disease. Neurology 2017, 88, 1273–1281.
- Hoglinger, G.U.; Respondek, G.; Stamelou, M.; Kurz, C.; Josephs, K.A.; Lang, A.E.; Mollenhauer, B.; Muller, U.; Nilsson, C.; Whitwell, J.L.; et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov. Disord. 2017, 32, 853–864.
- Spinelli, E.G.; Mandelli, M.L.; Miller, Z.A.; Santos-Santos, M.A.; Wilson, S.M.; Agosta, F.; Grinberg, L.T.; Huang, E.J.; Trojanowski, J.Q.; Meyer, M.; et al. Typical and atypical pathology in primary progressive aphasia variants. Ann. Neurol. 2017, 81, 430–443.
- Kovacs, G.G.; Robinson, J.L.; Xie, S.X.; Lee, E.B.; Grossman, M.; Wolk, D.A.; Irwin, D.J.; Weintraub, D.; Kim, C.F.; Schuck, T.; et al. Evaluating the Patterns of Aging-Related Tau Astrogliopathy Unravels Novel Insights into Brain Aging and Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2017, 76, 270–288.
- Rodriguez, R.D.; Suemoto, C.K.; Molina, M.; Nascimento, C.F.; Leite, R.E.; de Lucena Ferretti-Rebustini, R.E.; Farfel, J.M.; Heinsen, H.; Nitrini, R.; Ueda, K.; et al. Argyrophilic Grain Disease: Demographics, Clinical, and Neuropathological Features from a Large Autopsy Study. J. Neuropathol. Exp. Neurol. 2016, 75, 628–635.
- Arena, J.D.; Smith, D.H.; Lee, E.B.; Gibbons, G.S.; Irwin, D.J.; Robinson, J.L.; Lee, V.M.; Trojanowski, J.Q.; Stewart, W.; Johnson, V.E. Tau immunophenotypes in chronic traumatic encephalopathy recapitulate those of ageing and Alzheimer’s disease. Brain 2020, 143, 1572–1587.
- Hutton, M. Molecular genetics of chromosome 17 tauopathies. Ann. N. Y. Acad. Sci. 2000, 920, 63–73.
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21.
- Furman, J.L.; Vaquer-Alicea, J.; White, C.L., 3rd; Cairns, N.J.; Nelson, P.T.; Diamond, M.I. Widespread tau seeding activity at early Braak stages. Acta Neuropathol. 2017, 133, 91–100.
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349.
- Kuret, J.; Congdon, E.E.; Li, G.; Yin, H.; Yu, X.; Zhong, Q. Evaluating triggers and enhancers of tau fibrillization. Microsc. Res. Tech. 2005, 67, 141–155.
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247.
- von Bergen, M.; Barghorn, S.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim. Biophys. Acta 2005, 1739, 158–166.
- Sillen, A.; Leroy, A.; Wieruszeski, J.M.; Loyens, A.; Beauvillain, J.C.; Buee, L.; Landrieu, I.; Lippens, G. Regions of tau implicated in the paired helical fragment core as defined by NMR. Chembiochem 2005, 6, 1849–1856.
- Kovacs, G.G.; Lutz, M.I.; Ricken, G.; Strobel, T.; Hoftberger, R.; Preusser, M.; Regelsberger, G.; Honigschnabl, S.; Reiner, A.; Fischer, P.; et al. Dura mater is a potential source of Abeta seeds. Acta Neuropathol. 2016, 131, 911–923.
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622.
- Mez, J.; Daneshvar, D.H.; Kiernan, P.T.; Abdolmohammadi, B.; Alvarez, V.E.; Huber, B.R.; Alosco, M.L.; Solomon, T.M.; Nowinski, C.J.; McHale, L.; et al. Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. JAMA 2017, 318, 360–370.
- Jadhav, S.; Avila, J.; Scholl, M.; Kovacs, G.G.; Kovari, E.; Skrabana, R.; Evans, L.D.; Kontsekova, E.; Malawska, B.; de Silva, R.; et al. A walk through tau therapeutic strategies. Acta Neuropathol. Commun. 2019, 7, 22.
- Khanna, M.R.; Kovalevich, J.; Lee, V.M.; Trojanowski, J.Q.; Brunden, K.R. Therapeutic strategies for the treatment of tauopathies: Hopes and challenges. Alzheimers Dement. 2016, 12, 1051–1065.
- Feinstein, S.C.; Wilson, L. Inability of tau to properly regulate neuronal microtubule dynamics: A loss-of-function mechanism by which tau might mediate neuronal cell death. Biochim. Biophys. Acta 2005, 1739, 268–279.
- Trojanowski, J.Q.; Lee, V.M. Pathological tau: A loss of normal function or a gain in toxicity? Nat. Neurosci. 2005, 8, 1136–1137.
- Cripps, D.; Thomas, S.N.; Jeng, Y.; Yang, F.; Davies, P.; Yang, A.J. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J. Biol. Chem. 2006, 281, 10825–10838.
- Ramachandran, G.; Udgaonkar, J.B. Mechanistic studies unravel the complexity inherent in tau aggregation leading to Alzheimer’s disease and the tauopathies. Biochemistry 2013, 52, 4107–4126.
- Tepper, K.; Biernat, J.; Kumar, S.; Wegmann, S.; Timm, T.; Hubschmann, S.; Redecke, L.; Mandelkow, E.M.; Muller, D.J.; Mandelkow, E. Oligomer formation of tau protein hyperphosphorylated in cells. J. Biol. Chem. 2014, 289, 34389–34407.

